
RT-qPCR Detection of SARS-CoV-2: No Need for a Dedicated Reverse Transcription Step

 ,
,
Abstract
:1. Introduction
2. Results
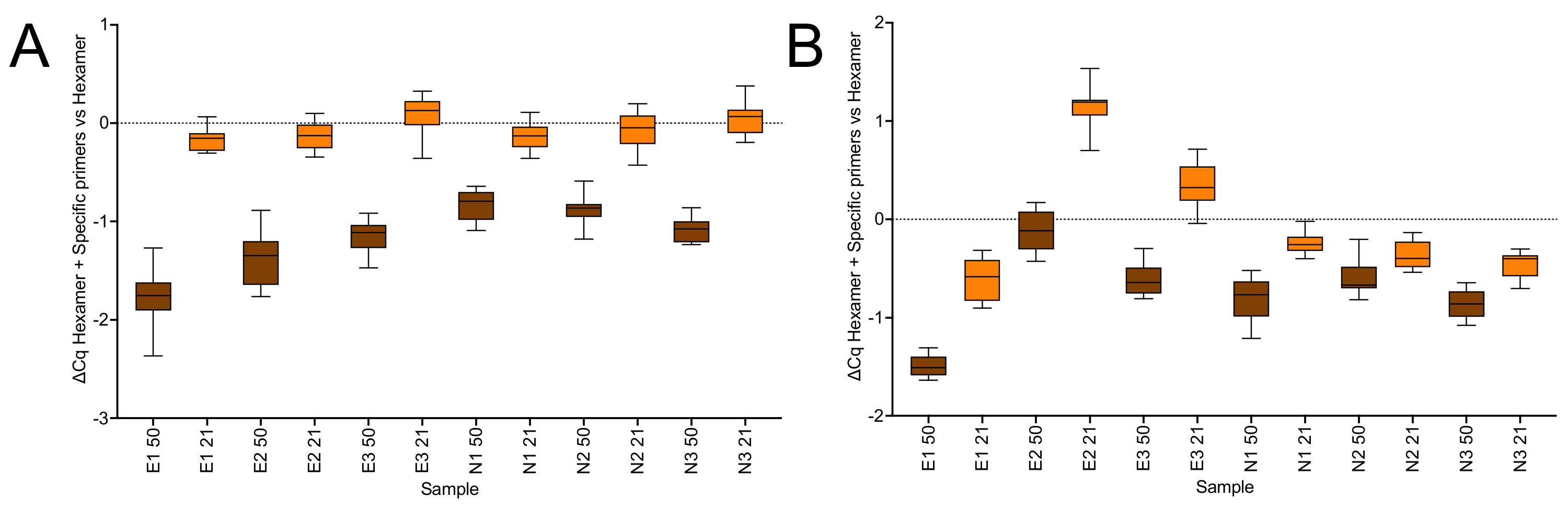
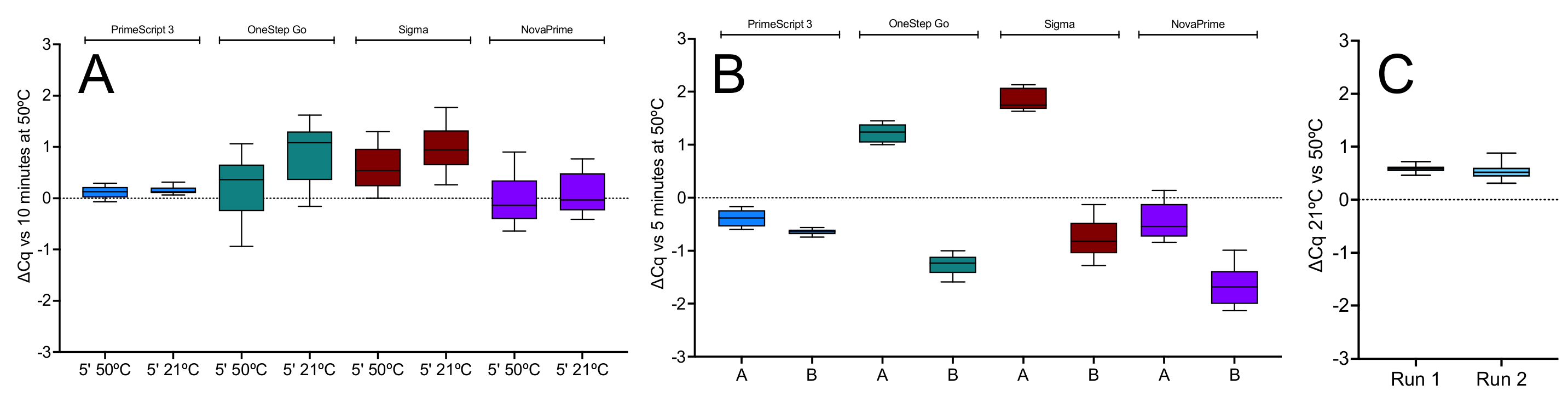
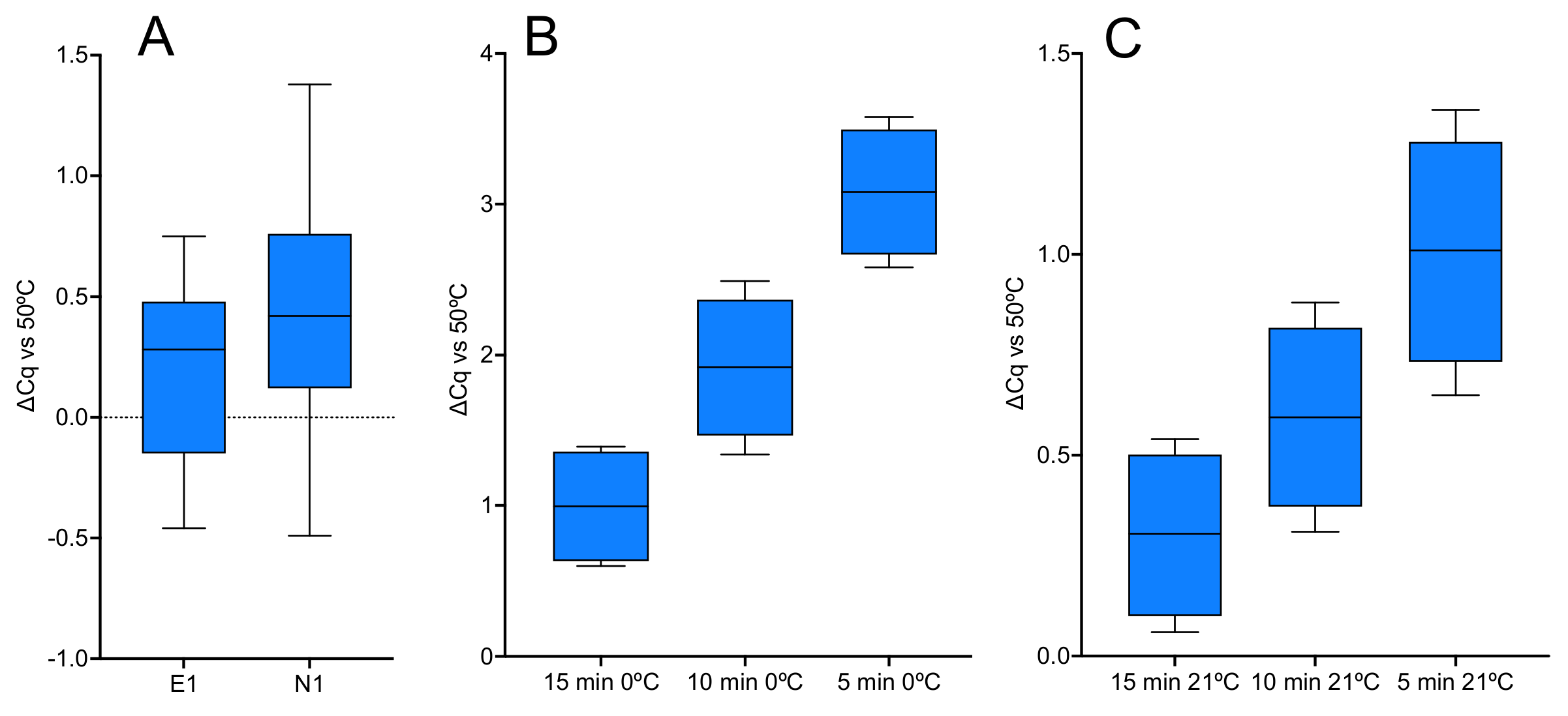
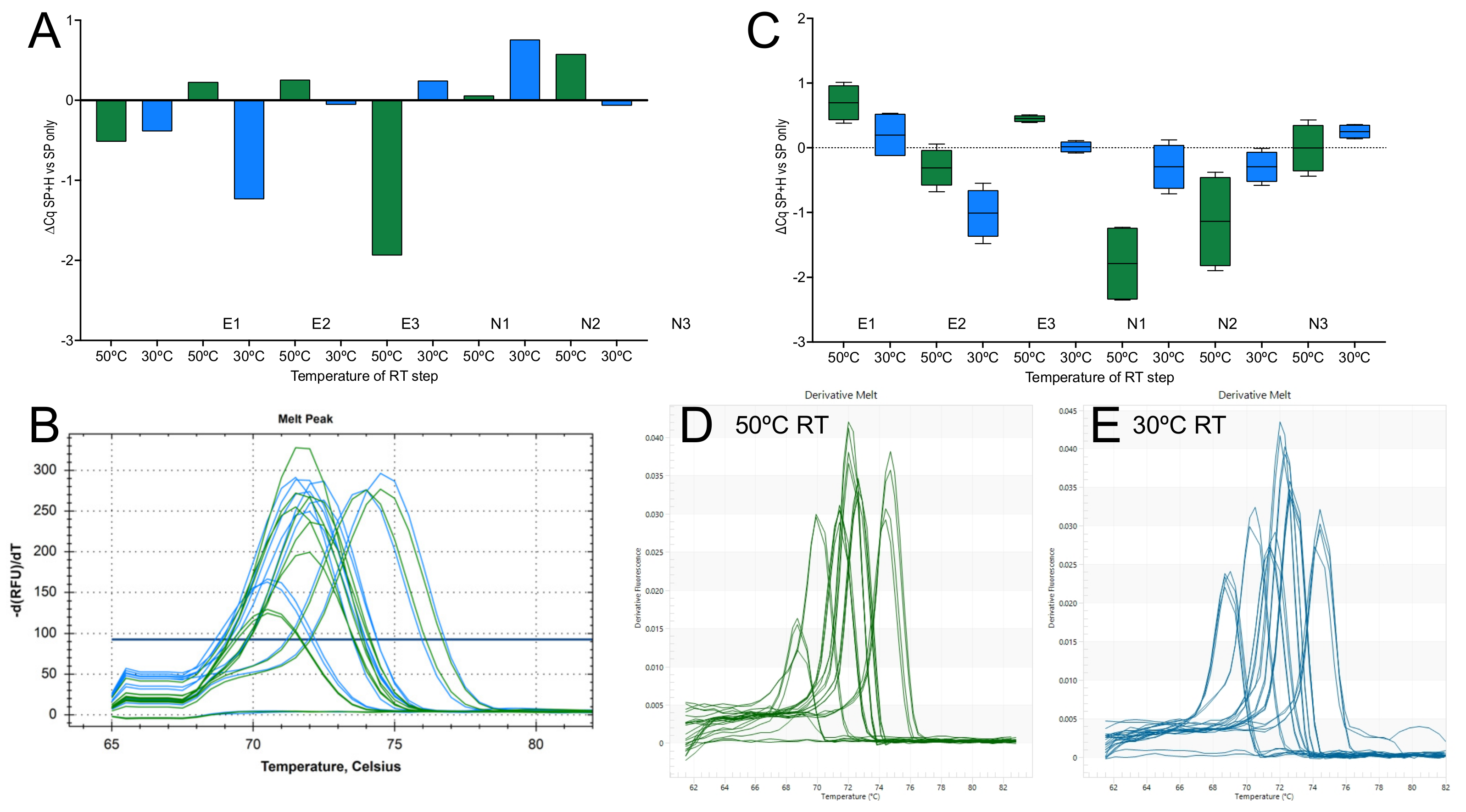
2.1. 2-Step RT-qPCR
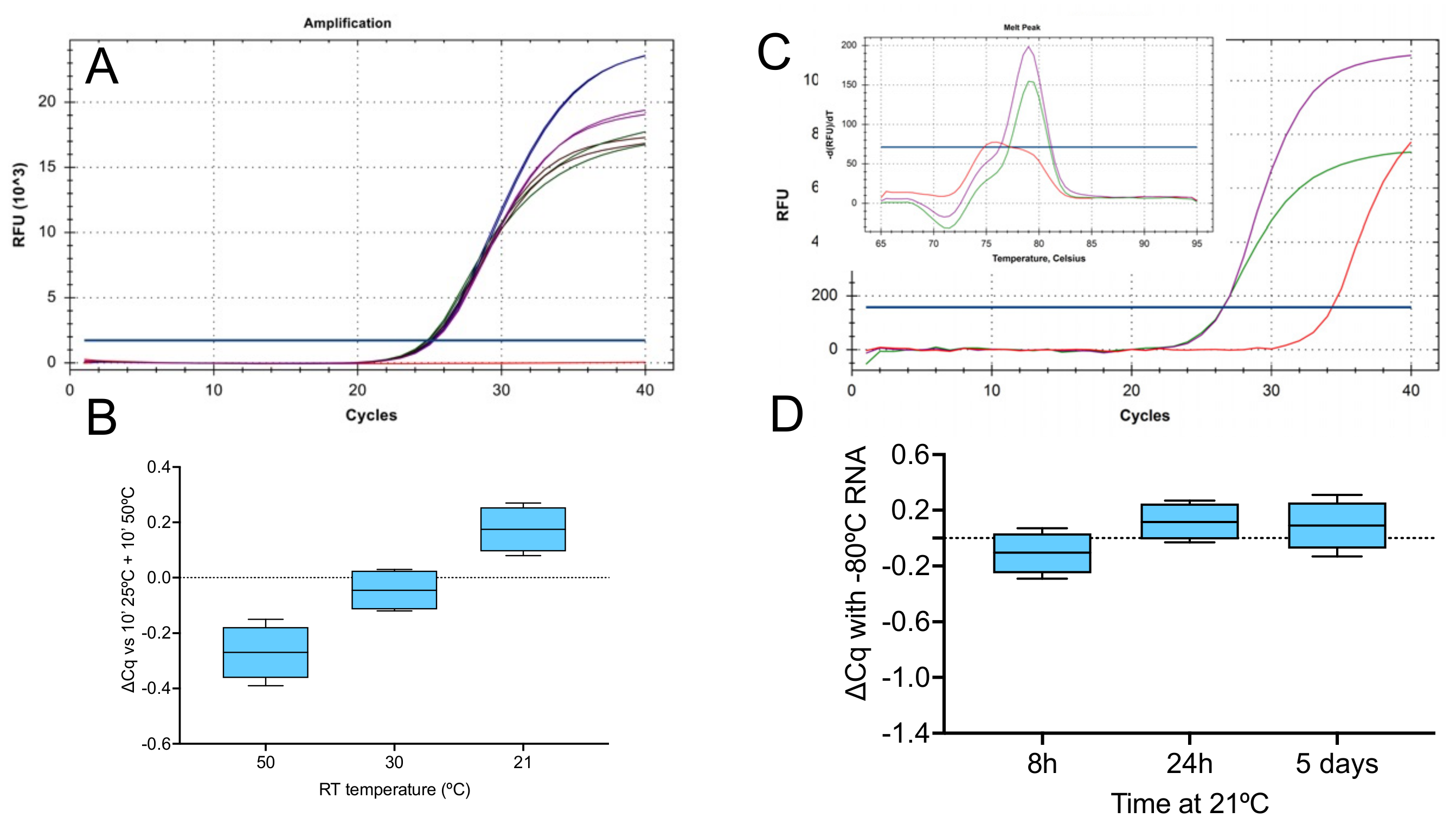
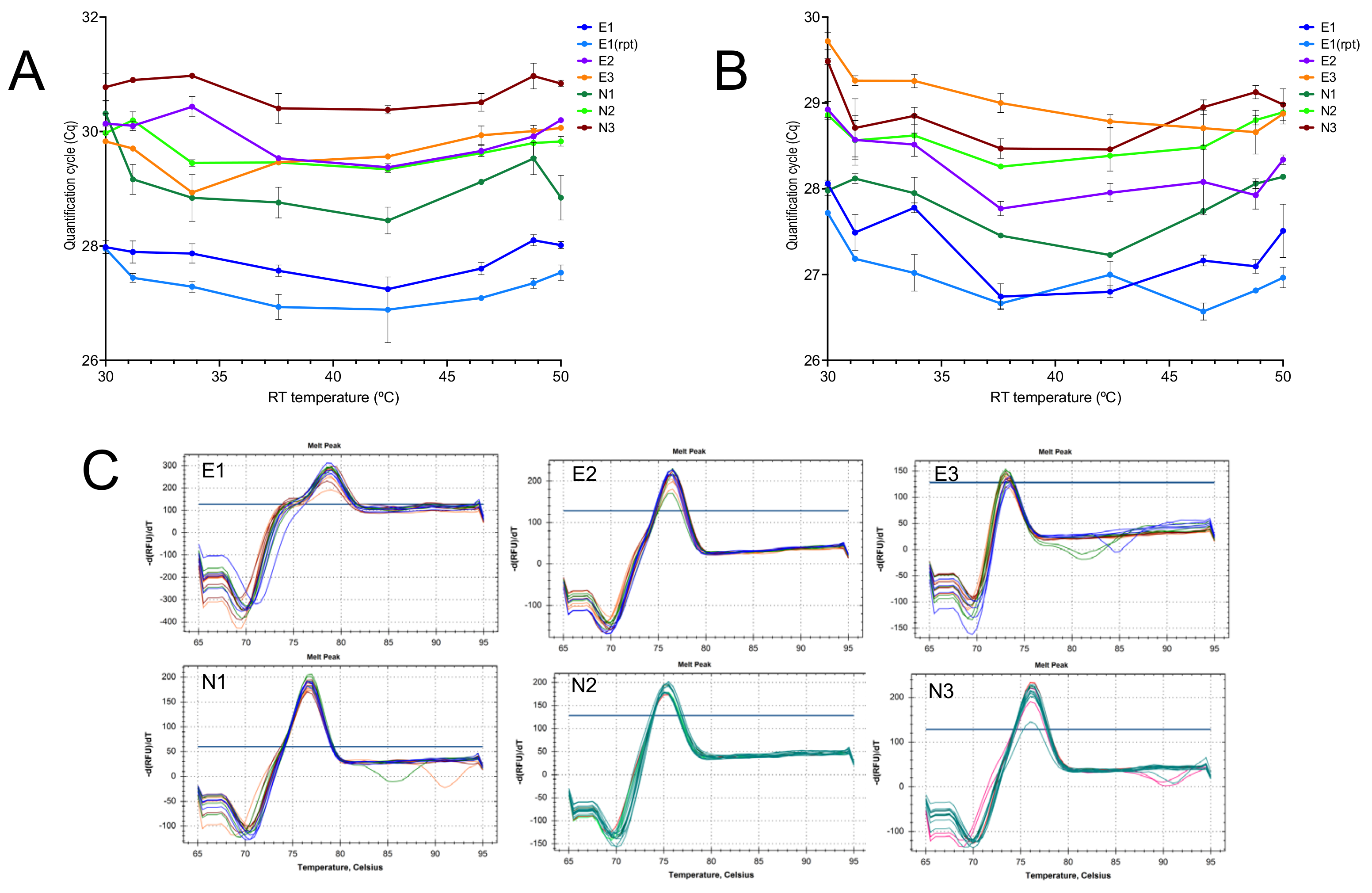
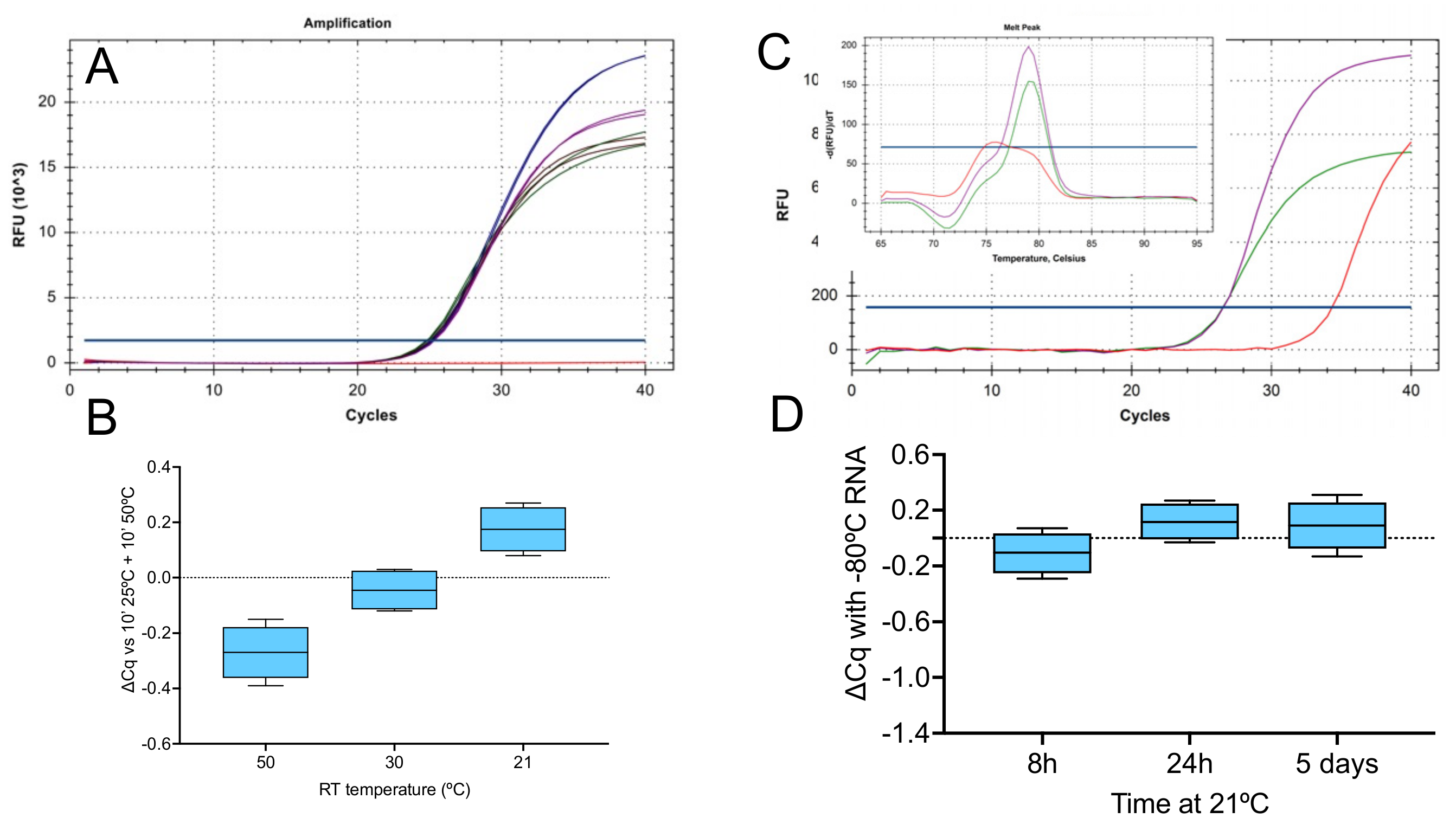
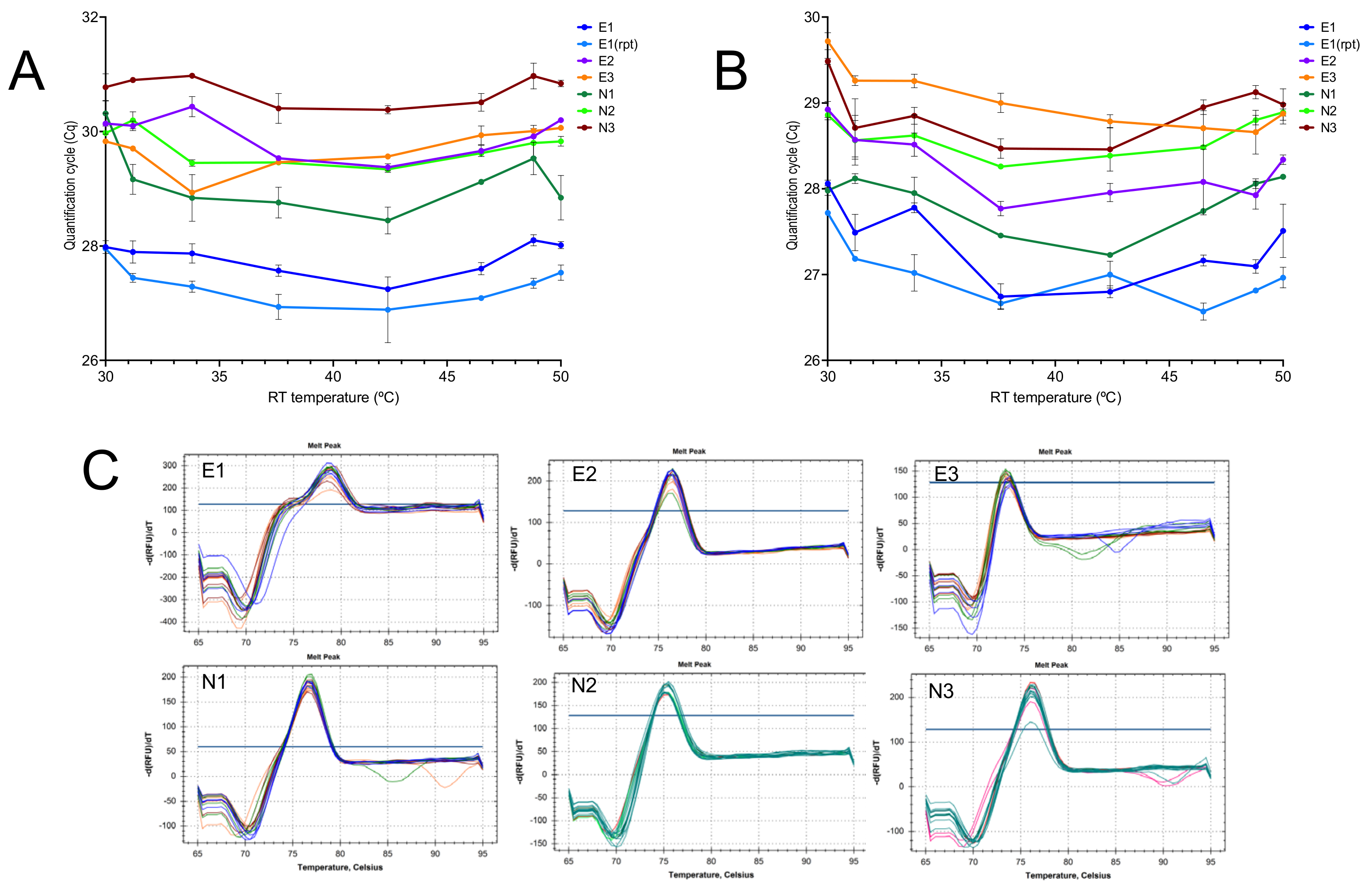
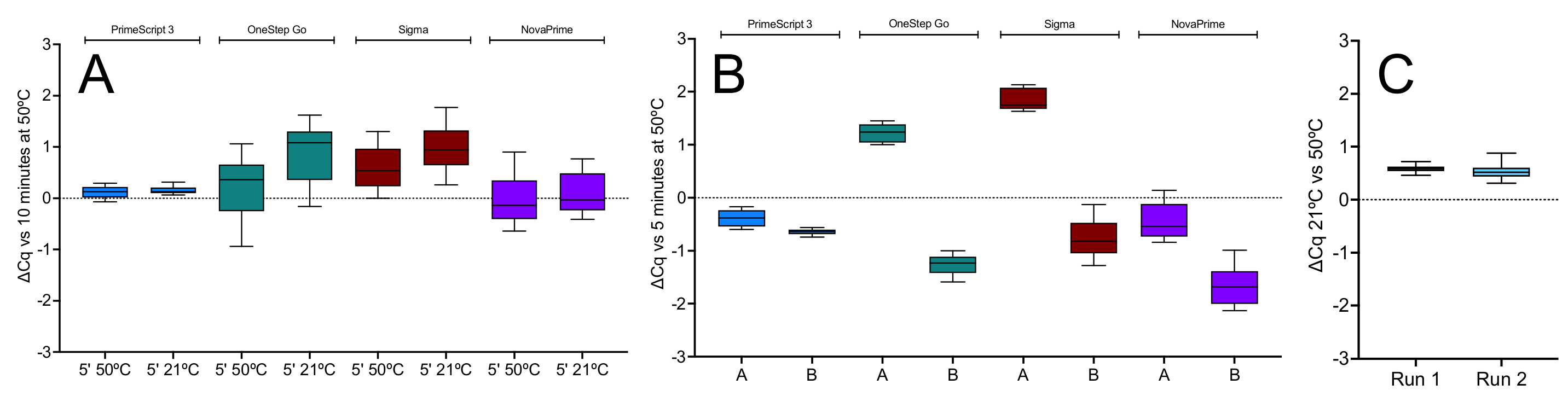
2.1.1. Effect of Temperature
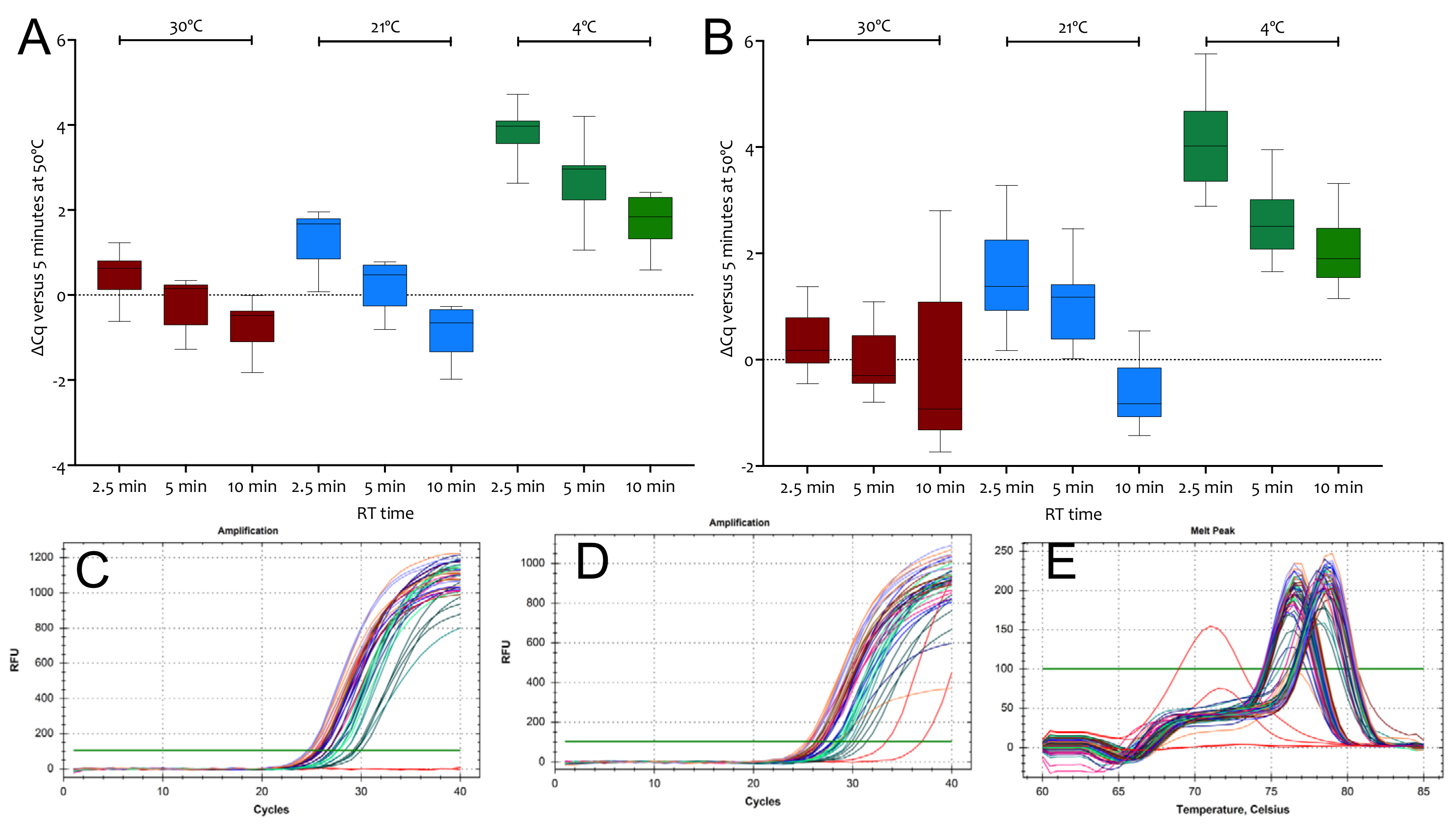
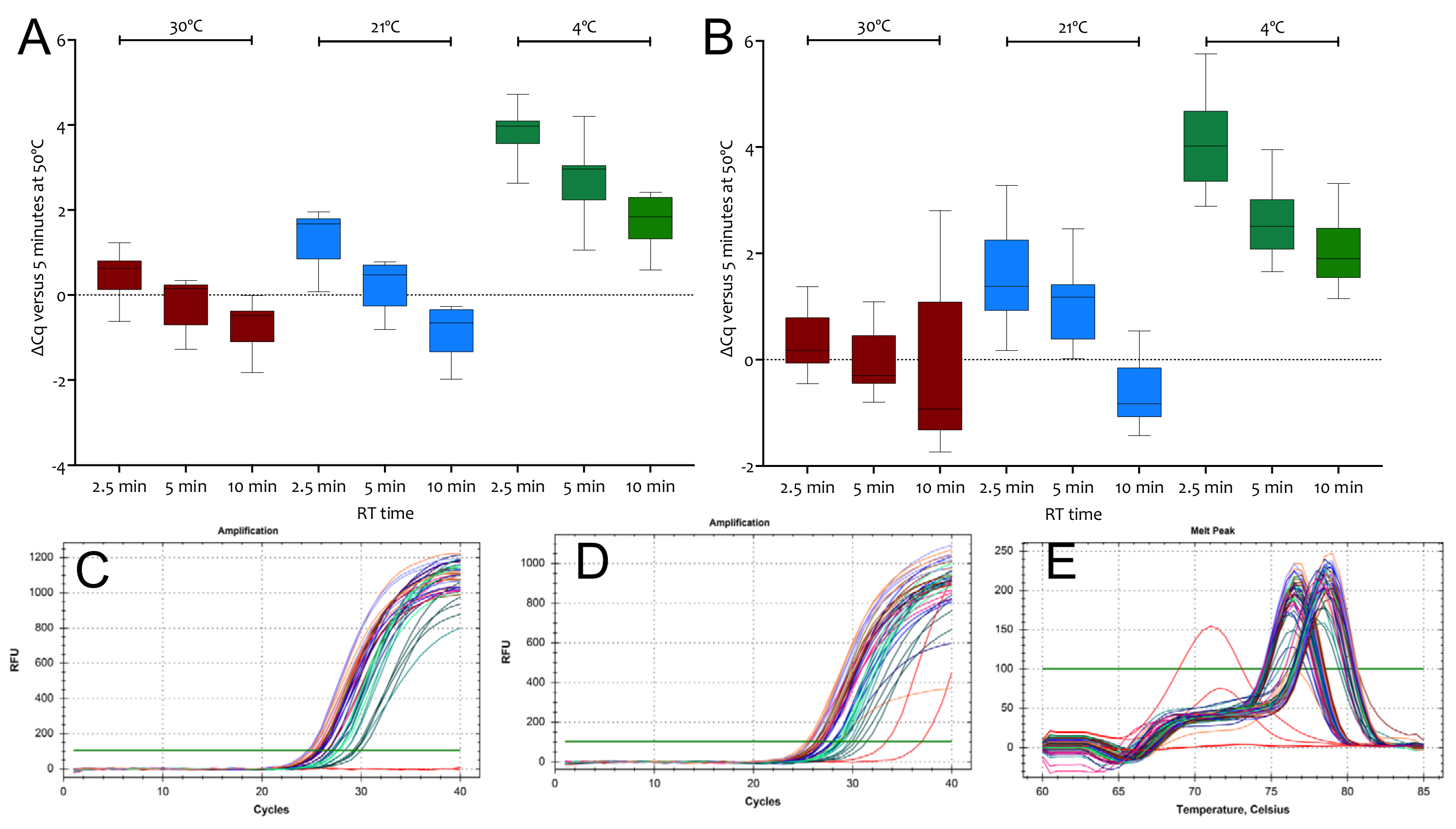
2.1.2. Priming on Ice
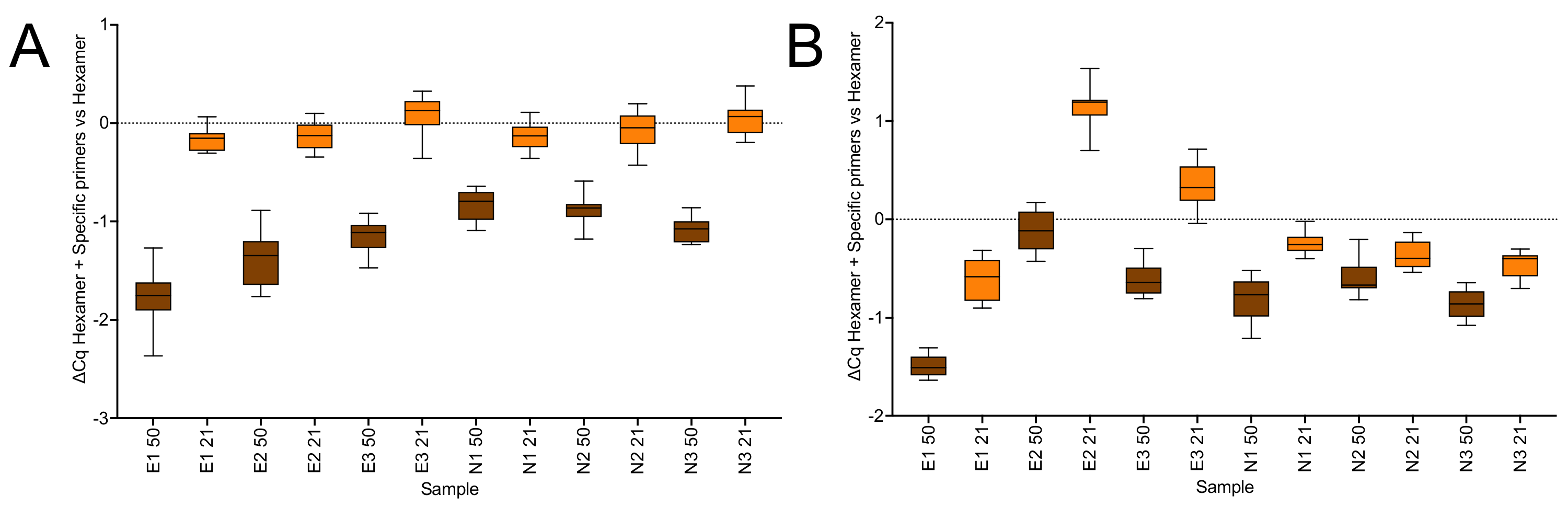
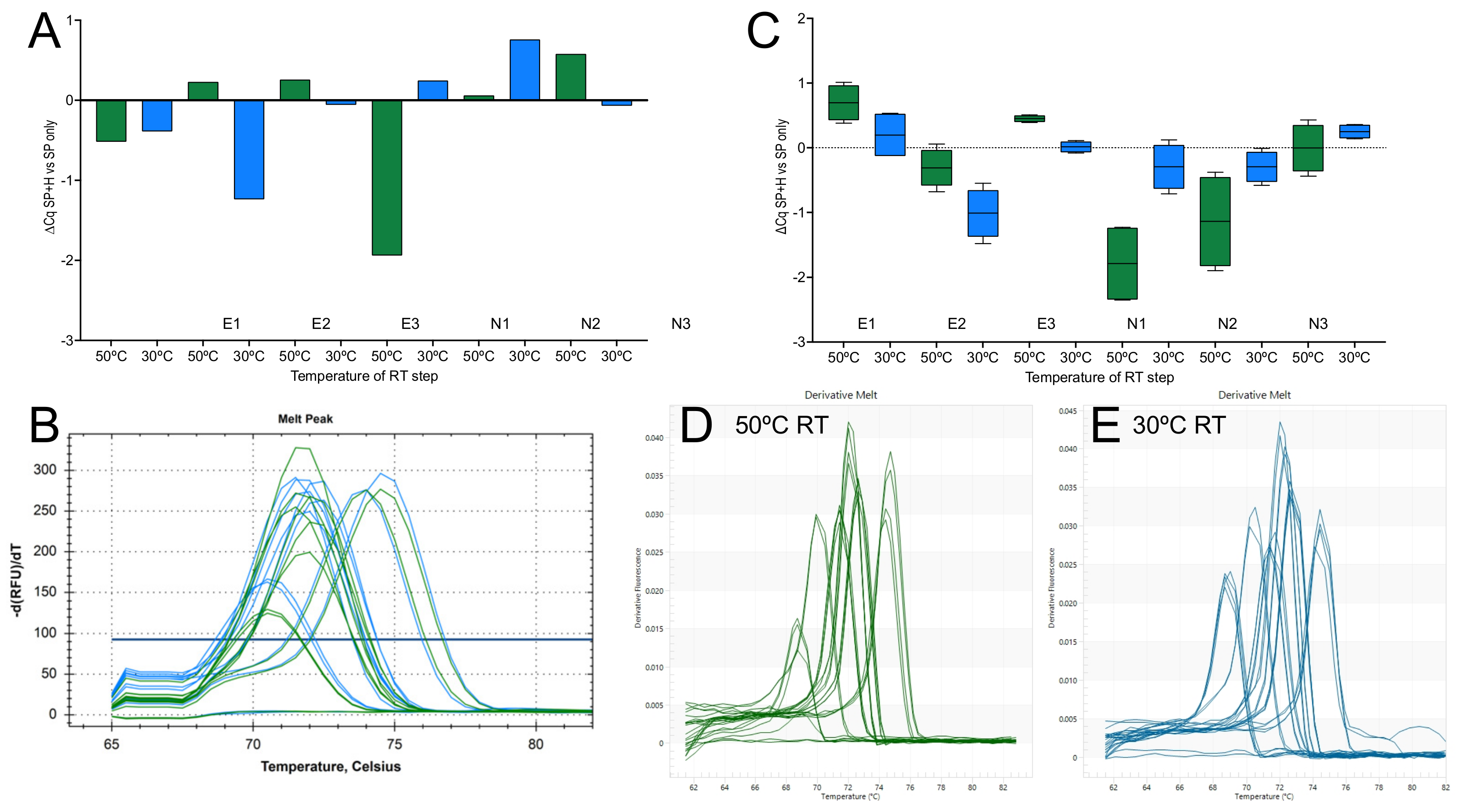
2.1.3. Priming Effects
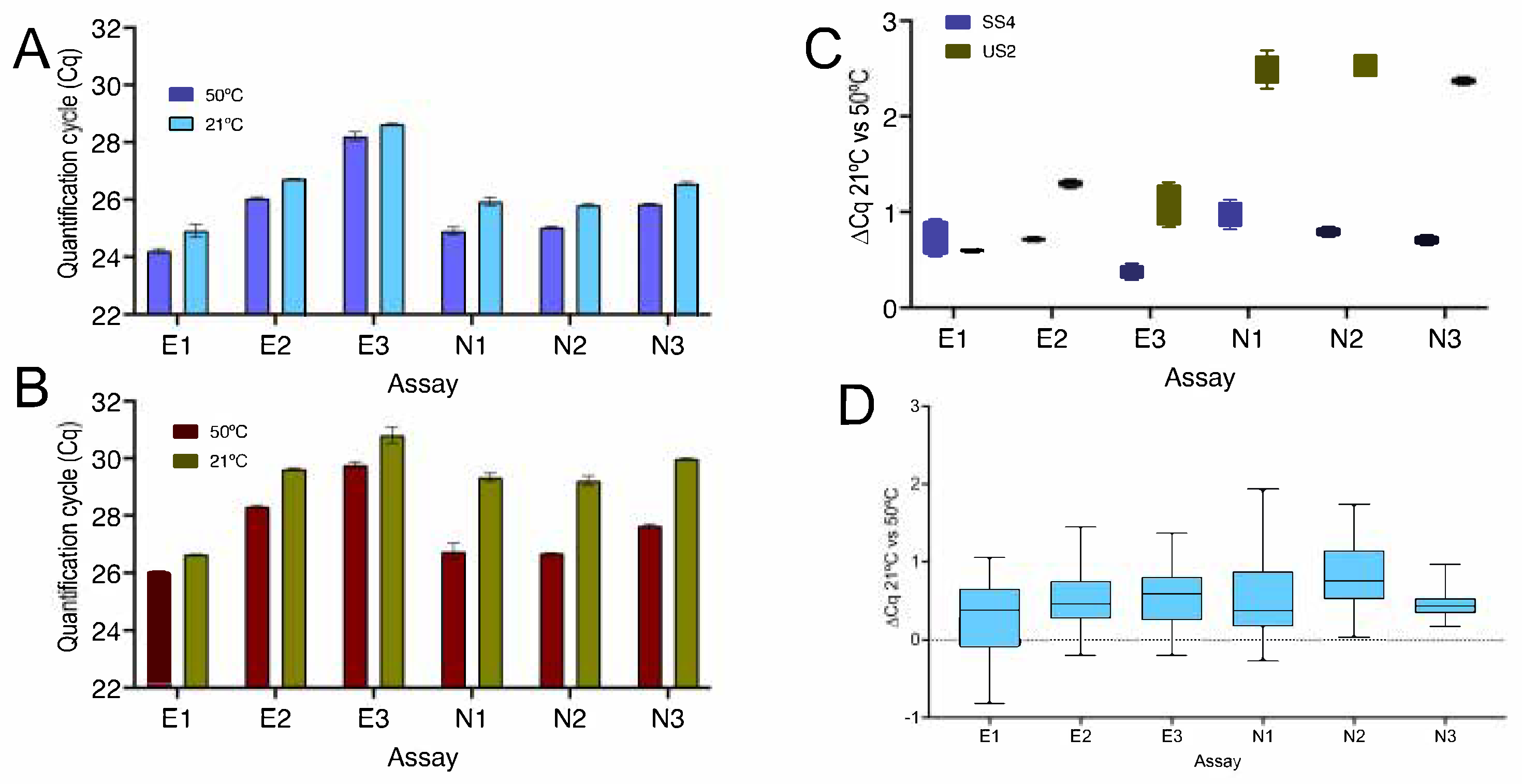
2.2. 1-Step RT-qPCR Cqs
3. Discussion
4. Materials and Methods
4.1. Instruments, Reagents and Analysis
4.2. RNA
4.3. Primer and Probe Design
4.4. 2-Step RT-qPCR Reactions
4.5. 1-Step RT-qPCR Reactions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baltimore, D. RNA-dependent DNA Polymerase in Virions of RNA Tumour Viruses. Nature 1970, 226, 1209–1211. [Google Scholar] [CrossRef] [PubMed]
- Temin, H.M.; Mizutani, S. RNA-dependent DNA Polymerase in Virions of Rous Sarcoma Virus. Nature 1970, 226, 1211–1213. [Google Scholar] [CrossRef] [PubMed]
- Chien, A.; Edgar, D.B.; Trela, J.M. Deoxyribonucleic acid polymerase from the extreme thermophile Thermus aquaticus. J. Bacteriol. 1976, 127, 1550–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleppe, K.; Ohtsuka, E.; Kleppe, R.; Molineux, I.; Khorana, H.G. Studies on polynucleotides. XCVI. Repair replications of short synthetic DNA’s as catalyzed by DNA polymerases. J. Mol. Biol. 1971, 56, 341–361. [Google Scholar] [CrossRef]
- Saiki, R.K.; Scharf, S.; Faloona, F.; Mullis, K.B.; Horn, G.T.; Erlich, H.A.; Arnheim, N. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 1985, 230, 1350–1354. [Google Scholar] [CrossRef]
- Mullis, K.; Faloona, F.; Scharf, S.; Saiki, R.; Horn, G.; Erlich, H. Specific enzymatic amplification of DNA in vitro: The polymerase chain reaction. Cold Spring Harb. Symp. Quant. Biol. 1986, 51 Pt 1, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Powell, L.M.; Wallis, S.C.; Pease, R.J.; Edwards, Y.H.; Knott, T.J.; Scott, J. A novel form of tissue-specific RNA processing produces apolipoprotein-B48 in intestine. Cell 1987, 50, 831–840. [Google Scholar] [CrossRef]
- Vrieling, H.; Simons, J.; van Zeeland, A. Nucleotide sequence determination of point mutations at the mouse HPRT locus using in vitro amplification of HPRT mRNA sequences. Mutat. Res. Mol. Mech. Mutagen. 1988, 198, 107–113. [Google Scholar] [CrossRef]
- Koos, R.D.; Olson, C.E. Expression of Basic Fibroblast Growth Factor in the Rat Ovary: Detection of mRNA Using Reverse Transcription-Polymerase Chain Reaction Amplification. Mol. Endocrinol. 1989, 3, 2041–2048. [Google Scholar] [CrossRef] [Green Version]
- Deng, K.; Uhlig, S.; Ip, H.S.; Killian, M.L.; Goodman, L.B.; Nemser, S.; Ulaszek, J.; Pickens, S.; Newkirk, R.; Kmet, M.; et al. Interlaboratory comparison of SARS-CoV2 molecular detection assays in use by U.S. veterinary diagnostic laboratories. J. Vet. Diagn. Investig. 2021, 33, 1039–1051. [Google Scholar] [CrossRef]
- Laconi, A.; Fortin, A.; Bedendo, G.; Shibata, A.; Sakoda, Y.; Awuni, J.A.; Go-Maro, E.; Arafa, A.; Ali, A.S.M.; Terregino, C.; et al. Detection of avian influenza virus: A comparative study of the in silico and in vitro performances of current RT-qPCR assays. Sci. Rep. 2020, 10, 8441. [Google Scholar] [CrossRef]
- Zhao, F.; Maren, N.A.; Kosentka, P.Z.; Liao, Y.-Y.; Lu, H.; Duduit, J.R.; Huang, D.; Ashrafi, H.; Zhao, T.; Huerta, A.I.; et al. An optimized protocol for stepwise optimization of real-time RT-PCR analysis. Hortic. Res. 2021, 8, 179. [Google Scholar] [CrossRef]
- Al-Ramadhani, S.; Sai-Giridhar, P.; George, D.; Gopinath, P.; Arkoumani, E.; Jader, S.; Sundaresan, M.; Salgado, R.; Larsimont, D.; Bustin, S.A.; et al. Metasin—An Intra-Operative RT-qPCR Assay to Detect Metastatic Breast Cancer in Sentinel Lymph Nodes. Int. J. Mol. Sci. 2013, 14, 12931–12952. [Google Scholar] [CrossRef] [Green Version]
- Bustin, S.A.; Nolan, T. RT-qPCR Testing of SARS-CoV-2: A Primer. Int. J. Mol. Sci. 2020, 21, 3004. [Google Scholar] [CrossRef]
- Wong, C.L.; Yong, C.Y.; Ong, H.K.; Ho, K.L.; Tan, W.S. Advances in the Diagnosis of Foot-and-Mouth Disease. Front. Vet. Sci. 2020, 7, 477. [Google Scholar] [CrossRef]
- Bustin, S.; Coward, A.; Sadler, G.; Teare, L.; Nolan, T. CoV2-ID, a MIQE-compliant sub-20-min 5-plex RT-PCR assay targeting SARS-CoV-2 for the diagnosis of COVID-19. Sci. Rep. 2020, 10, 22214. [Google Scholar] [CrossRef]
- Mallet, F.; Oriol, G.; Mary, C.; Verrier, B.; Mandrand, B. Continuous RT-PCR using AMV-RT and Taq DNA polymerase: Characterization and comparison to uncoupled procedures. BioTechniques 1995, 18, 678–687. [Google Scholar]
- Bustin, S.; Dhillon, H.S.; Kirvell, S.; Greenwood, C.; Parker, M.; Shipley, G.L.; Nolan, T. Variability of the Reverse Transcription Step: Practical Implications. Clin. Chem. 2015, 61, 202–212. [Google Scholar] [CrossRef] [Green Version]
- Zucha, D.; Androvic, P.; Kubista, M.; Valihrach, L. Performance Comparison of Reverse Transcriptases for Single-Cell Studies. Clin. Chem. 2019, 66, 217–228. [Google Scholar] [CrossRef]
- Malboeuf, C.M.; Isaacs, S.J.; Tran, N.H.; Kim, B. Thermal Effects on Reverse Transcription: Improvement of Accuracy and Processivity in cDNA Synthesis. BioTechniques 2001, 30, 1074–1084. [Google Scholar] [CrossRef] [Green Version]
- Rejali, N.A.; Zuiter, A.M.; Quackenbush, J.F.; Wittwer, C.T. Reverse transcriptase kinetics for one-step RT-PCR. Anal. Biochem. 2020, 601, 113768. [Google Scholar] [CrossRef] [PubMed]
- Baranauskas, A.; Paliksa, S.; Alzbutas, G.; Vaitkevicius, M.; Lubiene, J.; Letukiene, V.; Burinskas, S.; Sasnauskas, G.; Skirgaila, R. Generation and characterization of new highly thermostable and processive M-MuLV reverse transcriptase variants. Protein Eng. Des. Sel. 2012, 25, 657–668. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.D.; Bebenek, K.; Kunkel, T.A. The Accuracy of Reverse Transcriptase from HIV-1. Science 1988, 242, 1171–1173. [Google Scholar] [CrossRef]
- Yasukawa, K.; Yanagihara, I.; Fujiwara, S. Alteration of enzymes and their application to nucleic acid amplification (Review). Int. J. Mol. Med. 2020, 46, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Arezi, B.; Hogrefe, H. Novel mutations in Moloney Murine Leukemia Virus reverse transcriptase increase thermostability through tighter binding to template-primer. Nucleic Acids Res. 2008, 37, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Yasukawa, K.; Mizuno, M.; Konishi, A.; Inouye, K. Increase in thermal stability of Moloney murine leukaemia virus reverse transcriptase by site-directed mutagenesis. J. Biotechnol. 2010, 150, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, C.; Chung, S.; Crissy, K.; Dumas, K.; Schuster, D.; Schoenfeld, T.W. Engineering of a thermostable viral polymerase using metagenome-derived diversity for highly sensitive and specific RT-PCR. Nucleic Acids Res. 2019, 47, 3619–3630. [Google Scholar] [CrossRef] [Green Version]
- Battula, N.; Loeb, L.A. The Infidelity of Avian Myeloblastosis Virus Deoxyribonucleic Acid Polymerase in Polynucleotide Replication. J. Biol. Chem. 1974, 249, 4086–4093. [Google Scholar] [CrossRef]
- Verma, I.M.; Baltimore, D. Purification of the RNA-directed DNA polymerase from avian myeloblastosis virus and its assay with polynucleotide templates. Methods Enzymol. 1974, 29, 125–130. [Google Scholar] [CrossRef]
- Myers, T.W.; Gelfand, D.H. Reverse transcription and DNA amplification by a Thermus thermophilus DNA polymerase. Biochemistry 1991, 30, 7661–7666. [Google Scholar] [CrossRef]
- Nakura, Y.; Wu, H.N.; Okamoto, Y.; Takeuchi, M.; Suzuki, K.; Tamura, Y.; Oba, Y.; Nishiumi, F.; Hatori, N.; Fujiwara, S.; et al. Development of an efficient one-step real-time reverse transcription polymerase chain reaction method for severe acute respiratory syndrome-coronavirus-2 detection. PLoS ONE 2021, 16, e0252789. [Google Scholar] [CrossRef]
- Bhadra, S.; Maranhao, A.C.; Paik, I.; Ellington, A.D. One-Enzyme Reverse Transcription qPCR Using Taq DNA Polymerase. Biochemistry 2020, 59, 4638–4645. [Google Scholar] [CrossRef]
- Carter, J.G.; Iturbe, L.O.; Duprey, J.-L.H.A.; Carter, I.R.; Southern, D.D.; Rana, M.; Whalley, C.M.; Bosworth, A.; Beggs, A.D.; Hicks, M.R. Ultrarapid detection of SARS-CoV-2 RNA using a reverse transcription–free exponential amplification reaction, RTF-EXPAR. Proc. Natl. Acad. Sci. USA 2021, 118, e2100347118. [Google Scholar] [CrossRef]
- Brooks, E.M.; Sheflin, L.G.; Spaulding, S.W. Secondary structure in the 3′ UTR of EGF and the choice of reverse transcriptases affect the detection of message diversity by RT-PCR. BioTechniques 1995, 19, 806–812. [Google Scholar]
- Kuo, K.-W.; Leung, M.F.; Leung, W.-C. Intrinsic secondary structure of human TNFR-I mRNA influences the determination of gene expression by RT-PCR. Mol. Cell. Biochem. 1997, 177, 1–6. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Pan, H.-Y.; Gao, S.-J. Reverse Transcription Slippage over the mRNA Secondary Structure of the LIP1 Gene. BioTechniques 2001, 31, 1286–1294. [Google Scholar] [CrossRef]
- Oisen, D.B.; Carroll, S.S.; Culberson, J.; Shafer, J.A.; Kuo, L.C. Effect of template secondary structure on the inhibition of HIV-1 reverse transcriptase by a pyridinone non-nucleoside inhibitor. Nucleic Acids Res. 1994, 22, 1437–1443. [Google Scholar] [CrossRef] [Green Version]
- Robertson, J.M.; Walsh-Weller, J.; Lincoln, P.J.; Thomson, J. An Introduction to PCR Primer Design and Optimization of Amplification Reactions. Forensic DNA Profiling Protoc. 2003, 98, 121–154. [Google Scholar] [CrossRef]
- Bustin, S.; Huggett, J. qPCR primer design revisited. Biomol. Detect. Quantif. 2017, 14, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Byrne-Steele, M.; Wang, C.; Lu, S.; Clemmons, S.; Zahorchak, R.J.; Han, J. DNA polymerase preference determines PCR priming efficiency. BMC Biotechnol. 2014, 14, 10. [Google Scholar] [CrossRef] [Green Version]
- Cao, C.; Cai, Z.; Xiao, X.; Rao, J.; Chen, J.; Hu, N.; Yang, M.; Xing, X.; Wang, Y.; Li, M.; et al. The architecture of the SARS-CoV-2 RNA genome inside virion. Nature communications. Nat. Commun. 2021, 12, 3917. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.; Lorenz, R.; Bernhart, S.H.F.; Neuböck, R.; Hofacker, I.L. The Vienna RNA Websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellaousov, S.; Reuter, J.S.; Seetin, M.G.; Mathews, D.H. RNAstructure: Web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res. 2013, 41, W471–W474. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E-gene 26,305–26,429 (NC_045512.2) | ||||||
|---|---|---|---|---|---|---|
| Assay | Primers (5′-3′) | Tm (°C) | R primer position | FAM Probe (5′-3′) | Tm (°C) | Amplicon (bp) |
| E1-F | CTTGCTTTCGTGGTATTCTTG | 63.5 | actAgcCatCctTactgcg | 71.1 | 69 | |
| E1-R | GCAGTACGCACACAATCG | 65.2 | 26,356–26,373 | |||
| E2-F | CTTCGATTGTGTGCGTAC | 62.0 | tgaGtcTtgTaaAaccttc | 64.9 | 77 | |
| E2-R | ACACGAGAGTAAACGTAAAAAG | 63.0 | 26,408–26,429 | |||
| E3-F | TGCTGCAATATTGTTAAC | 57.0 | 56 | |||
| E3-R | CGAGAGTAAACGTAAAAAG | 57.0 | 26,408–26,426 | |||
| Nsp10 13,025–13,094 (NC_045512.2) | ||||||
| Primers (5′-3′) | Tm (°C) | R primer position | FAM Probe (5′-3′) | Tm (°C) | Amplicon (bp) | |
| N1-F | CTGGTAATGCAACAGAAG | 57.1 | ctgCcaAttCaaCtgta | 65.2 | 69 | |
| N1-R | CAGCATCTACAGCAAAAG | 57.5 | 13,077–13,094 | |||
| N2-F | CTGGTAATGCAACAGAAG | 57.1 | 59 | |||
| N2-R | AGCAAAAGCACAGAAAG | 56.8 | 13,068–13,084 | |||
| N3-F | GCTGGTAATGCAACAGAAGTG | 63.9 | 61 | |||
| N3-R | CAGCAAAAGCACAGAAAGATAAT | 63.7 | 13,063–13,085 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bustin, S.A.; Shipley, G.L.; Kirvell, S.; Mueller, R.; Nolan, T. RT-qPCR Detection of SARS-CoV-2: No Need for a Dedicated Reverse Transcription Step. Int. J. Mol. Sci. 2022, 23, 1303. https://doi.org/10.3390/ijms23031303
Bustin SA, Shipley GL, Kirvell S, Mueller R, Nolan T. RT-qPCR Detection of SARS-CoV-2: No Need for a Dedicated Reverse Transcription Step. International Journal of Molecular Sciences. 2022; 23(3):1303. https://doi.org/10.3390/ijms23031303
Chicago/Turabian StyleBustin, Stephen A., Gregory L. Shipley, Sara Kirvell, Reinhold Mueller, and Tania Nolan. 2022. "RT-qPCR Detection of SARS-CoV-2: No Need for a Dedicated Reverse Transcription Step" International Journal of Molecular Sciences 23, no. 3: 1303. https://doi.org/10.3390/ijms23031303
APA StyleBustin, S. A., Shipley, G. L., Kirvell, S., Mueller, R., & Nolan, T. (2022). RT-qPCR Detection of SARS-CoV-2: No Need for a Dedicated Reverse Transcription Step. International Journal of Molecular Sciences, 23(3), 1303. https://doi.org/10.3390/ijms23031303