
Prion Protein: The Molecule of Many Forms and Faces


{kind=link}
{kind=link}
Abstract
1. Introduction
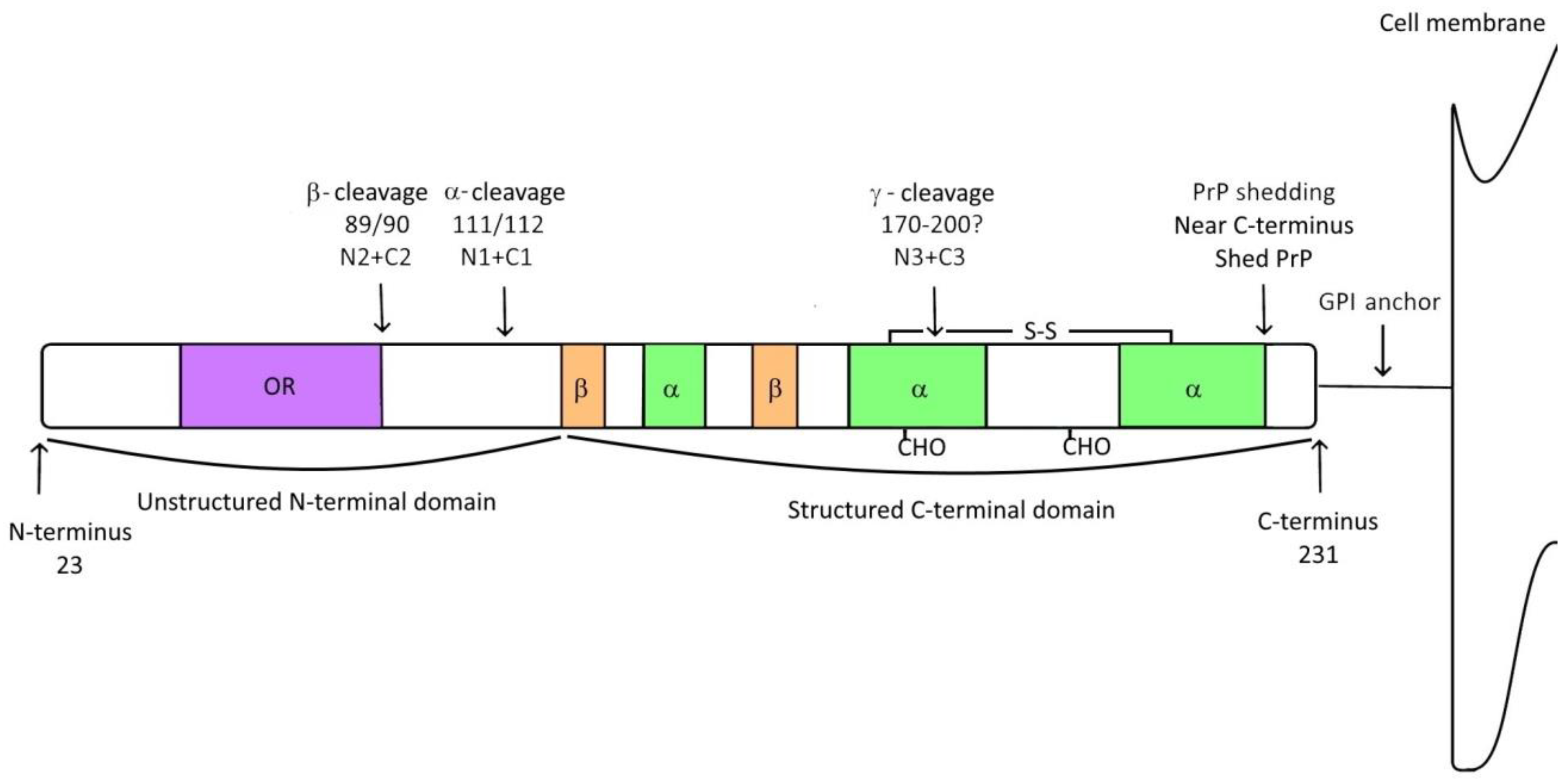
2. Prion Protein
3. Prion Protein Fragments
3.1. α-Cleavage
3.2. β-Cleavage
3.3. γ-Cleavage
3.4. Shedding of Prion Protein
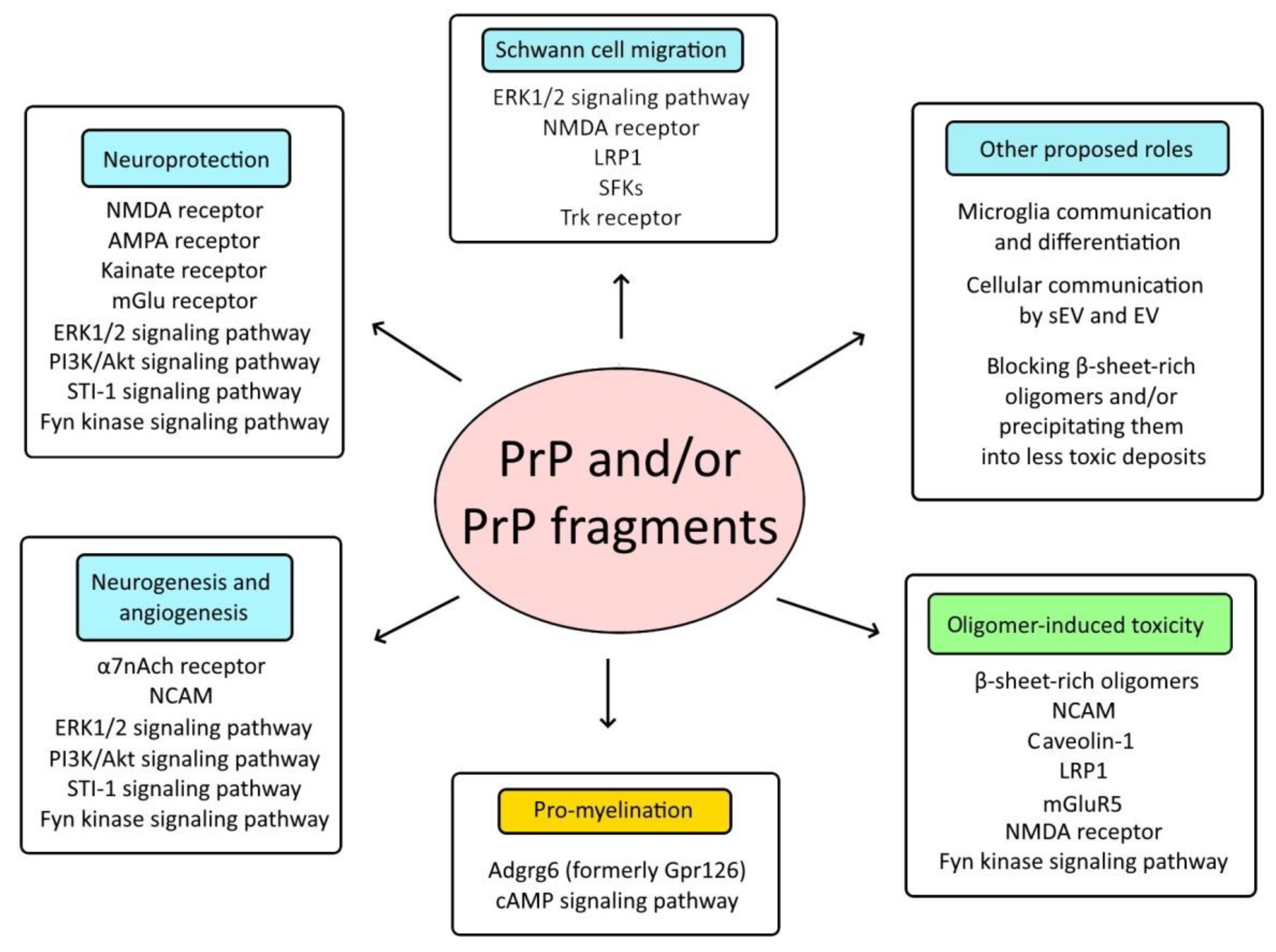
4. Prion Protein and Myelination
5. Prion Protein and Ischemic Strokes
6. Prion Protein and Neurodegeneration
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef] [PubMed]
- O’Carroll, A.; Coyle, J.; Gambin, Y. Prions and Prion-like assemblies in neurodegeneration and immunity: The emergence of universal mechanisms across health and disease. Semin. Cell Dev. Biol. 2020, 99, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.L.; Barria, M.A. Prion Diseases: A Unique Transmissible Agent or a Model for Neurodegenerative Diseases? Biomolecules 2021, 11, 207. [Google Scholar] [CrossRef] [PubMed]
- Herms, J.; Tings, T.; Gall, S.; Madlung, A.; Giese, A.; Siebert, H.; Schurmann, P.; Windl, O.; Brose, N.; Kretzschmar, H. Evidence of presynaptic location and function of the prion protein. J. Neurosci. 1999, 19, 8866–8875. [Google Scholar] [CrossRef]
- Bendheim, P.E.; Brown, H.R.; Rudelli, R.D.; Scala, L.J.; Goller, N.L.; Wen, G.Y. Nearly ubiquitous tissue distribution of the scrapie agent precursor protein. Neurology 1992, 42, 149. [Google Scholar] [CrossRef]
- Wulf, M.-A.; Senatore, A.; Aguzzi, A. The biological function of the cellular prion protein: An update. BMC Biol. 2017, 15, 34. [Google Scholar] [CrossRef]
- Kuffer, A.; Lakkaraju, A.K.; Mogha, A.; Petersen, S.C.; Airich, K.; Doucerain, C.; Marpakwar, R.; Bakirci, P.; Senatore, A.; Monnard, A.; et al. The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature 2016, 536, 464–468. [Google Scholar] [CrossRef]
- Carulla, P.; Bribián, A.; Rangel, A.; Gavín, R.; Ferrer, I.; Caelles, C. Neuroprotective role of PrPC against kainate-induced epileptic seizures and cell death depends on the modulation of JNK3 activation by GluR6/7–PSD-95 binding. Mol. Biol. Cell 2011, 22, 3041–3054. [Google Scholar] [CrossRef]
- Carulla, P.; Llorens, F.; Matamoros-Angles, A.; Aguilar-Calvo, P.; Espinosa, J.C.; Gavín, R. Involvement of PrPC in kainate-induced excitotoxicity in several mouse strains. Sci. Rep. 2015, 5, srep11971. [Google Scholar] [CrossRef]
- Collins, S.; McLean, C.A.; Masters, C.L. Gerstmann–Sträussler–Scheinker syndrome, fatal familial insomnia, and kuru: A review ofthese less common human transmissiblespongiform encephalopathies. J. Clin. Neurosci. 2001, 8, 387–397. [Google Scholar] [CrossRef]
- Dibner, C.; Schibler, U.; Albrecht, U. The Mammalian Circadian Timing System: Organization and Coordination of Central and Peripheral Clocks. Annu. Rev. Physiol. 2010, 72, 517–549. [Google Scholar] [CrossRef] [PubMed]
- Cingaram, P.K.R.; Nyeste, A.; Dondapati, D.T.; Fodor, E.; Welker, E. Prion Protein Does Not Confer Resistance to Hippocampus-Derived Zpl Cells against the Toxic Effects of Cu2+, Mn2+, Zn2+ and Co2+ Not Supporting a General Protective Role for PrP in Transition Metal Induced Toxicity. PLoS ONE 2015, 10, e0139219. [Google Scholar] [CrossRef] [PubMed]
- Gasperini, L.; Meneghetti, E.; Pastore, B.; Benetti, F.; Legname, G. Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid. Redox Signal. 2015, 22, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Faris, R.; Moore, R.A.; Ward, A.; Race, B.; Dorward, D.W.; Hollister, J.R.; Fischer, E.R.; Priola, S.A. Cellular prion protein is present in mitochondria of healthy mice. Sci. Rep. 2017, 7, 41556. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.A. Cellular biology of prion diseases. Clin. Microbiol. Rev. 1999, 12, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Slapšak, U.; Salzano, G.; Amin, L.; Abskharon, R.N.; Ilc, G.; Zupančič, B.; Biljan, I.; Plavec, J.; Giachin, G.; Legname, G. The N terminus of the prion protein mediates functional interactions with the neuronal cell adhesion molecule (NCAM) fibronectin domain. J. Biol. Chem. 2016, 291, 21857–21868. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Koliatsos, V.E.; Guarnieri, M.; Pardo, C.A.; Sisodia, S.S.; Price, D.L. Rapid anterograde axonal transport of the cellular prion glycoprotein in the peripheral and central nervous systems. J. Biol. Chem. 1994, 269, 14711–14714. [Google Scholar] [CrossRef]
- Moya, K.L.; Hässig, R.; Créminon, C.; Laffont, I.; Giamberardino, L. Enhanced detection and retrograde axonal transport of PrPc in peripheral nerve: Cellular prion protein in peripheral nerve. J. Neurochem. 2003, 88, 155–160. [Google Scholar] [CrossRef]
- Haeberle, A.M.; Ribaut-Barassin, C.; Bombarde, G.; Mariani, J.; Hunsmann, G.; Grassi, J. Synaptic prion protein immuno-reactivity in the rodent cerebellum. Microsc. Res. Tech. 2000, 50, 66–75. [Google Scholar] [CrossRef]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef]
- Abskharon, R.; Wang, F.; Wohlkonig, A.; Ruan, J.; Soror, S.; Giachin, G.; Pardon, E.; Zou, W.; Legname, G.; Ma, J.; et al. Structural evidence for the critical role of the prion protein hydrophobic region in forming an infectious prion. PLoS Pathog. 2019, 15, e1008139. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Zahn, R.; Liu, A.; Luhrs, T.; Riek, R.; von Schroetter, C.; Lopez Garcia, F.; Billeter, M.; Calzolai, L.; Wider, G.; Wuthrich, K. NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. USA 2000, 97, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Dexter, E.; Kong, Q. Neuroprotective effect and potential of cellular prion protein and its cleavage products for treatment of neurodegenerative disorders part I. A literature review. Expert Rev. Neurother. 2021, 21, 969–982. [Google Scholar] [CrossRef]
- Castle, A.R.; Gill, A.C. Physiological Functions of the Cellular Prion Protein. Front. Mol. Biosci. 2017, 4, 19. [Google Scholar] [CrossRef]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.W.; Fletterick, R.J.; Cohen, F.E.; et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef]
- Walker, L.C.; Jucker, M. Neurodegenerative diseases: Expanding the prion concept. Annu. Rev. Neurosci. 2015, 38, 87–103. [Google Scholar] [CrossRef]
- Amin, L.; Harris, D.A. Aβ receptors specifically recognize molecular features displayed by fibril ends and neurotoxic oligomers. Nat. Commun. 2021, 12, 3451. [Google Scholar] [CrossRef]
- Castillo-Carranza, D.L.; Gerson, J.E.; Sengupta, U.; Guerrero-Muñoz, M.J.; Lasagna-Reeves, C.A.; Kayed, R. Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J. Alzheimer’s Dis. JAD 2014, 40 (Suppl. S1), S97–S111. [Google Scholar] [CrossRef]
- Corbett, G.T.; Wang, Z.; Hong, W.; Colom-Cadena, M.; Rose, J.; Liao, M.; Asfaw, A.; Hall, T.C.; Ding, L.; DeSousa, A.; et al. PrP is a central player in toxicity mediated by soluble aggregates of neurodegeneration-causing proteins. Acta Neuropathol. 2020, 139, 503–526. [Google Scholar] [CrossRef]
- Salazar, S.V.; Strittmatter, S.M. Cellular prion protein as a receptor for amyloid-beta oligomers in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- Vascellari, S.; Manzin, A. Parkinson’s Disease: A Prionopathy? Int. J. Mol. Sci. 2021, 22, 8022. [Google Scholar] [CrossRef] [PubMed]
- La Vitola, P.; Beeg, M.; Balducci, C.; Santamaria, G.; Restelli, E.; Colombo, L.; Caldinelli, L.; Pollegioni, L.; Gobbi, M.; Chiesa, R.; et al. Cellular prion protein neither binds to alpha-synuclein oligomers nor mediates their detrimental effects. Brain J. Neurol. 2019, 142, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Borchelt, D.R.; Prusiner, S.B. Differential release of cellular and scrapie prion proteins from cellular membranes by phosphatidylinositol-specific phospholipase C. Biochemistry 1990, 29, 5405–5412. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Rogers, M.; Stahl, N.; Telling, G.; Prusiner, S.B. Release of the cellular prion protein from cultured cells after loss of its glycoinositol phospholipid anchor. Glycobiology 1993, 3, 319–329. [Google Scholar] [CrossRef]
- Chen, S.G.; Teplow, D.B.; Parchi, P.; Teller, J.K.; Gambetti, P.; Autilio-Gambetti, L. Truncated forms of the human prion protein in normal brain and in prion diseases. J. Biol. Chem. 1995, 270, 19173–19180. [Google Scholar] [CrossRef]
- Mange, A.; Beranger, F.; Peoc’h, K.; Onodera, T.; Frobert, Y.; Lehmann, S. Alpha- and beta-cleavages of the amino-terminus of the cellular prion protein. Biol. Cell 2004, 96, 125–132. [Google Scholar] [CrossRef]
- Oliveira-Martins, J.B.; Yusa, S.-I.; Calella, A.M.; Bridel, C.; Baumann, F.; Dametto, P.; Aguzzi, A. Unexpected Tolerance of α-Cleavage of the Prion Protein to Sequence Variations. PLoS ONE 2010, 5, e9107. [Google Scholar] [CrossRef]
- Harris, D.A.; Huber, M.T.; Dijken, P.; Shyng, S.L.; Chait, B.T.; Wang, R. Processing of a cellular prion protein: Identification of N-and C-terminal cleavage sites. Biochemistry 1993, 32, 1009–1016. [Google Scholar] [CrossRef]
- Linsenmeier, L.; Altmeppen, H.C.; Wetzel, S.; Mohammadi, B.; Saftig, P.; Glatzel, M. Diverse functions of the prion protein—Does proteolytic processing hold the key? Biochim. Biophys. Acta 2017, 1864, 2128–2137. [Google Scholar] [CrossRef]
- Laffont-Proust, I.; Faucheux, B.A.; Hässig, R.; Sazdovitch, V.; Simon, S.; Grassi, J.; Hauw, J.-J.; Moya, K.L.; Haïk, S. The N-terminal cleavage of cellular prion protein in the human brain. FEBS Lett. 2005, 579, 6333–6337. [Google Scholar] [CrossRef]
- Pietri, M.; Dakowski, C.; Hannaoui, S.; Alleaume-Butaux, A.; Hernandez-Rapp, J.; Ragagnin, A.; Mouillet-Richard, S.; Haik, S.; Bailly, Y.; Peyrin, J.-M.; et al. PDK1 decreases TACE-mediated α-secretase activity and promotes disease progression in prion and Alzheimer’s diseases. Nat. Med. 2013, 19, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Alleaume-Butaux, A.; Nicot, S.; Pietri, M.; Baudry, A.; Dakowski, C.; Tixador, P.; Ardila-Osorio, H.; Haeberlé, A.-M.; Bailly, Y.; Peyrin, J.-M.; et al. Double-Edge Sword of Sustained ROCK Activation in Prion Diseases through Neuritogenesis Defects and Prion Accumulation. PLoS Pathog. 2015, 11, e1005073. [Google Scholar] [CrossRef] [PubMed]
- Vincent, B.; Paitel, E.; Frobert, Y.; Lehmann, S.; Grassi, J.; Checler, F. Phorbol ester-regulated cleavage of normal prion protein in HEK293 human cells and murine neurons. J. Biol. Chem. 2000, 275, 35612–35616. [Google Scholar] [CrossRef] [PubMed]
- Vincent, B.; Paitel, E.; Saftig, P.; Frobert, Y.; Hartmann, D.; De Strooper, B.; Grassi, J.; Lopez-Perez, E.; Checler, F. The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J. Biol. Chem. 2001, 276, 37743–37746. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Wang, W.; Sorensen, D.; Medina, S.; Ilchenko, S.; Kiselar, J.; Surewicz, W.K.; Booth, S.A.; Kong, Q. Cellular Prion Protein Regulates Its Own α-Cleavage through ADAM8 in Skeletal Muscle*. J. Biol. Chem. 2012, 287, 16510–16520. [Google Scholar] [CrossRef]
- McDonald, A.J.; Dibble, J.P.; Evans, E.G.B.; Millhauser, G.L. A New Paradigm for Enzymatic Control of α-Cleavage and β-Cleavage of the Prion Protein. J. Biol. Chem. 2014, 289, 803–813. [Google Scholar] [CrossRef]
- Jimenez-Huete, A.; Lievens, P.M.; Vidal, R.; Piccardo, P.; Ghetti, B.; Tagliavini, F.; Frangione, B.; Prelli, F. Endogenous proteolytic cleavage of normal and disease-associated isoforms of the human prion protein in neural and non-neural tissues. Am. J. Pathol. 1998, 153, 1561–1572. [Google Scholar] [CrossRef]
- Kuczius, T.; Grassi, J.; Karch, H.; Groschup, M.H. Binding of N- and C-terminal anti-prion protein antibodies generates distinct phenotypes of cellular prion proteins (PrPC) obtained from human, sheep, cattle and mouse. FEBS J. 2007, 274, 1492–1502. [Google Scholar] [CrossRef]
- Shyng, S.L.; Huber, M.T.; Harris, D.A. A prion protein cycles between the cell surface and an endocytic compartment in cultured neuroblastoma cells. J. Biol. Chem. 1993, 268, 15922–15928. [Google Scholar] [CrossRef]
- Taraboulos, A.; Scott, M.; Semenov, A.; Avrahami, D.; Laszlo, L.; Prusiner, S.B. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J. Cell Biol. 1995, 129, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Klingeborn, M.; Simonsson, M.; Linné, T. Proteolytic cleavage and shedding of the bovine prion protein in two cell culture systems. Virus Res. 2006, 115, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Walmsley, A.R.; Watt, N.T.; Taylor, D.R.; Perera, W.S.S.; Hooper, N.M. α-cleavage of the prion protein occurs in a late compartment of the secretory pathway and is independent of lipid rafts. Mol. Cell Neurosci. 2009, 40, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Nunziante, M.; Gilch, S.; Schätzl, H.M. Essential Role of the Prion Protein N Terminus in Subcellular Trafficking and Half-life of Cellular Prion Protein. J. Biol. Chem. 2003, 278, 3726–3734. [Google Scholar] [CrossRef]
- Béland, M.; Roucou, X. The prion protein unstructured N-terminal region is a broad-spectrum molecular sensor with diverse and contrasting potential functions. J. Neurochem. 2012, 120, 853–868. [Google Scholar] [CrossRef]
- Nieznanski, K.; Rutkowski, M.; Dominik, M.; Stepkowski, D. Proteolytic processing and glycosylation influence formation of porcine prion protein complexes. Biochem. J. 2005, 387, 93–100. [Google Scholar] [CrossRef][Green Version]
- Wik, L.; Klingeborn, M.; Willander, H.; Linné, T. Separate mechanisms act concurrently to shed and release the prion protein from the cell. Prion 2012, 6, 498–509. [Google Scholar] [CrossRef]
- Lewis, V.; Hill, A.F.; Haigh, C.L.; Klug, G.M.; Masters, C.L.; Lawson, V.A.; Collins, S.J. Increased proportions of C1 truncated prion protein protect against cellular M1000 prion infection. J. Neuropathol. Exp. Neurol. 2009, 68, 1125–1135. [Google Scholar] [CrossRef]
- Westergard, L.; Turnbaugh, J.A.; Harris, D.A. A naturally occurring C-terminal fragment of the prion protein (PrP) delays disease and acts as a dominant-negative inhibitor of PrPSc formation. J. Biol. Chem. 2011, 286, 44234–44242. [Google Scholar] [CrossRef]
- Guillot-Sestier, M.V.; Sunyach, C.; Druon, C.; Scarzello, S.; Checler, F. The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J. Biol. Chem. 2009, 284, 35973–35986. [Google Scholar] [CrossRef]
- Guillot-Sestier, M.V.; Checler, F. Cellular prion and its catabolites in the brain: Production and function. Curr. Mol. Med. 2012, 12, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Yusa, S.; Oliveira-Martins, J.B.; Sugita-Konishi, Y.; Kikuchi, Y. Cellular prion protein: From physiology to pathology. Viruses 2012, 4, 3109–3131. [Google Scholar] [CrossRef] [PubMed]
- Altmeppen, H.C.; Puig, B.; Dohler, F.; Thurm, D.K.; Falker, C.; Krasemann, S.; Glatzel, M. Proteolytic processing of the prion protein in health and disease. Am. J. Neurodegener. Dis. 2012, 1, 15–31. [Google Scholar] [PubMed]
- McMahon, H.E.M.; Mangé, A.; Nishida, N.; Créminon, C.; Casanova, D.; Lehmann, S. Cleavage of the Amino Terminus of the Prion Protein by Reactive Oxygen Species*. J. Biol. Chem. 2001, 276, 2286–2291. [Google Scholar] [CrossRef]
- Watt, N.T.; Taylor, D.R.; Gillott, A.; Thomas, D.A.; Perera, W.S.; Hooper, N.M. Reactive oxygen species-mediated beta-cleavage of the prion protein in the cellular response to oxidative stress. J. Biol. Chem. 2005, 280, 35914–35921. [Google Scholar] [CrossRef] [PubMed]
- Pushie, M.J.; Vogel, H.J. Modeling by Assembly and Molecular Dynamics Simulations of the Low Cu2+ Occupancy Form of the Mammalian Prion Protein Octarepeat Region: Gaining Insight into Cu2+-Mediated β-Cleavage. Biophys. J. 2008, 95, 5084–5091. [Google Scholar] [CrossRef] [PubMed]
- Engelke, A.D.; Gonsberg, A.; Thapa, S.; Jung, S.; Ulbrich, S.; Seidel, R.; Basu, S.; Multhaup, G.; Baier, M.; Engelhard, M.; et al. Dimerization of the cellular prion protein inhibits propagation of scrapie prions. J. Biol. Chem. 2018, 293, 8020–8031. [Google Scholar] [CrossRef]
- Benestad, S.L.; Austbø, L.; Tranulis, M.A.; Espenes, A.; Olsaker, I. Healthy goats naturally devoid of prion protein. Vet. Res. 2012, 43, 87. [Google Scholar] [CrossRef]
- Meier, P.; Genoud, N.; Prinz, M.; Maissen, M.; Rülicke, T.; Zurbriggen, A.; Raeber, A.J.; Aguzzi, A. Soluble dimeric prion protein binds PrP(Sc) in vivo and antagonizes prion disease. Cell 2003, 113, 49–60. [Google Scholar] [CrossRef]
- Parizek, P. Similar turnover and shedding of the cellular prion protein in primary lymphoid and neuronal cells. J. Biol. Chem. 2001, 276, 44627–44632. [Google Scholar] [CrossRef]
- Oesch, B.; Westaway, D.; Walchli, M.; McKinley, M.P.; Kent, S.B.; Aebersold, R.; Barry, R.A.; Tempst, P.; Teplow, D.B.; Hood, L.E.; et al. A cellular gene encodes scrapie PrP 27-30 protein. Cell 1985, 40, 735–746. [Google Scholar] [CrossRef]
- Rogers, M.; Yehiely, F.; Scott, M.; Prusiner, S.B. Conversion of truncated and elongated prion proteins into the scrapie isoform in cultured cells. Proc. Natl. Acad. Sci. USA 1993, 90, 3182–3186. [Google Scholar] [CrossRef] [PubMed]
- Lewis, V.; Johanssen, V.A.; Crouch, P.J.; Klug, G.M.; Hooper, N.M.; Collins, S.J. Prion protein “gamma-cleavage”: Characterizing a novel endoproteolytic processing event. Cell Mol. Life Sci. 2016, 73, 667–683. [Google Scholar] [CrossRef] [PubMed]
- Haigh, C.; Collins, S. Endoproteolytic cleavage as a molecular switch regulating and diversifying prion protein function. Neural Regen. Res. 2016, 11, 238–239. [Google Scholar] [CrossRef] [PubMed]
- Kojima, A.; Konishi, M.; Akizawa, T. Prion Fragment Peptides Are Digested with Membrane Type Matrix Metalloproteinases and Acquire Enzyme Resistance through Cu2+-Binding. Biomolecules 2014, 4, 510–526. [Google Scholar] [CrossRef]
- Stahl, N.; Baldwin, M.A.; Burlingame, A.L.; Prusiner, S.B. Identification of glycoinositol phospholipid linked and truncated forms of the scrapie prion protein. Biochemistry 1990, 29, 8879–8884. [Google Scholar] [CrossRef]
- Tagliavini, F.; Prelli, F.; Porro, M.; Salmona, M.; Bugiani, O.; Frangione, B. A soluble form of prion protein in human cerebrospinal fluid: Implications for prion-related encephalopathies. Biochem. Biophys. Res. Commun. 1992, 184, 1398–1404. [Google Scholar] [CrossRef]
- Altmeppen, H.C.; Prox, J.; Krasemann, S.; Puig, B.; Kruszewski, K.; Dohler, F.; Bernreuther, C.; Hoxha, A.; Linsenmeier, L.; Sikorska, B.; et al. The sheddase ADAM10 is a potent modulator of prion disease. Elife 2015, 4, e04260. [Google Scholar] [CrossRef]
- Glatzel, M.; Linsenmeier, L.; Dohler, F.; Krasemann, S.; Puig, B.; Altmeppen, H.C. Shedding light on prion disease. Prion 2015, 9, 244–256. [Google Scholar] [CrossRef][Green Version]
- Altmeppen, H.C.; Prox, J.; Puig, B.; Dohler, F.; Falker, C.; Krasemann, S.; Glatzel, M. Roles of endoproteolytic α-cleavage and shedding of the prion protein in neurodegeneration. FEBS J. 2013, 280, 4338–4347. [Google Scholar] [CrossRef]
- Linsenmeier, L.; Mohammadi, B.; Shafiq, M.; Frontzek, K.; Bär, J.; Shrivastava, A.N.; Damme, M.; Song, F.; Schwarz, A.; Da Vela, S.; et al. Ligands binding to the prion protein induce its proteolytic release with therapeutic potential in neurodegenerative proteinopathies. Sci. Adv. 2021, 7, eabj1826. [Google Scholar] [CrossRef] [PubMed]
- Jarosz-Griffiths, H.H.; Corbett, N.J.; Rowland, H.A.; Fisher, K.; Jones, A.C.; Baron, J.; Howell, G.J.; Cowley, S.A.; Chintawar, S.; Cader, M.Z.; et al. Proteolytic shedding of the prion protein via activation of metallopeptidase ADAM10 reduces cellular binding and toxicity of amyloid-β; oligomers. J. Biol. Chem. 2019, 294, 7085–7097. [Google Scholar] [CrossRef] [PubMed]
- Megra, B.W.; Eugenin, E.A.; Berman, J.W. The Role of Shed PrP(C) in the Neuropathogenesis of HIV Infection. J. Immunol. 2017, 199, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.R.; Parkin, E.T.; Cocklin, S.L.; Ault, J.R.; Ashcroft, A.E.; Turner, A.J. Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protein. J. Biol. Chem. 2009, 284, 22590–22600. [Google Scholar] [CrossRef] [PubMed]
- Altmeppen, H.C.; Prox, J.; Puig, B.; Kluth, M.A.; Bernreuther, C.; Thurm, D.; Jorissen, E.; Petrowitz, B.; Bartsch, U.; De Strooper, B.; et al. Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo. Mol. Neurodegener. 2011, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- McDonald, A.J.; Millhauser, G.L. PrP overdrive: Does inhibition of α-cleavage contribute to PrP(C) toxicity and prion disease? Prion 2014, 8, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Tucher, J.; Linke, D.; Koudelka, T.; Cassidy, L.; Tredup, C.; Wichert, R.; Pietrzik, C.; Becker-Pauly, C.; Tholey, A. LC-MS based cleavage site profiling of the proteases ADAM10 and ADAM17 using proteome-derived peptide libraries. J. Proteome Res. 2014, 13, 2205–2214. [Google Scholar] [CrossRef] [PubMed]
- Jansen, C.; Parchi, P.; Capellari, S.; Vermeij, A.J.; Corrado, P.; Baas, F.; Strammiello, R.; van Gool, W.A.; van Swieten, J.C.; Rozemuller, A.J. Prion protein amyloidosis with divergent phenotype associated with two novel nonsense mutations in PRNP. Acta Neuropathol. 2010, 119, 189–197. [Google Scholar] [CrossRef]
- Čurin Šerbec, V.; Bresjanac, M.; Popović, M.; Pretnar Hartman, K.; Galvani, V.; Rupreht, R.; Černilec, M.; Vranac, T.; Hafner, I.; Jerala, R. Monoclonal antibody against a peptide of human prion protein discriminates between Creutzfeldt-Jacob’s disease-affected and normal brain tissue. J. Biol. Chem. 2004, 279, 3694–3698. [Google Scholar] [CrossRef]
- Dvorakova, E.; Vranac, T.; Janouskova, O.; Černilec, M.; Koren, S.; Lukan, A.; Novakova, J.; Matej, R.; Holada, K.; Čurin Šerbec, V. Detection of the GPI-anchorless prion protein fragment PrP226* in human brain. BMC Neurol. 2013, 13, 126. [Google Scholar] [CrossRef]
- Koren, S.; Kosmač, M.; Colja Venturini, A.; Montanič, S.; Čurin Šerbec, V. Antibody variable-region sequencing as a method for hybridoma cell-line authentication. Appl. Microbiol. Biotechnol. 2008, 78, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Kovač, V.; Hafner-Bratkovič, I.; Čurin Šerbec, V. Anchorless forms of prion protein—Impact of truncation on structure destabilization and prion protein conversion. Biochem. Biophys. Res. Commun. 2016, 481, 1–6. [Google Scholar] [CrossRef][Green Version]
- Kovač, V.; Zupančič, B.; Ilc, G.; Plavec, J.; Čurin Šerbec, V. Truncated prion protein PrP226*—A structural view on its role in amyloid disease. Biochem. Biophys. Res. Commun. 2017, 484, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Lukan, A.; Černilec, M.; Vranac, T.; Popović, M.; Čurin Šerbec, V. Regional distribution of anchorless prion protein, PrP226 *, in the human brain. Prion 2014, 8, 203–209. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kovač, V.; Čurin Šerbec, V. Prion Proteins Without the Glycophosphatidylinositol Anchor: Potential Biomarkers in Neurodegenerative Diseases. Biomark. Insights 2018, 13, 1177271918756648. [Google Scholar] [CrossRef] [PubMed]
- Salès, N.; Hässig, R.; Rodolfo, K.; Giamberardino, L.; Traiffort, E.; Ruat, M. Developmental expression of the cellular prion protein in elongating axons. Eur. J. Neurosci. 2002, 15, 1163–1177. [Google Scholar] [CrossRef]
- Salès, N.; Rodolfo, K.; Hässig, R.; Faucheux, B.; Giamberardino, L.; Moya, K.L. Cellular prion protein localization in rodent and primate brain. Eur. J. Neurosci. 1998, 10, 2464–2471. [Google Scholar] [CrossRef]
- Mironov, A.; Latawiec, D.; Wille, H.; Bouzamondo-Bernstein, E.; Legname, G.; Williamson, R.A. Cytosolic prion protein in neurons. J. Neurosci. 2003, 23, 7183–7193. [Google Scholar] [CrossRef]
- Baumann, F.; Tolnay, M.; Brabeck, C.; Pahnke, J.; Kloz, U.; Niemann, H.H. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 2007, 26, 538–547. [Google Scholar] [CrossRef]
- Radovanovic, I. Truncated prion protein and Doppel are myelinotoxic in the absence of oligodendrocytic PrPC. J. Neurosci. 2005, 25, 4879–4888. [Google Scholar] [CrossRef]
- Bremer, J.; Baumann, F.; Tiberi, C.; Wessig, C.; Fischer, H.; Schwarz, P. Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci. 2010, 13, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Nuvolone, M.; Hermann, M.; Sorce, S.; Russo, G.; Tiberi, C.; Schwarz, P.; Minikel, E.; Sanoudou, D.; Pelczar, P.; Aguzzi, A. Strictly co-isogenic C57BL/6J-Prnp−/−mice: A rigorous resource for prion science. J. Exp. Med. 2016, 213, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Tremblay, P.; Sugimoto, T.; Shigematsu, K.; Shirabe, S.; Petromilli, C.; Erpel, S.P.; Nakaoke, R.; Atarashi, R.; Houtani, T.; et al. A mouse prion protein transgene rescues mice deficient for the prion protein gene from purkinje cell degeneration and demyelination. Lab. Investig. A J. Tech. Methods Pathol. 1999, 79, 689–697. [Google Scholar]
- Henzi, A.; Senatore, A.; Lakkaraju, A.K.K.; Scheckel, C.; Mühle, J.; Reimann, R.; Sorce, S.; Schertler, G.; Toyka, K.V.; Aguzzi, A. Soluble dimeric prion protein ligand activates Adgrg6 receptor but does not rescue early signs of demyelination in PrP-deficient mice. PLoS ONE 2020, 15, e0242137. [Google Scholar] [CrossRef]
- Henzi, A.; Aguzzi, A. The prion protein is not required for peripheral nerve de- and remyelination after crush injury. PLoS ONE 2021, 16, e0245944. [Google Scholar] [CrossRef]
- Büeler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.P.; DeArmond, S.J.; Prusiner, S.B.; Aguet, M.; Weissmann, C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992, 356, 577–582. [Google Scholar] [CrossRef]
- Manson, J.C.; Clarke, A.R.; Hooper, M.L.; Aitchison, L.; McConnell, I.; Hope, J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol. Neurobiol. 1994, 8, 121–127. [Google Scholar] [CrossRef]
- Steele, A.D.; Lindquist, S.; Aguzzi, A. The prion protein knockout mouse: A phenotype under challenge. Prion 2007, 1, 83–93. [Google Scholar] [CrossRef]
- Rossi, D.; Cozzio, A.; Flechsig, E.; Klein, M.A.; Rülicke, T.; Aguzzi, A.; Weissmann, C. Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. EMBO J. 2001, 20, 694–702. [Google Scholar] [CrossRef]
- Richt, J.A.; Kasinathan, P.; Hamir, A.N.; Castilla, J.; Sathiyaseelan, T.; Vargas, F. Production of cattle lacking prion protein. Nat. Biotechnol. 2007, 25, 132–138. [Google Scholar] [CrossRef]
- Skedsmo, F.S.; Malachin, G.; Våge, D.I.; Hammervold, M.M.; Salvesen, Ø.; Ersdal, C.; Ranheim, B.; Stafsnes, M.H.; Bartosova, Z.; Bruheim, P.; et al. Demyelinating polyneuropathy in goats lacking prion protein. FASEB J. 2020, 34, 2359–2375. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, Ø.; Tatzelt, J.; Tranulis, M.A. The prion protein in neuroimmune crosstalk. Neurochem. Int. 2019, 130, 104335. [Google Scholar] [CrossRef] [PubMed]
- Criado, J.R.; Sánchez-Alavez, M.; Conti, B.; Giacchino, J.L.; Wills, D.N.; Henriksen, S.J. Mice devoid of prion protein have cognitive deficits that are rescued by reconstitution of PrP in neurons. Neurobiol. Dis. 2005, 19, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Katamine, S.; Nishida, N.; Moriuchi, R.; Shigematsu, K.; Sugimoto, T.; Nakatani, A.; Kataoka, Y.; Houtani, T.; Shirabe, S.; et al. Loss of cerebellar Purkinje cells in aged mice homozygous for a disrupted PrP gene. Nature 1996, 380, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.C.; Lee, I.Y.; Silverman, G.L.; Harrison, P.M.; Strome, R.; Heinrich, C.; Karunaratne, A.; Pasternak, S.H.; Chishti, M.A.; Liang, Y.; et al. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J. Mol. Biol. 1999, 292, 797–817. [Google Scholar] [CrossRef] [PubMed]
- Mallucci, G.R.; Ratté, S.; Asante, E.A.; Linehan, J.; Gowland, I.; Jefferys, J.G.; Collinge, J. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. Embo J. 2002, 21, 202–210. [Google Scholar] [CrossRef]
- White, A.R.; Collins, S.J.; Maher, F.; Jobling, M.F.; Stewart, L.R.; Thyer, J.M. Prion protein-deficient neurons reveal lower glutathione reductase activity and increased susceptibility to hydrogen peroxide toxicity. Am. J. Pathol. 1999, 155, 1723–1730. [Google Scholar] [CrossRef]
- Hutter, G.; Heppner, F.L.; Aguzzi, A. No Superoxide Dismutase Activity of Cellular Prion Protein in vivo. Biol. Chem. 2003, 384, 1279–1285. [Google Scholar] [CrossRef]
- Davies, P.; Brown, D.R. The chemistry of copper binding to PrP: Is there sufficient evidence to elucidate a role for copper in protein function? Biochem. J. 2008, 410, 237–244. [Google Scholar] [CrossRef]
- Brown, D.R.; Nicholas, R.S.; Canevari, L. Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J. Neurosci. Res. 2002, 67, 211–224. [Google Scholar] [CrossRef]
- Brown, D.R.; Boon-Seng, W.; Hafiz, F.; Clive, C.; Haswell, S.J.; Jones, I.M. Normal prion protein has an activity like that of superoxide dismutase. Biochem. J. 1999, 344, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Schulz-Schaeffer, W.J.; Schmidt, B.; Kretzschmar, H.A. Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Exp. Neurol. 1997, 146, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Sakudo, A.; Lee, D.-c.; Saeki, K.; Nakamura, Y.; Inoue, K.; Matsumoto, Y.; Itohara, S.; Onodera, T. Impairment of superoxide dismutase activation by N-terminally truncated prion protein (PrP) in PrP-deficient neuronal cell line. Biochem. Biophys. Res. Commun. 2003, 308, 660–667. [Google Scholar] [CrossRef]
- Guentchev, M.; Voigtländer, T.; Haberler, C.; Groschup, M.H.; Budka, H. Evidence for oxidative stress in experimental prion disease. Neurobiol. Dis. 2000, 7, 270–273. [Google Scholar] [CrossRef]
- Klamt, F.; Dal-Pizzol, F.; Conte da Frota, M.L.; Walz, R.; Andrades, M.E.; Silva, E.G. Imbalance of antioxidant defense in mice lacking cellular prion protein. Free Radic. Biol. Med. 2001, 30, 1137–1144. [Google Scholar] [CrossRef]
- Wong, B.S.; Liu, T.; Li, R.; Pan, T.; Petersen, R.B.; Smith, M.A.; Gambetti, P.; Perry, G.; Manson, J.C.; Brown, D.R.; et al. Increased levels of oxidative stress markers detected in the brains of mice devoid of prion protein. J. Neurochem. 2001, 76, 565–572. [Google Scholar] [CrossRef]
- McLennan, N.F.; Brennan, P.M.; McNeill, A.; Davies, I.; Fotheringham, A.; Rennison, K.A.; Ritchie, D.; Brannan, F.; Head, M.W.; Ironside, J.W. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am. J. Pathol. 2004, 165, 227–235. [Google Scholar] [CrossRef]
- Brenna, S.; Altmeppen, H.C.; Mohammadi, B.; Rissiek, B.; Schlink, F.; Ludewig, P.; Krisp, C.; Schlüter, H.; Failla, A.V.; Schneider, C.; et al. Characterization of brain-derived extracellular vesicles reveals changes in cellular origin after stroke and enrichment of the prion protein with a potential role in cellular uptake. J. Extracell. Vesicles 2020, 9, 1809065. [Google Scholar] [CrossRef]
- Puig, B.; Yang, D.; Brenna, S.; Altmeppen, H.C.; Magnus, T. Show Me Your Friends and I Tell You Who You Are: The Many Facets of Prion Protein in Stroke. Cells 2020, 9, 1609. [Google Scholar] [CrossRef]
- Zeng, L.; Zou, W.; Wang, G. Cellular prion protein (PrP(C)) and its role in stress responses. Int. J. Clin. Exp. Med. 2015, 8, 8042–8050. [Google Scholar]
- Mitsios, N.; Saka, M.; Krupinski, J.; Pennucci, R.; Sanfeliu, C.; Miguel Turu, M.; Gaffney, J.; Kumar, P.; Kumar, S.; Sullivan, M.; et al. Cellular prion protein is increased in the plasma and peri-infarcted brain tissue after acute stroke. J. Neurosci. Res. 2007, 85, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Pham, N.; Dhar, A.; Khalaj, S.; Desai, K.; Taghibiglou, C. Down regulation of brain cellular prion protein in an animal model of insulin resistance: Possible implication in increased prevalence of stroke in pre-diabetics/diabetics. Biochem. Biophys. Res. Commun. 2014, 448, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Shyu, W.-C. Overexpression of PrPC by adenovirus-mediated gene targeting reduces ischemic injury in a stroke rat model. J. Neurosci. 2005, 25, 8967–8977. [Google Scholar] [CrossRef] [PubMed]
- Spudich, A.; Frigg, R.; Kilic, E.; Kilic, Ü.; Oesch, B.; Raeber, A.; Bassetti, C.L.; Hermann, D.M. Aggravation of ischemic brain injury by prion protein deficiency: Role of ERK-1/-2 and STAT-1. Neurobiol. Dis. 2005, 20, 442–449. [Google Scholar] [CrossRef]
- Weise, J.; Crome, O.; Sandau, R.; Schulz-Schaeffer, W.; Bähr, M.; Zerr, I. Upregulation of cellular prion protein (PrPc) after focal cerebral ischemia and influence of lesion severity. Neurosci. Lett. 2004, 372, 146–150. [Google Scholar] [CrossRef]
- Weise, J.; Sandau, R.; Schwarting, S.; Crome, O.; Wrede, A.; Schulz-Schaeffer, W. Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke 2006, 37, 1296–1300. [Google Scholar] [CrossRef]
- Wang, V.; Chuang, T.-C.; Hsu, Y.-D.; Chou, W.-Y.; Kao, M.-C. Nitric oxide induces prion protein via MEK and p38 MAPK signaling. Biochem. Biophys. Res. Commun. 2005, 333, 95–100. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. Pi3k-pkb/akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef]
- Brazil, D.P.; Hemmings, B.A. Ten years of protein kinase B signalling: A hard Akt to follow. Trends Biochem. Sci. 2001, 26, 657–664. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Mitteregger, G.; Vosko, M.; Krebs, B.; Xiang, W.; Kohlmannsperger, V.; Nölting, S. The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol. 2007, 17, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Vassallo, N.; Herms, J.; Behrens, C.; Krebs, B.; Saeki, K.; Onodera, T.; Windl, O.; Kretzschmar, H.A. Activation of phosphatidylinositol 3-kinase by cellular prion protein and its role in cell survival. Biochem. Biophys. Res. Commun. 2005, 332, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Black, S.A.G.; Stys, P.K.; Zamponi, G.W.; Tsutsui, S. Cellular prion protein and NMDA receptor modulation: Protecting against excitotoxicity. Front. Cell Dev. Biol. 2014, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, L.B.; Freitas, A.R.O.; Zanata, S.M.; Brentani, R.R.; Martins, V.R.; Linden, R. Cellular prion protein transduces neuroprotective signals. EMBO J. 2002, 21, 3317–3326. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Mangé, A.; Dong, L.; Lehmann, S.; Schachner, M. Prion protein as trans-interacting partner for neurons is involved in neurite outgrowth and neuronal survival. Mol. Cell. Neurosci. 2003, 22, 227–233. [Google Scholar] [CrossRef]
- Lopes, M.H.; Hajj, G.N.; Muras, A.G.; Mancini, G.L.; Castro, R.M.; Ribeiro, K.C.; Brentani, R.R.; Linden, R.; Martins, V.R. Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J. Neurosci. 2005, 25, 11330–11339. [Google Scholar] [CrossRef] [PubMed]
- Crestini, A.; Santilli, F.; Martellucci, S.; Carbone, E.; Sorice, M.; Piscopo, P.; Mattei, V. Prions and Neurodegenerative Diseases: A Focus on Alzheimer’s Disease. J. Alzheimer’s Dis. 2022, 85, 503–518. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Paudel, Y.N.; Julian, T.; Shaikh, M.F.; Piperi, C. Pivotal Role of Fyn Kinase in Parkinson’s Disease and Levodopa-Induced Dyskinesia: A Novel Therapeutic Target? Mol. Neurobiol. 2021, 58, 1372–1391. [Google Scholar] [CrossRef]
- Grayson, J.D.; Baumgartner, M.P.; Santos Souza, C.D.; Dawes, S.J.; El Idrissi, I.G.; Louth, J.C.; Stimpson, S.; Mead, E.; Dunbar, C.; Wolak, J.; et al. Amyloid binding and beyond: A new approach for Alzheimer’s disease drug discovery targeting Aβo–PrPC binding and downstream pathways. Chem. Sci. 2021, 12, 3768–3785. [Google Scholar] [CrossRef]
- Briner, A.; Götz, J.; Polanco, J.C. Fyn Kinase Controls Tau Aggregation In Vivo. Cell Rep. 2020, 32, 108045. [Google Scholar] [CrossRef]
- Santuccione, A.; Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59 fyn and to enhance neurite outgrowth. J. Cell Biol. 2005, 169, 341–354. [Google Scholar] [CrossRef]
- Cheung, H.H.; Takagi, N.; Teves, L.; Logan, R.; Wallace, M.C.; Gurd, J.W. Altered association of protein tyrosine kinases with postsynaptic densities after transient cerebral ischemia in the rat brain. J. Cereb. Blood Flow Metab. 2000, 20, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Takagi, N.; Cheung, H.H.; Bissoon, N.; Teves, L.; Wallace, M.C.; Gurd, J.W. The effect of transient global ischemia on the interaction of Src and Fyn with the N-methyl-D-aspartate receptor and postsynaptic densities: Possible involvement of Src homology 2 domains. J. Cereb. Blood Flow Metab. 1999, 19, 880–888. [Google Scholar] [CrossRef]
- Knox, R.; Jiang, X. Fyn in Neurodevelopment and Ischemic Brain Injury. Dev. Neurosci. 2015, 37, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Zhang, Z.G.; Eliceiri, B.P.; Jiang, Q.; Boccia, A.D.; Zhang, R.L.; Chopp, M.; Cheresh, D.A. Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat. Med. 2001, 7, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.-Y.; Liu, Y.; Zhang, G.-Y. PP2, a potent inhibitor of Src family kinases, protects against hippocampal CA1 pyramidal cell death after transient global brain ischemia. Neurosci. Lett. 2007, 420, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Knox, R.; Brennan-Minnella, A.M.; Lu, F.; Yang, D.; Nakazawa, T.; Yamamoto, T.; Swanson, R.A.; Ferriero, D.M.; Jiang, X. NR2B Phosphorylation at Tyrosine 1472 Contributes to Brain Injury in a Rodent Model of Neonatal Hypoxia-Ischemia. Stroke 2014, 45, 3040–3047. [Google Scholar] [CrossRef]
- Du, C.-P.; Gao, J.; Tai, J.-M.; Liu, Y.; Qi, J.; Wang, W.; Hou, X.-Y. Increased tyrosine phosphorylation of PSD-95 by Src family kinases after brain ischaemia. Biochem. J. 2008, 417, 277–285. [Google Scholar] [CrossRef][Green Version]
- Knox, R.; Zhao, C.; Miguel-Perez, D.; Wang, S.; Yuan, J.; Ferriero, D.; Jiang, X. Enhanced NMDA receptor tyrosine phosphorylation and increased brain injury following neonatal hypoxia–ischemia in mice with neuronal Fyn overexpression. Neurobiol. Dis. 2013, 51, 113–119. [Google Scholar] [CrossRef]
- Haigh, C.L.; Drew, S.C.; Boland, M.P.; Masters, C.L.; Barnham, K.J.; Lawson, V.A.; Collins, S.J. Dominant roles of the polybasic proline motif and copper in the PrP23-89-mediated stress protection response. J. Cell Sci. 2009, 122, 1518–1528. [Google Scholar] [CrossRef]
- Haigh, C.L.; McGlade, A.R.; Collins, S.J. MEK1 transduces the prion protein N2 fragment antioxidant effects. Cell. Mol. Life Sci. 2015, 72, 1613–1629. [Google Scholar] [CrossRef] [PubMed]
- Haigh, C.L.; Tumpach, C.; Drew, S.C.; Collins, S.J. The Prion Protein N1 and N2 Cleavage Fragments Bind to Phosphatidylserine and Phosphatidic Acid; Relevance to Stress-Protection Responses. PLoS ONE 2015, 10, e0134680. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.J.; Tumpach, C.; Groveman, B.R.; Drew, S.C.; Haigh, C.L. Prion protein cleavage fragments regulate adult neural stem cell quiescence through redox modulation of mitochondrial fission and SOD2 expression. Cell. Mol. Life Sci. 2018, 75, 3231–3249. [Google Scholar] [CrossRef] [PubMed]
- Sunyach, C.; Cisse, M.A.; da Costa, C.A.; Vincent, B.; Checler, F. The C-terminal Products of Cellular Prion Protein Processing, C1 and C2, Exert Distinct Influence on p53-dependent Staurosporine-induced Caspase-3 Activation *. J. Biol. Chem. 2007, 282, 1956–1963. [Google Scholar] [CrossRef] [PubMed]
- Pager, C.T.; Craft, W.W.; Patch, J.; Dutch, R.E. A mature and fusogenic form of the Nipah virus fusion protein requires proteolytic processing by cathepsin L. Virology 2006, 346, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Diederich, S.; Sauerhering, L.; Weis, M.; Altmeppen, H.; Schaschke, N.; Reinheckel, T.; Erbar, S.; Maisner, A. Activation of the Nipah Virus Fusion Protein in MDCK Cells Is Mediated by Cathepsin B within the Endosome-Recycling Compartment. J. Virol. 2012, 86, 3736–3745. [Google Scholar] [CrossRef]
- Carroll, J.A.; Groveman, B.R.; Williams, K.; Moore, R.; Race, B.; Haigh, C.L. Prion protein N1 cleavage peptides stimulate microglial interaction with surrounding cells. Sci. Rep. 2020, 10, 6654. [Google Scholar] [CrossRef]
- Martellucci, S.; Santacroce, C.; Santilli, F.; Piccoli, L.; Delle Monache, S.; Angelucci, A.; Misasi, R.; Sorice, M.; Mattei, V. Cellular and Molecular Mechanisms Mediated by recPrPC Involved in the Neuronal Differentiation Process of Mesenchymal Stem Cells. Int. J. Mol. Sci. 2019, 20, 345. [Google Scholar] [CrossRef]
- Amin, L.; Nguyen, X.T.; Rolle, I.G.; D’Este, E.; Giachin, G.; Tran, T.H.; Serbec, V.C.; Cojoc, D.; Legname, G. Characterization of prion protein function by focal neurite stimulation. J. Cell Sci. 2016, 129, 3878–3891. [Google Scholar] [CrossRef]
- Mantuano, E.; Azmoon, P.; Banki, M.A.; Lam, M.S.; Sigurdson, C.J.; Gonias, S.L. A soluble derivative of PrP(C) activates cell-signaling and regulates cell physiology through LRP1 and the NMDA receptor. J. Biol. Chem. 2020, 295, 14178–14188. [Google Scholar] [CrossRef]
- Shi, Y.; Mantuano, E.; Inoue, G.; Campana, W.M.; Gonias, S.L. Ligand binding to LRP1 transactivates Trk receptors by a Src family kinase-dependent pathway. Sci. Signal. 2009, 2, ra18. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Borchelt, D.R.; Hsiao, K.; Prusiner, S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51, 229–240. [Google Scholar] [CrossRef]
- Desplats, P.; Lee, H.-J.; Bae, E.-J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.-J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.G.; Temido-Ferreira, M.; Vicente Miranda, H.; Batalha, V.L.; Coelho, J.E.; Szegö, É.M.; Marques-Morgado, I.; Vaz, S.H.; Rhee, J.S.; Schmitz, M.; et al. α-synuclein interacts with PrPC to induce cognitive impairment through mGluR5 and NMDAR2B. Nat. Neurosci. 2017, 20, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- del Río, J.A.; Ferrer, I.; Gavín, R. Role of cellular prion protein in interneuronal amyloid transmission. Prog. Neurobiol. 2018, 165–167, 87–102. [Google Scholar] [CrossRef]
- Urrea, L.; Segura-Feliu, M.; Masuda-Suzukake, M.; Hervera, A.; Pedraz, L.; García Aznar, J.M.; Vila, M.; Samitier, J.; Torrents, E.; Ferrer, I.; et al. Involvement of Cellular Prion Protein in α-Synuclein Transport in Neurons. Mol. Neurobiol. 2018, 55, 1847–1860. [Google Scholar] [CrossRef]
- Domert, J.; Rao, S.B.; Agholme, L.; Brorsson, A.-C.; Marcusson, J.; Hallbeck, M.; Nath, S. Spreading of amyloid-β peptides via neuritic cell-to-cell transfer is dependent on insufficient cellular clearance. Neurobiol. Dis. 2014, 65, 82–92. [Google Scholar] [CrossRef]
- Eisele, Y.S.; Obermüller, U.; Heilbronner, G.; Baumann, F.; Kaeser, S.A.; Wolburg, H.; Walker, L.C.; Staufenbiel, M.; Heikenwalder, M.; Jucker, M. Peripherally applied Aβ-containing inoculates induce cerebral β-amyloidosis. Science 2010, 330, 980–982. [Google Scholar] [CrossRef]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef]
- Sydow, A.; Mandelkow, E.M. ‘Prion-Like’ Propagation of Mouse and Human Tau Aggregates in an Inducible Mouse Model of Tauopathy. Neurodegener. Dis. 2010, 7, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Lee, V.M.-Y. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 2011, 286, 15317–15331. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. USA 2013, 110, 9535–9540. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.H.; Yokotsuka, M.; Tobiume, M.; Sato, Y.; Hasegawa, H.; Nagao, T.; Gouras, G.K. Accumulation of cellular prion protein within β-amyloid oligomer plaques in aged human brains. Brain Pathol. 2021, 31, e12941. [Google Scholar] [CrossRef]
- Aulić, S.; Masperone, L.; Narkiewicz, J.; Isopi, E.; Bistaffa, E.; Ambrosetti, E.; Pastore, B.; De Cecco, E.; Scaini, D.; Zago, P. α-Synuclein amyloids hijack prion protein to gain cell entry, facilitate cell-to-cell spreading and block prion replication. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Haas, L.T.; Salazar, S.V.; Kostylev, M.A.; Um, J.W.; Kaufman, A.C.; Strittmatter, S.M. Metabotropic glutamate receptor 5 couples cellular prion protein to intracellular signalling in Alzheimer’s disease. Brain A J. Neurol. 2016, 139, 526–546. [Google Scholar] [CrossRef]
- Hachiya, N.; Fułek, M.; Zajączkowska, K.; Kurpas, D.; Trypka, E.; Leszek, J. Cellular Prion Protein and Amyloid–β Oligomers in Alzheimer’s Disease–There Are Connections? Preprints 2021, 2021050032. [Google Scholar] [CrossRef]
- Kostylev, M.A.; Tuttle, M.D.; Lee, S.; Klein, L.E.; Takahashi, H.; Cox, T.O.; Gunther, E.C.; Zilm, K.W.; Strittmatter, S.M. Liquid and Hydrogel Phases of PrPC Linked to Conformation Shifts and Triggered by Alzheimer’s Amyloid-β; Oligomers. Mol. Cell 2018, 72, 426–443.e412. [Google Scholar] [CrossRef]
- Um, J.W.; Kaufman, A.C.; Kostylev, M.; Heiss, J.K.; Stagi, M.; Takahashi, H. Metabotropic glutamate receptor 5 is a coreceptor for alzheimer Aβ oligomer bound to cellular prion protein. Neuron 2013, 79, 887–902. [Google Scholar] [CrossRef]
- Rushworth, J.V.; Griffiths, H.H.; Watt, N.T.; Hooper, N.M. Prion Protein-mediated Toxicity of Amyloid–β Oligomers Requires Lipid Rafts and the Transmembrane LRP1. J. Biol. Chem. 2013, 288, 8935–8951. [Google Scholar] [CrossRef]
- Mattei, V.; Manganelli, V.; Martellucci, S.; Capozzi, A.; Mantuano, E.; Longo, A.; Ferri, A.; Garofalo, T.; Sorice, M.; Misasi, R. A multimolecular signaling complex including PrP(C) and LRP1 is strictly dependent on lipid rafts and is essential for the function of tissue plasminogen activator. J. Neurochem. 2020, 152, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Kessels, H.W.; Nguyen, L.N.; Nabavi, S.; Malinow, R. The prion protein as a receptor for amyloid-β. Nature 2010, 466, E3–E5. [Google Scholar] [CrossRef] [PubMed]
- Calella, A.M.; Farinelli, M.; Nuvolone, M.; Mirante, O.; Moos, R.; Falsig, J.; Mansuy, I.M.; Aguzzi, A. Prion protein and A-β-related synaptic toxicity impairment. EMBO Mol. Med. 2010, 2, 306–314. [Google Scholar] [CrossRef]
- Mengel, D.; Hong, W.; Corbett, G.T.; Liu, W.; DeSousa, A.; Solforosi, L.; Fang, C.; Frosch, M.P.; Collinge, J.; Harris, D.A.; et al. PrP-grafted antibodies bind certain amyloid β-protein aggregates, but do not prevent toxicity. Brain Res. 2019, 1710, 125–135. [Google Scholar] [CrossRef]
- Chen, S.; Yadav, S.P.; Surewicz, W.K. Interaction between human prion protein and amyloid-β (Aβ) oligomers: Role of N-terminal residues. J. Biol. Chem. 2010, 285, 26377–26383. [Google Scholar] [CrossRef]
- De Cecco, E.; Legname, G. The role of the prion protein in the internalization of α-synuclein amyloids. Prion 2018, 12, 23–27. [Google Scholar] [CrossRef]
- Gomes, L.A.; Hipp, S.A.; Upadhaya, A.R.; Balakrishnan, K.; Ospitalieri, S.; Koper, M.J.; Largo-Barrientos, P.; Uytterhoeven, V.; Reichwald, J.; Rabe, S. Aβ-induced acceleration of Alzheimer-related τ-pathology spreading and its association with prion protein. Acta Neuropathol. 2019, 138, 913–941. [Google Scholar] [CrossRef]
- Rösener, N.S.; Gremer, L.; Wördehoff, M.M.; Kupreichyk, T.; Etzkorn, M.; Neudecker, P.; Hoyer, W. Clustering of human prion protein and α-synuclein oligomers requires the prion protein N-terminus. Commun. Biol. 2020, 3, 365. [Google Scholar] [CrossRef]
- De Cecco, E.; Celauro, L.; Vanni, S.; Grandolfo, M.; Bistaffa, E.; Moda, F.; Aguzzi, A.; Legname, G. The uptake of tau amyloid fibrils is facilitated by the cellular prion protein and hampers prion propagation in cultured cells. J. Neurochem. 2020, 155, 577–591. [Google Scholar] [CrossRef]
- Resenberger, U.K.; Harmeier, A.; Woerner, A.C.; Goodman, J.L.; Muller, V.; Krishnan, R. The cellular prion protein mediates neurotoxic signalling of β-sheet-rich conformers independent of prion replication. EMBO J. 2011, 30, 2057–2070. [Google Scholar] [CrossRef] [PubMed]
- Nieznanski, K.; Choi, J.K.; Chen, S.; Surewicz, K.; Surewicz, W.K. Soluble prion protein inhibits amyloid-β (Aβ) fibrillization and toxicity. J. Biol. Chem. 2012, 287, 33104–33108. [Google Scholar] [CrossRef]
- Fluharty, B.R.; Biasini, E.; Stravalaci, M.; Sclip, A.; Diomede, L.; Balducci, C.; La Vitola, P.; Messa, M.; Colombo, L.; Forloni, G.; et al. An N-terminal Fragment of the Prion Protein Binds to Amyloid-β Oligomers and Inhibits Their Neurotoxicity in Vivo. J. Biol. Chem. 2013, 288, 7857–7866. [Google Scholar] [CrossRef] [PubMed]
- Nieznanska, H.; Bandyszewska, M.; Surewicz, K.; Zajkowski, T.; Surewicz, W.K.; Nieznanski, K. Identification of prion protein-derived peptides of potential use in Alzheimer’s disease therapy. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2143–2153. [Google Scholar] [CrossRef] [PubMed]
- Béland, M.; Bédard, M.; Tremblay, G.; Lavigne, P.; Roucou, X. Aβ induces its own prion protein N-terminal fragment (PrPN1)–mediated neutralization in amorphous aggregates. Neurobiol. Aging 2014, 35, 1537–1548. [Google Scholar] [CrossRef]
- Heiseke, A.; Schöbel, S.; Lichtenthaler, S.F.; Vorberg, I.; Groschup, M.H.; Kretzschmar, H.; Schätzl, H.M.; Nunziante, M. The Novel Sorting Nexin SNX33 Interferes with Cellular PrPSc Formation by Modulation of PrPc Shedding. Traffic 2008, 9, 1116–1129. [Google Scholar] [CrossRef] [PubMed]
- Kanaani, J.; Prusiner, S.B.; Diacovo, J.; Baekkeskov, S.; Legname, G. Recombinant prion protein induces rapid polarization and development of synapses in embryonic rat hippocampal neurons in vitro: Prion protein enhances neuronal polarization. J. Neurochem. 2005, 95, 1373–1386. [Google Scholar] [CrossRef]
- Bove-Fenderson, E.; Urano, R.; Straub, J.E.; Harris, D.A. Cellular prion protein targets amyloid-β fibril ends via its C-terminal domain to prevent elongation. J. Biol. Chem. 2017, 292, 16858–16871. [Google Scholar] [CrossRef]
- Roberts, T.K.; Eugenin, E.A.; Morgello, S.; Clements, J.E.; Zink, M.C.; Berman, J.W. PrP(C), the cellular isoform of the human prion protein, is a novel biomarker of HIV-associated neurocognitive impairment and mediates neuroinflammation. Am. J. Pathol. 2010, 177, 1848–1860. [Google Scholar] [CrossRef]
- Linsenmeier, L.; Mohammadi, B.; Wetzel, S.; Puig, B.; Jackson, W.S.; Hartmann, A.; Uchiyama, K.; Sakaguchi, S.; Endres, K.; Tatzelt, J.; et al. Structural and mechanistic aspects influencing the ADAM10-mediated shedding of the prion protein. Mol. Neurodegener. 2018, 13, 18. [Google Scholar] [CrossRef]
- Legname, G.; Scialò, C. On the role of the cellular prion protein in the uptake and signaling of pathological aggregates in neurodegenerative diseases. Prion 2020, 14, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Scott-McKean, J.J.; Surewicz, K.; Choi, J.-K.; Ruffin, V.A.; Salameh, A.I.; Nieznanski, K.; Costa, A.C.; Surewicz, W.K. Soluble prion protein and its N-terminal fragment prevent impairment of synaptic plasticity by Aβ oligomers: Implications for novel therapeutic strategy in Alzheimer’s disease. Neurobiol. Dis. 2016, 91, 124–131. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vella, L.J.; Greenwood, D.L.V.; Cappai, R.; Scheerlinck, J.-P.Y.; Hill, A.F. Enrichment of prion protein in exosomes derived from ovine cerebral spinal fluid. Vet. Immunol. Immunopathol. 2008, 124, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Falker, C.; Hartmann, A.; Guett, I.; Dohler, F.; Altmeppen, H.; Betzel, C.; Schubert, R.; Thurm, D.; Wegwitz, F.; Joshi, P.; et al. Exosomal cellular prion protein drives fibrillization of amyloid beta and counteracts amyloid beta-mediated neurotoxicity. J. Neurochem. 2016, 137, 88–100. [Google Scholar] [CrossRef]
- Yuyama, K.; Sun, H.; Sakai, S.; Mitsutake, S.; Okada, M.; Tahara, H.; Furukawa, J.-i.; Fujitani, N.; Shinohara, Y.; Igarashi, Y. Decreased amyloid-β pathologies by intracerebral loading of glycosphingolipid-enriched exosomes in Alzheimer model mice. J. Biol. Chem. 2014, 289, 24488–24498. [Google Scholar] [CrossRef]
- Yuyama, K.; Sun, H.; Usuki, S.; Sakai, S.; Hanamatsu, H.; Mioka, T.; Kimura, N.; Okada, M.; Tahara, H.; Furukawa, J.-i. A potential function for neuronal exosomes: Sequestering intracerebral amyloid-β peptide. FEBS Lett. 2015, 589, 84–88. [Google Scholar] [CrossRef]
- An, K.; Klyubin, I.; Kim, Y.; Jung, J.H.; Mably, A.J.; T O’Dowd, S.; Lynch, T.; Kanmert, D.; Lemere, C.A.; Finan, G.M. Exosomes neutralize synaptic-plasticity-disrupting activity of Aβ assemblies in vivo. Mol. Brain 2013, 6, 1–13. [Google Scholar] [CrossRef]
- Cervenakova, L.; Saá, P.; Yakovleva, O.; Vasilyeva, I.; de Castro, J.; Brown, P.; Dodd, R. Are prions transported by plasma exosomes? Transfus. Apher. Sci. 2016, 55, 70–83. [Google Scholar] [CrossRef]
- Vella, L.; Sharples, R.; Lawson, V.; Masters, C.; Cappai, R.; Hill, A. Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2007, 211, 582–590. [Google Scholar] [CrossRef]
- Alves, R.N.; Iglesia, R.P.; Prado, M.B.; Melo Escobar, M.I.; Boccacino, J.M.; Fernandes, C.F.d.L.; Coelho, B.P.; Fortes, A.C.; Lopes, M.H. A New Take on Prion Protein Dynamics in Cellular Trafficking. Int. J. Mol. Sci. 2020, 21, 7763. [Google Scholar] [CrossRef]
- Leblanc, P.; Arellano-Anaya, Z.E.; Bernard, E.; Gallay, L.; Provansal, M.; Lehmann, S.; Schaeffer, L.; Raposo, G.; Vilette, D. Isolation of Exosomes and Microvesicles from Cell Culture Systems to Study Prion Transmission. In Exosomes and Microvesicles: Methods and Protocols; Hill, A.F., Ed.; Springer: New York, NY, USA, 2017; pp. 153–176. [Google Scholar]
- Fevrier, B.; Vilette, D.; Archer, F.; Loew, D.; Faigle, W.; Vidal, M.; Laude, H.; Raposo, G. Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. USA 2004, 101, 9683–9688. [Google Scholar] [CrossRef] [PubMed]
- Rösener, N.S.; Gremer, L.; Reinartz, E.; König, A.; Brener, O.; Heise, H.; Hoyer, W.; Neudecker, P.; Willbold, D. A d-enantiomeric peptide interferes with heteroassociation of amyloid-β oligomers and prion protein. J. Biol. Chem. 2018, 293, 15748–15764. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovač, V.; Čurin Šerbec, V. Prion Protein: The Molecule of Many Forms and Faces. Int. J. Mol. Sci. 2022, 23, 1232. https://doi.org/10.3390/ijms23031232
Kovač V, Čurin Šerbec V. Prion Protein: The Molecule of Many Forms and Faces. International Journal of Molecular Sciences. 2022; 23(3):1232. https://doi.org/10.3390/ijms23031232
Chicago/Turabian StyleKovač, Valerija, and Vladka Čurin Šerbec. 2022. "Prion Protein: The Molecule of Many Forms and Faces" International Journal of Molecular Sciences 23, no. 3: 1232. https://doi.org/10.3390/ijms23031232
APA StyleKovač, V., & Čurin Šerbec, V. (2022). Prion Protein: The Molecule of Many Forms and Faces. International Journal of Molecular Sciences, 23(3), 1232. https://doi.org/10.3390/ijms23031232