
Kynurenic Acid and Its Analog SZR104 Exhibit Strong Antiinflammatory Effects and Alter the Intracellular Distribution and Methylation Patterns of H3 Histones in Immunochallenged Microglia-Enriched Cultures of Newborn Rat Brains


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
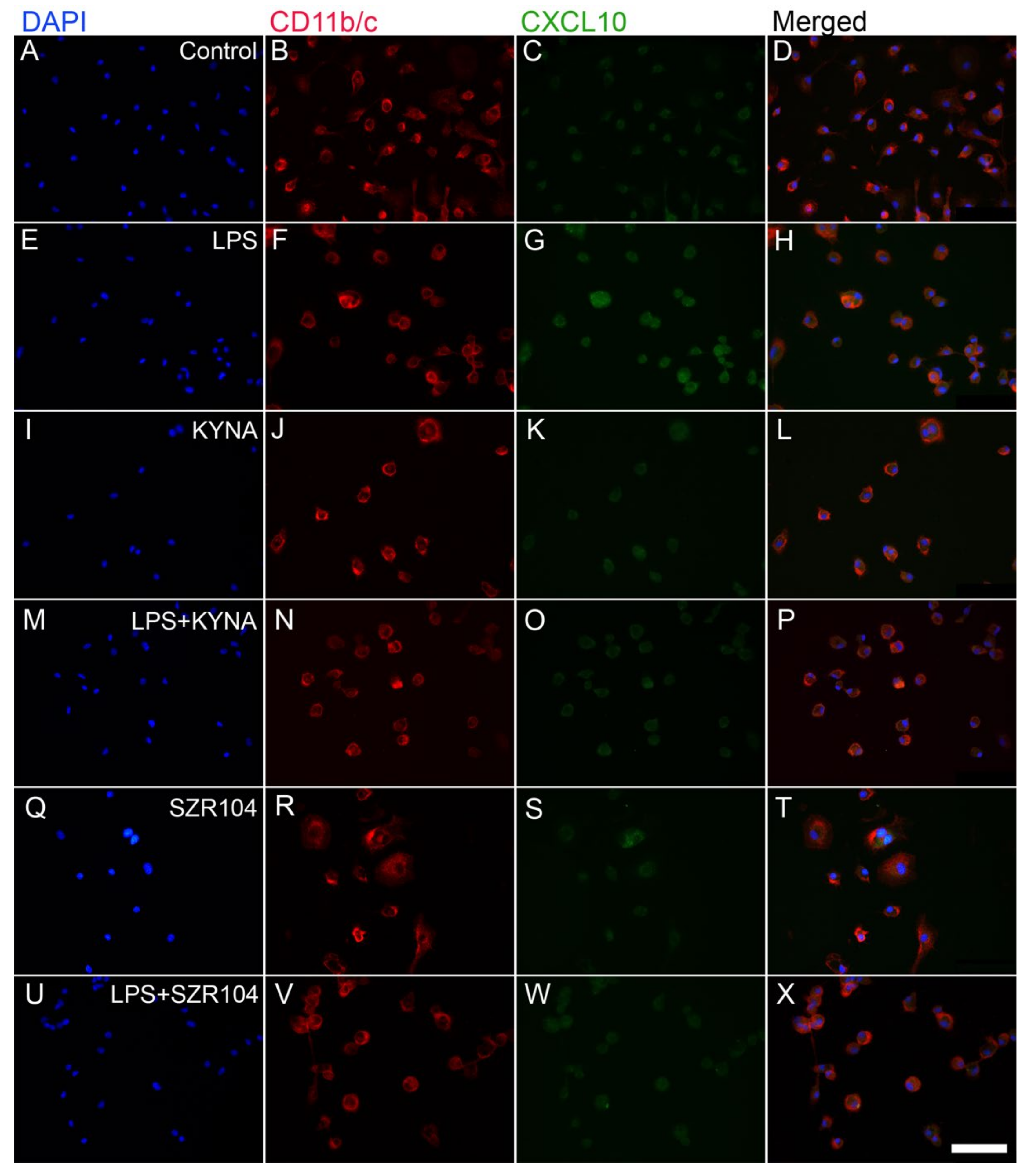
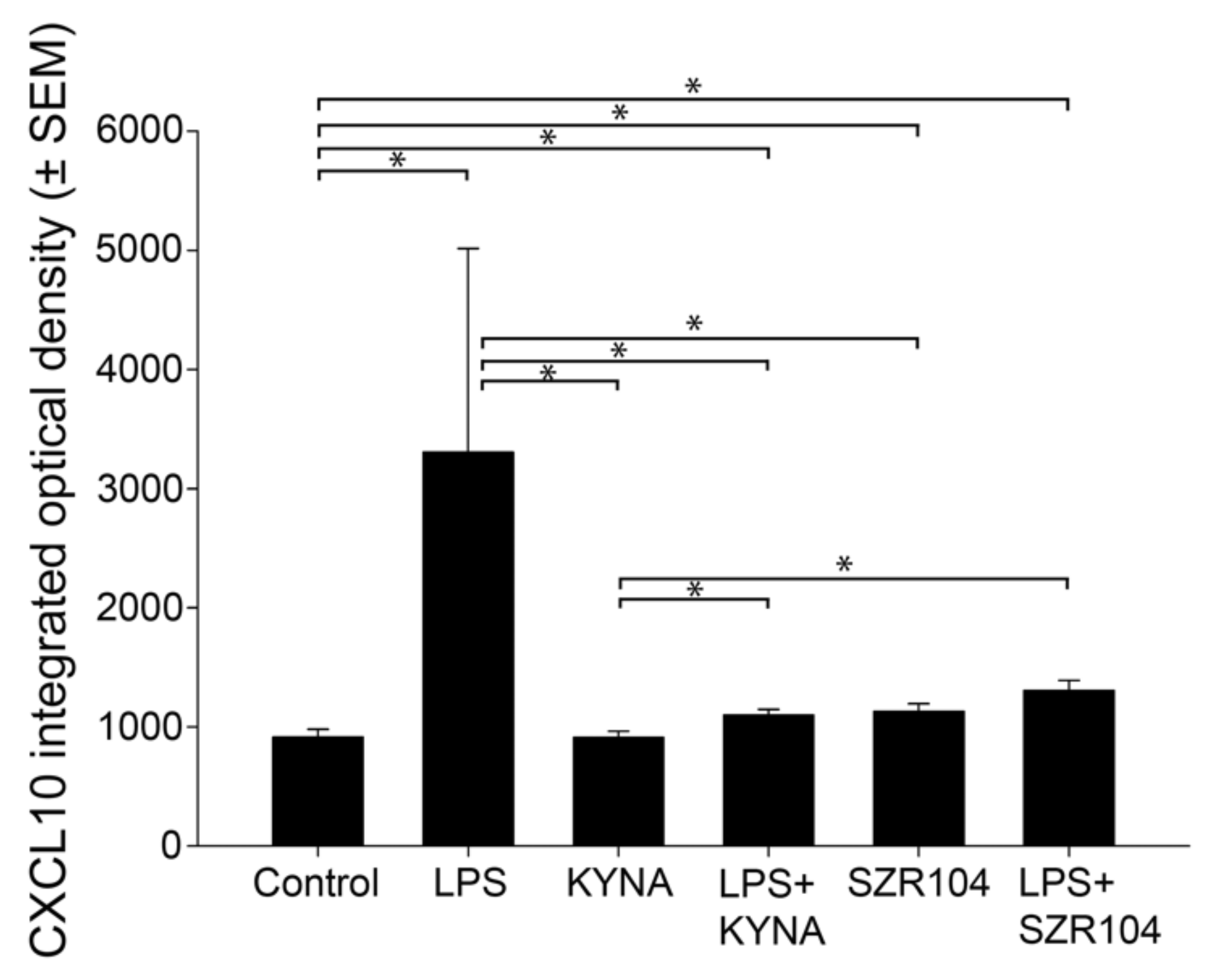
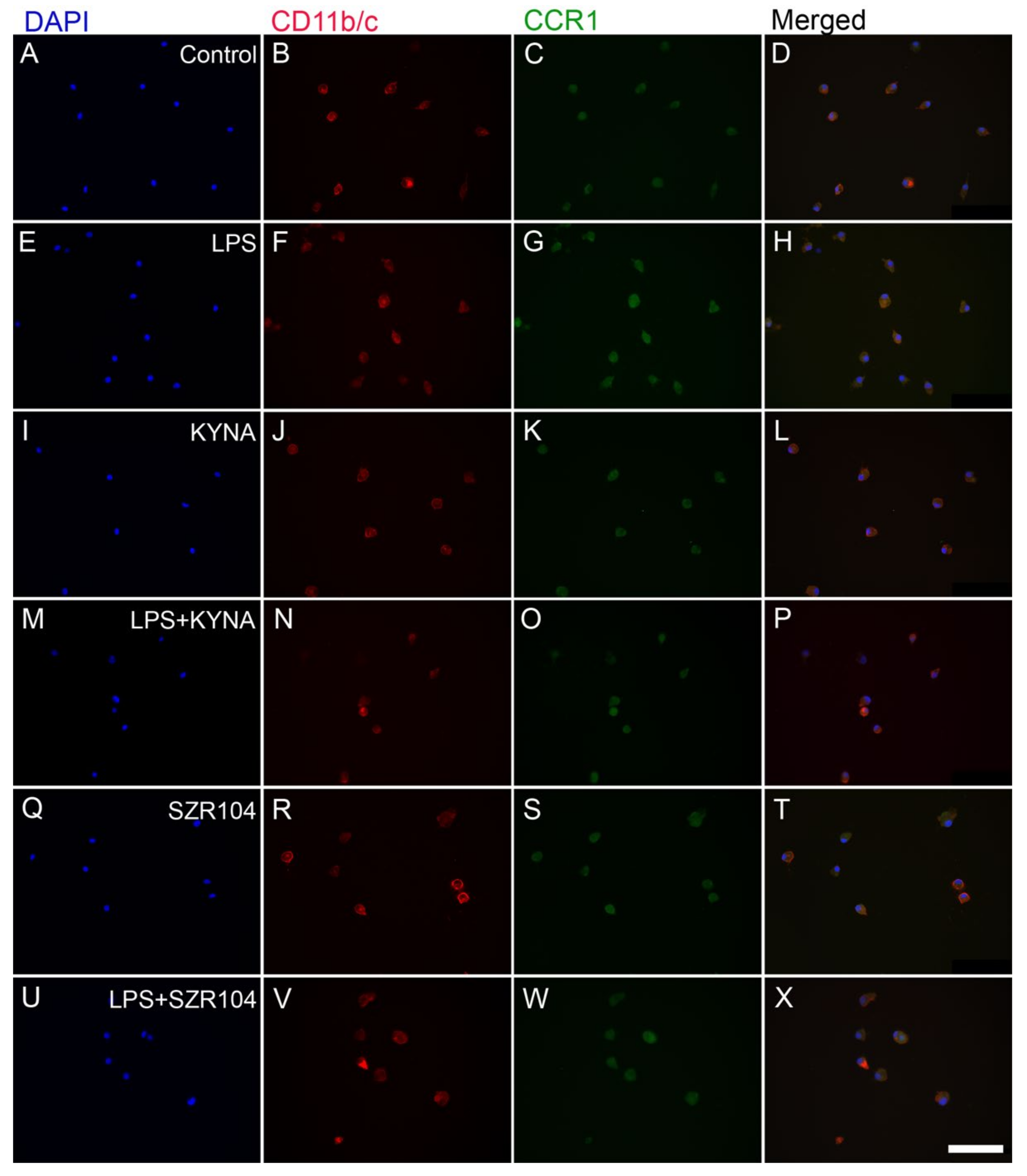
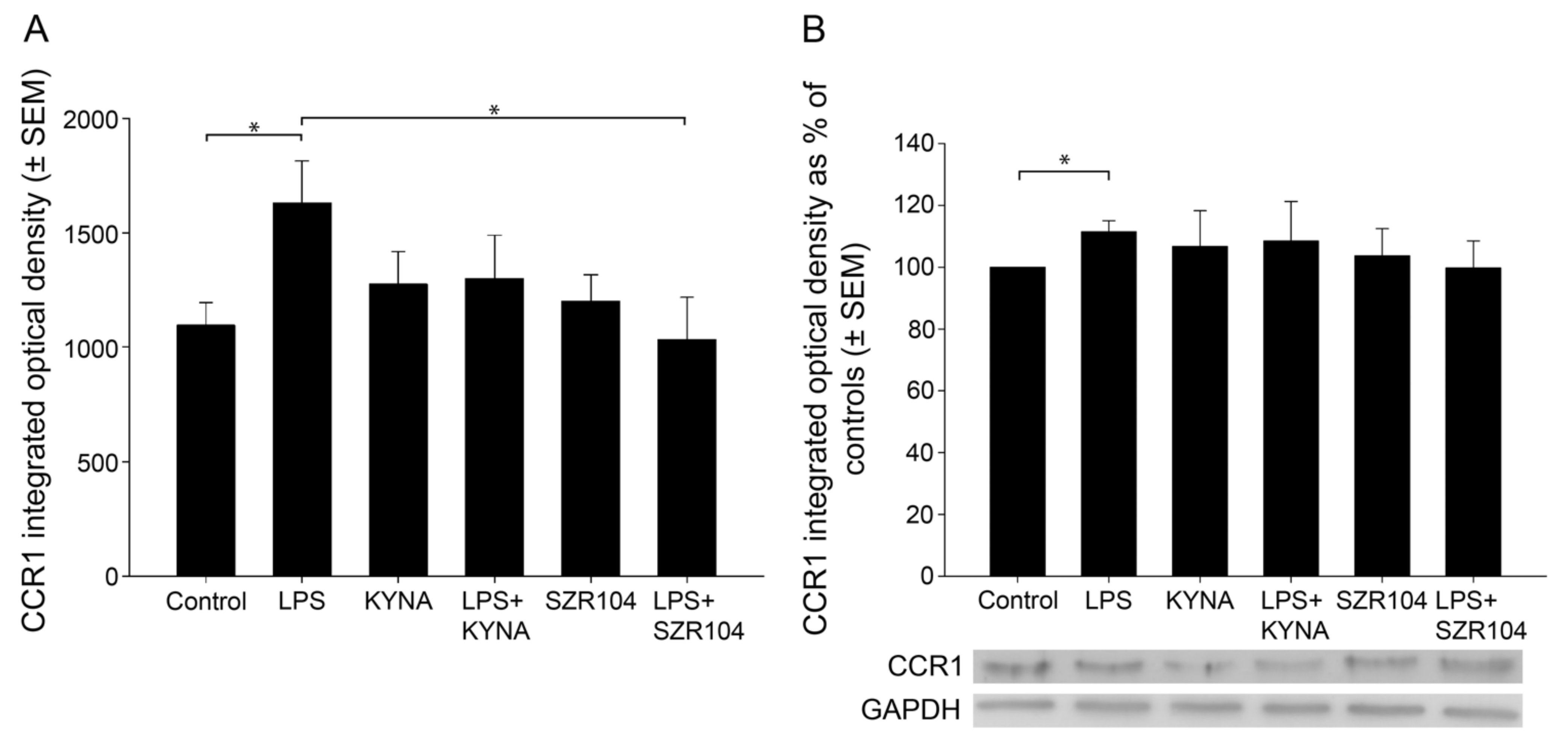
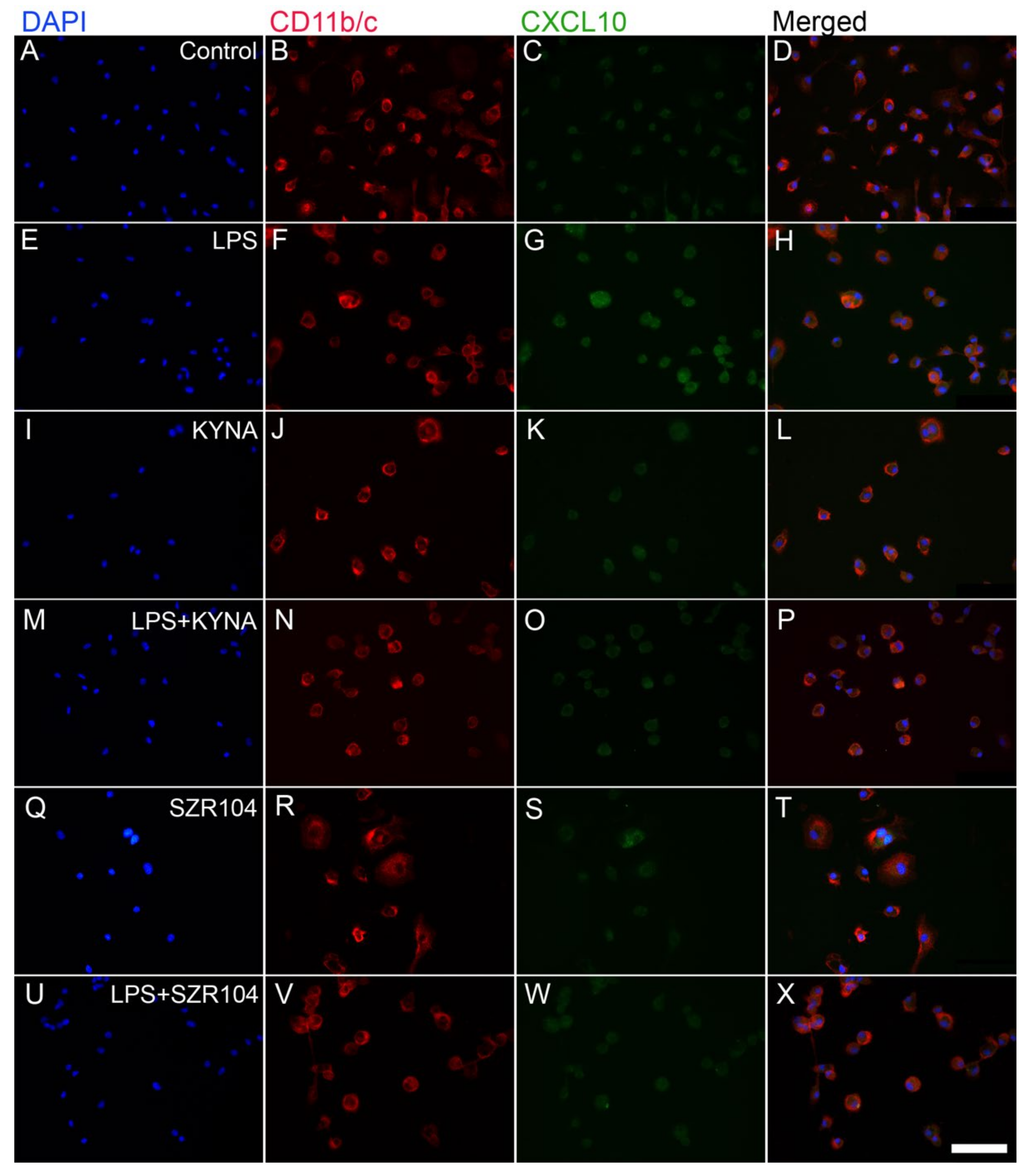
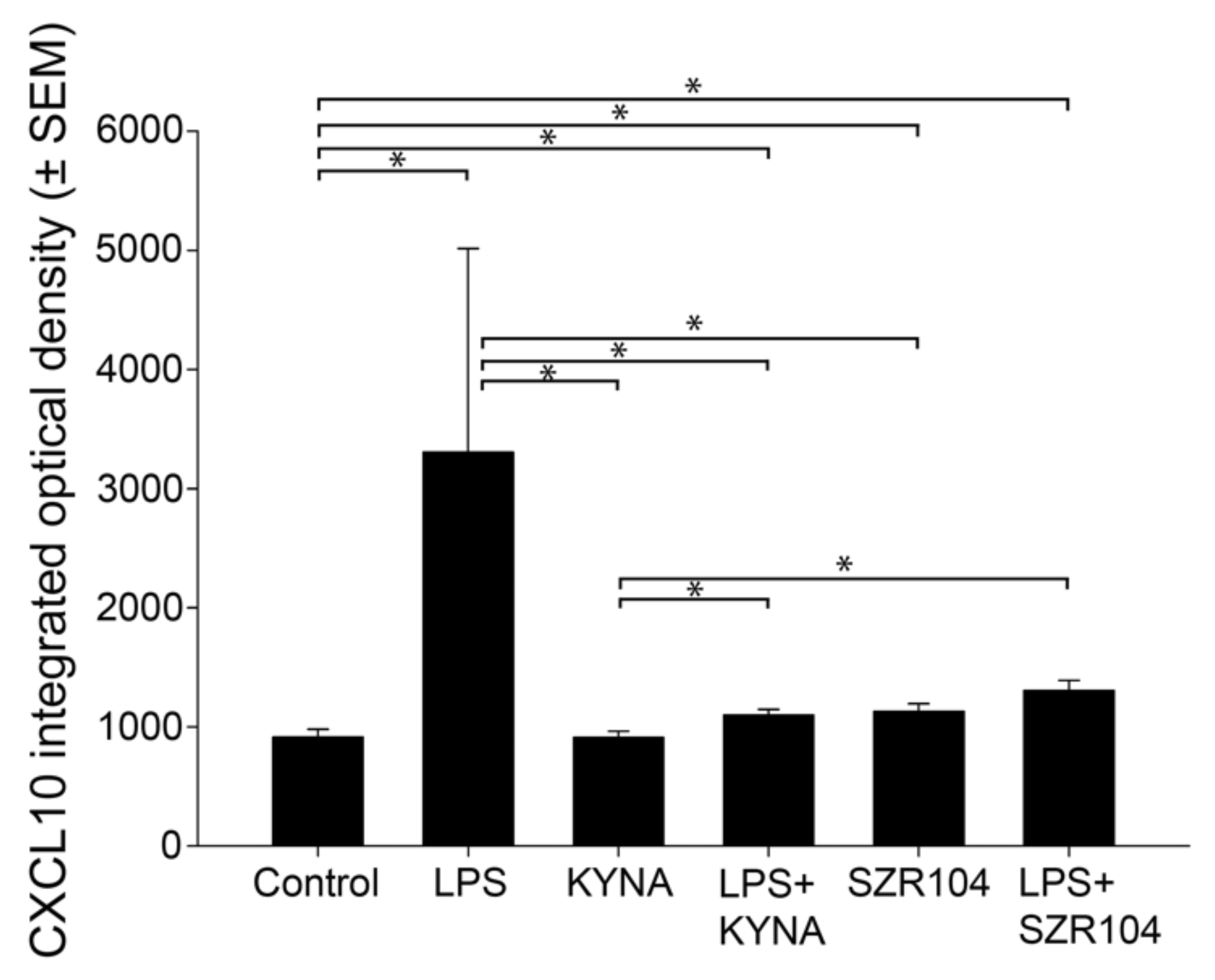
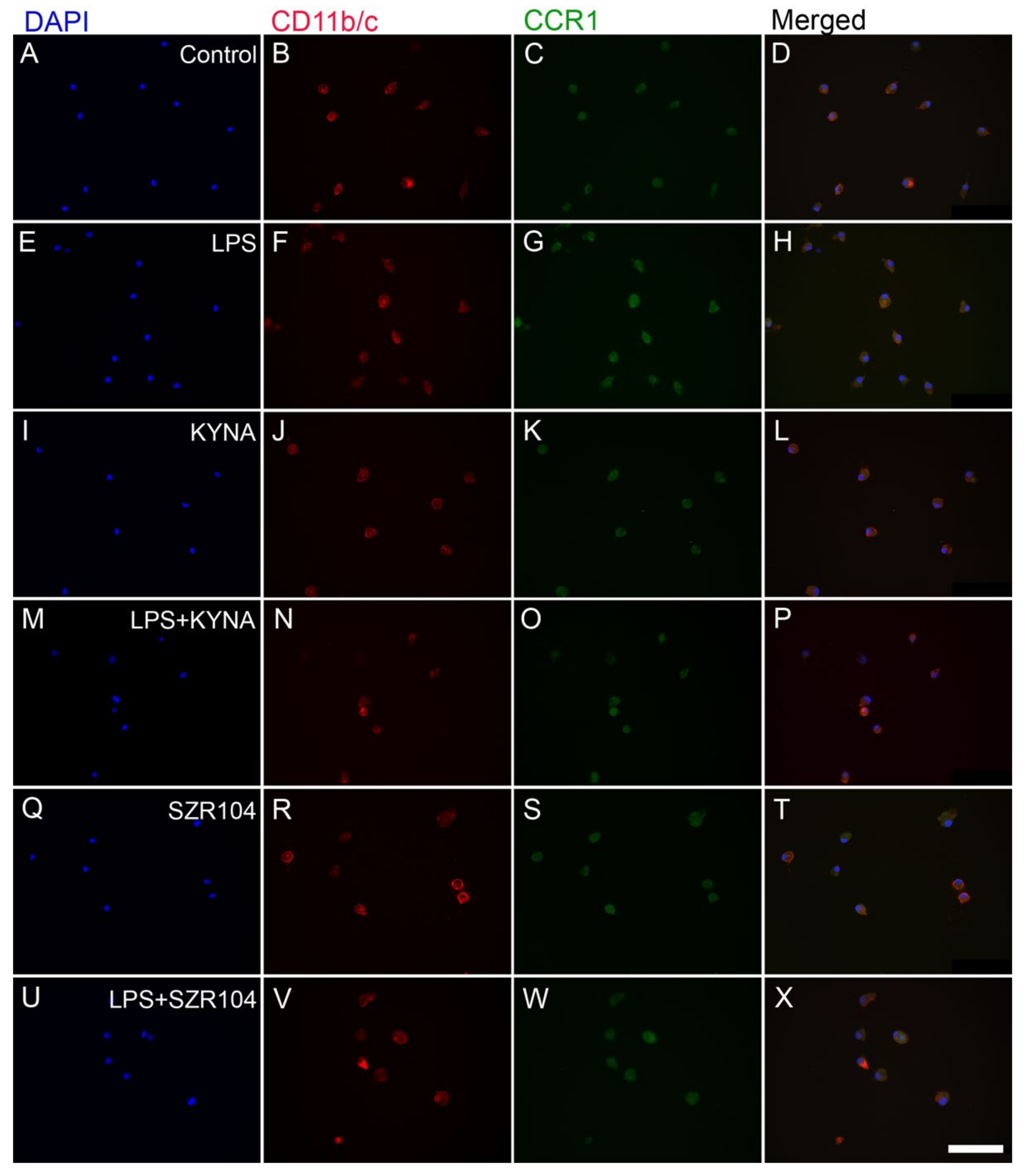
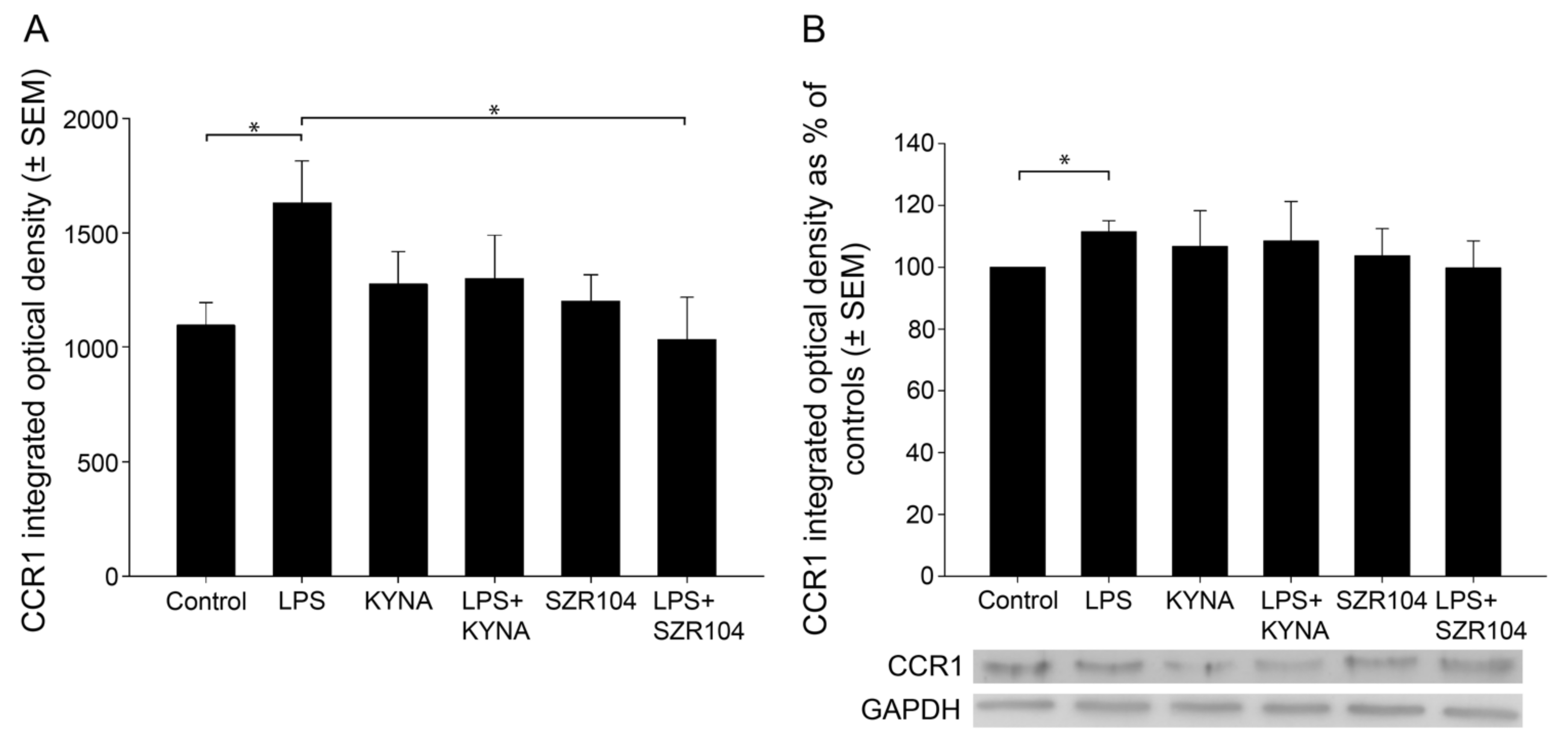
2.1. KYNA and SZR104 Downregulate LPS-Induced CXCL10 Levels While Differentially Altering CCR1 Levels
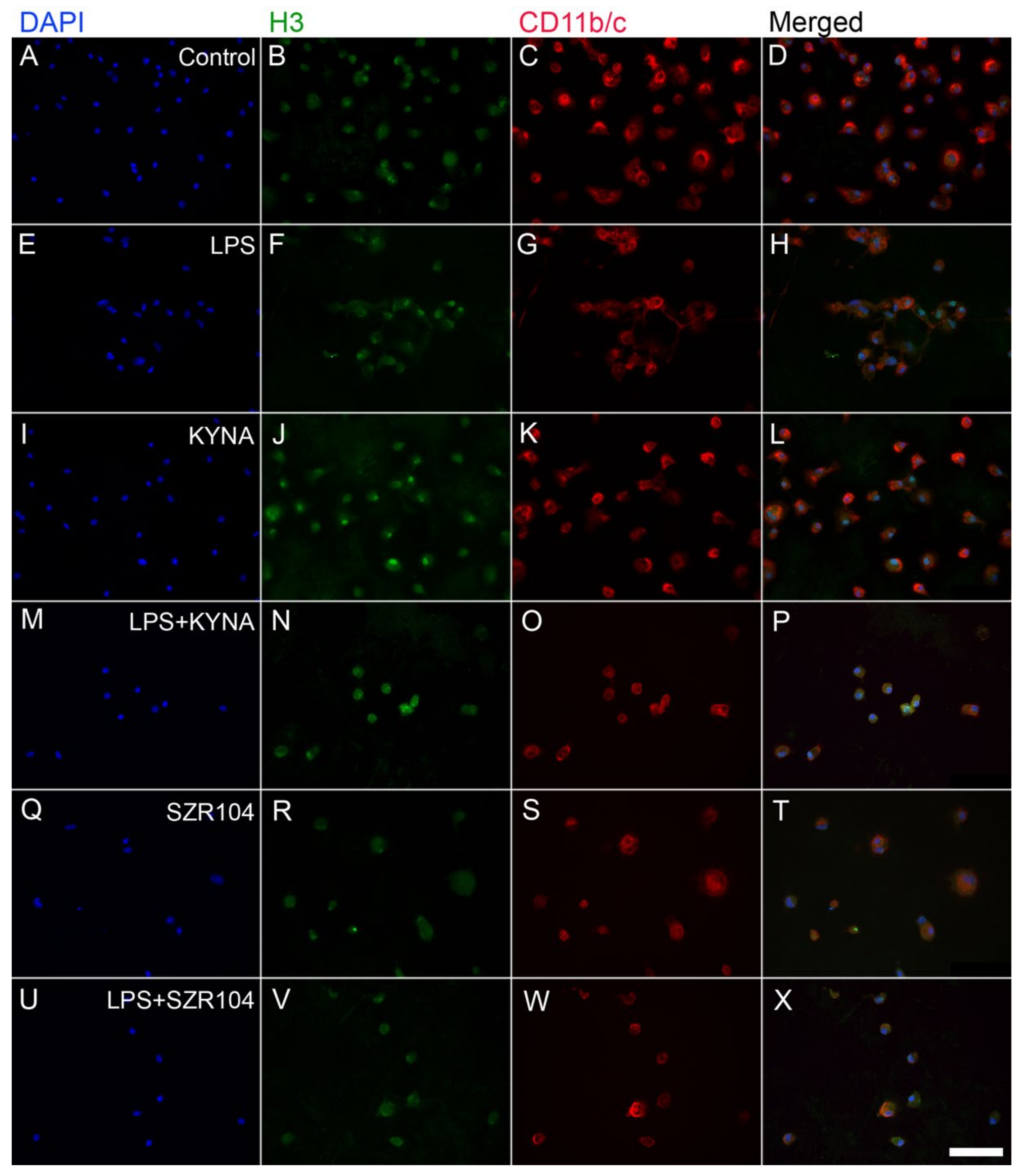
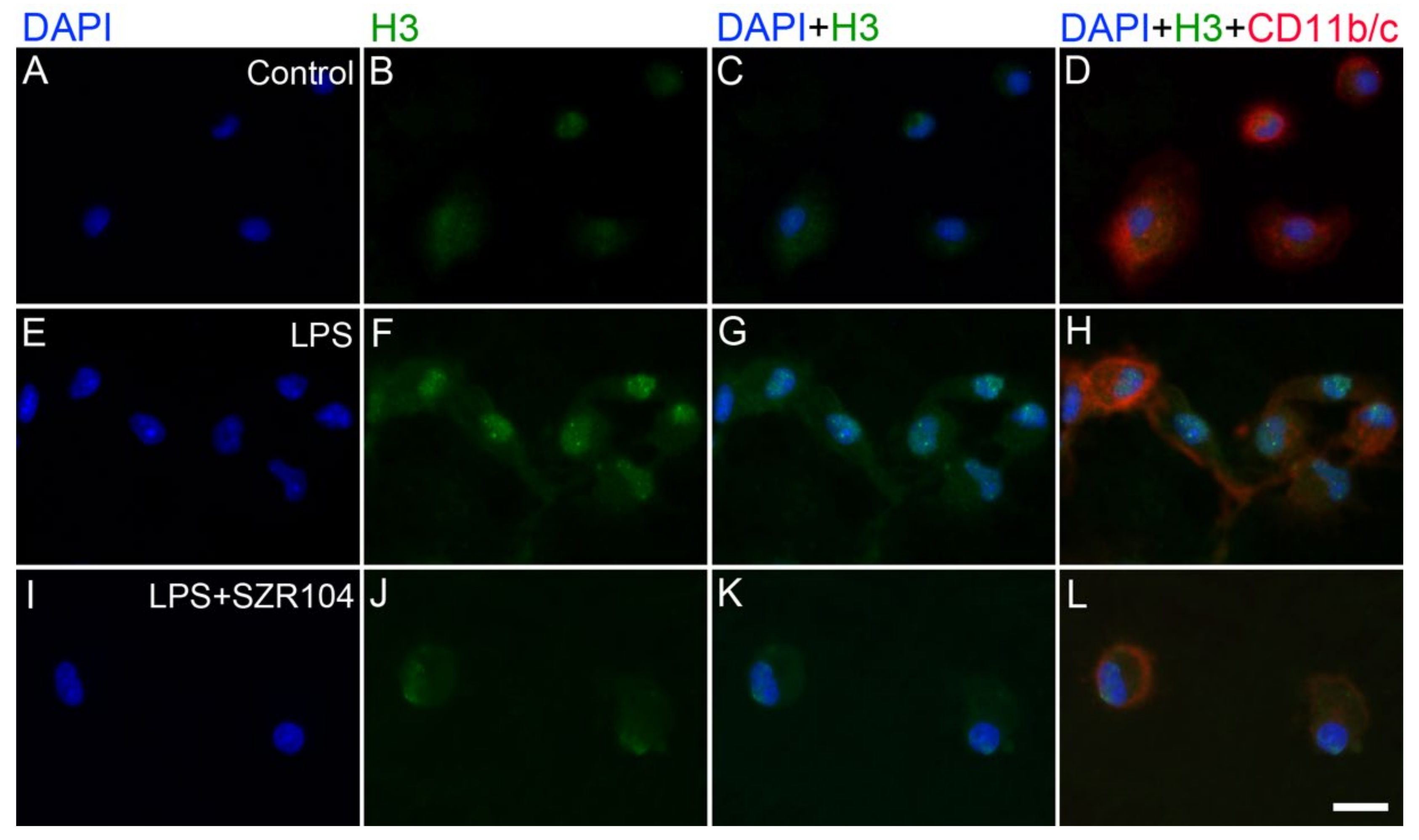
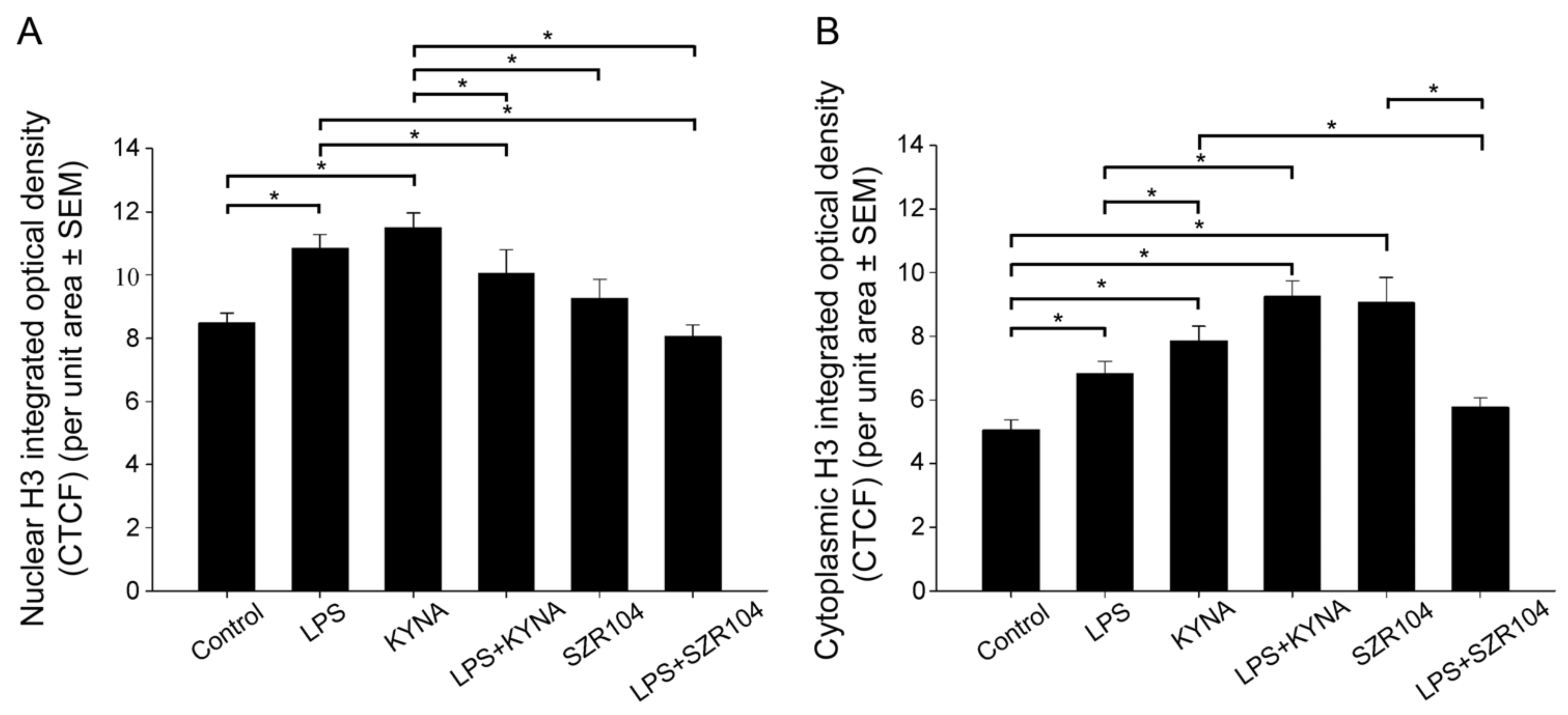
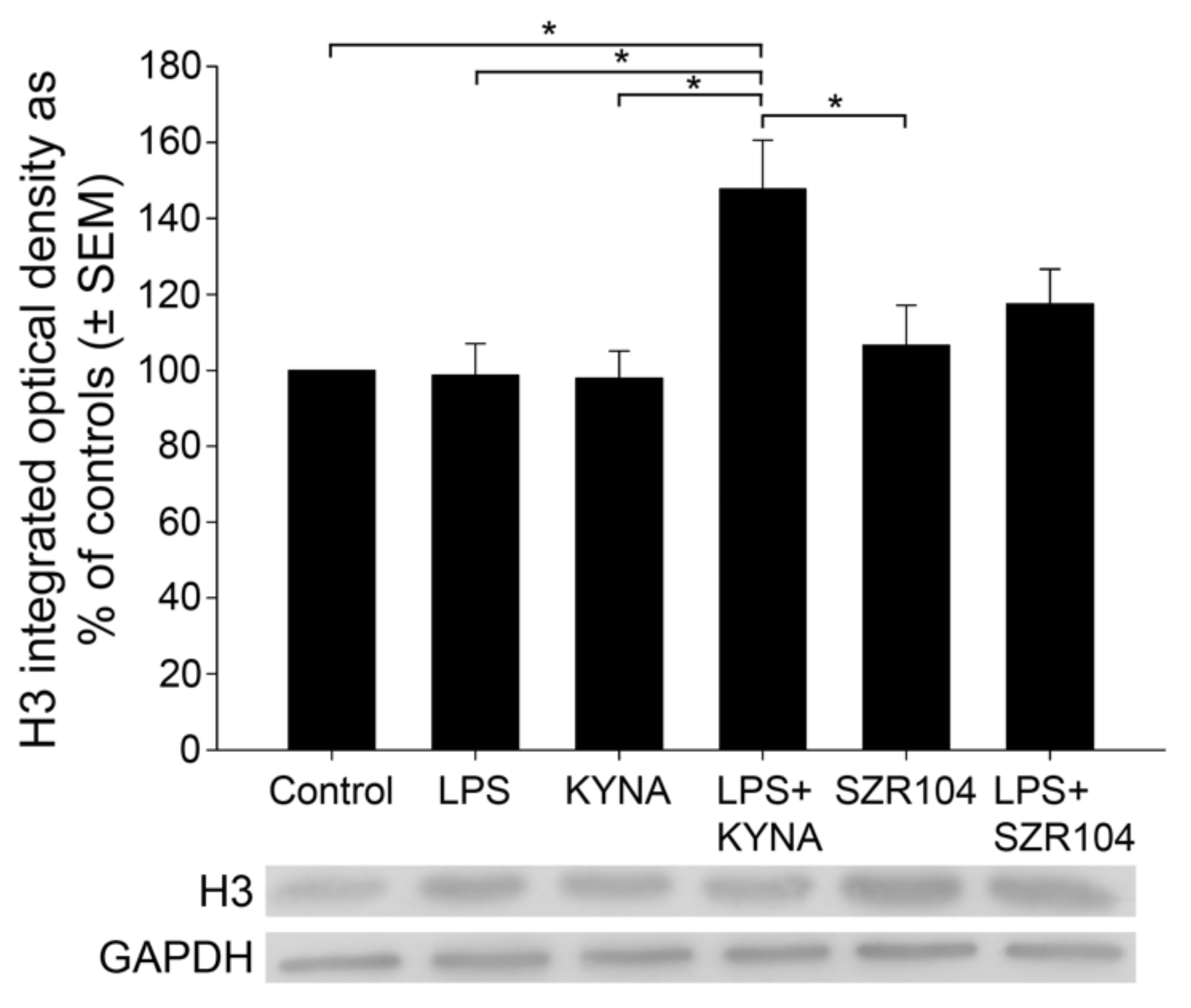
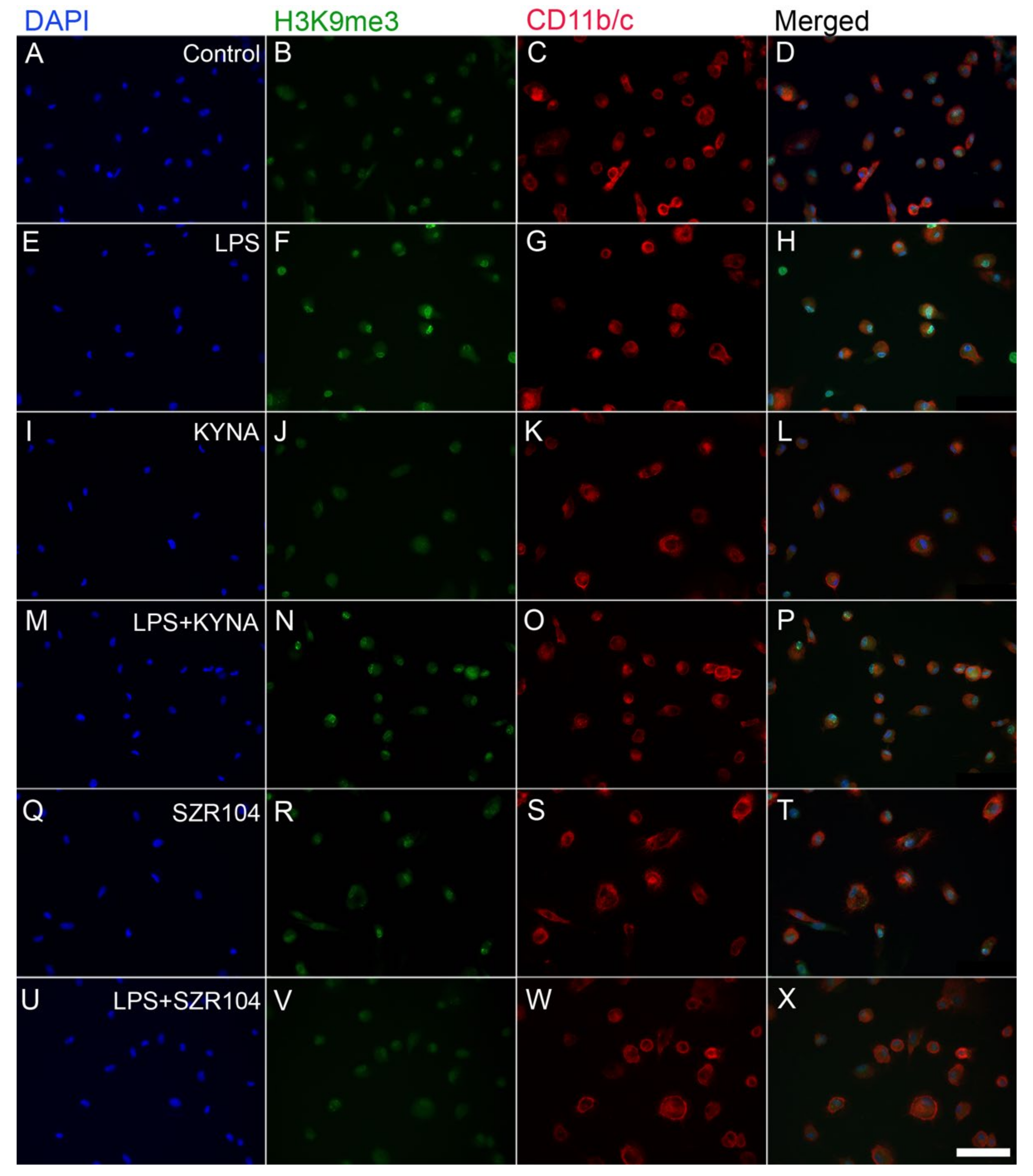
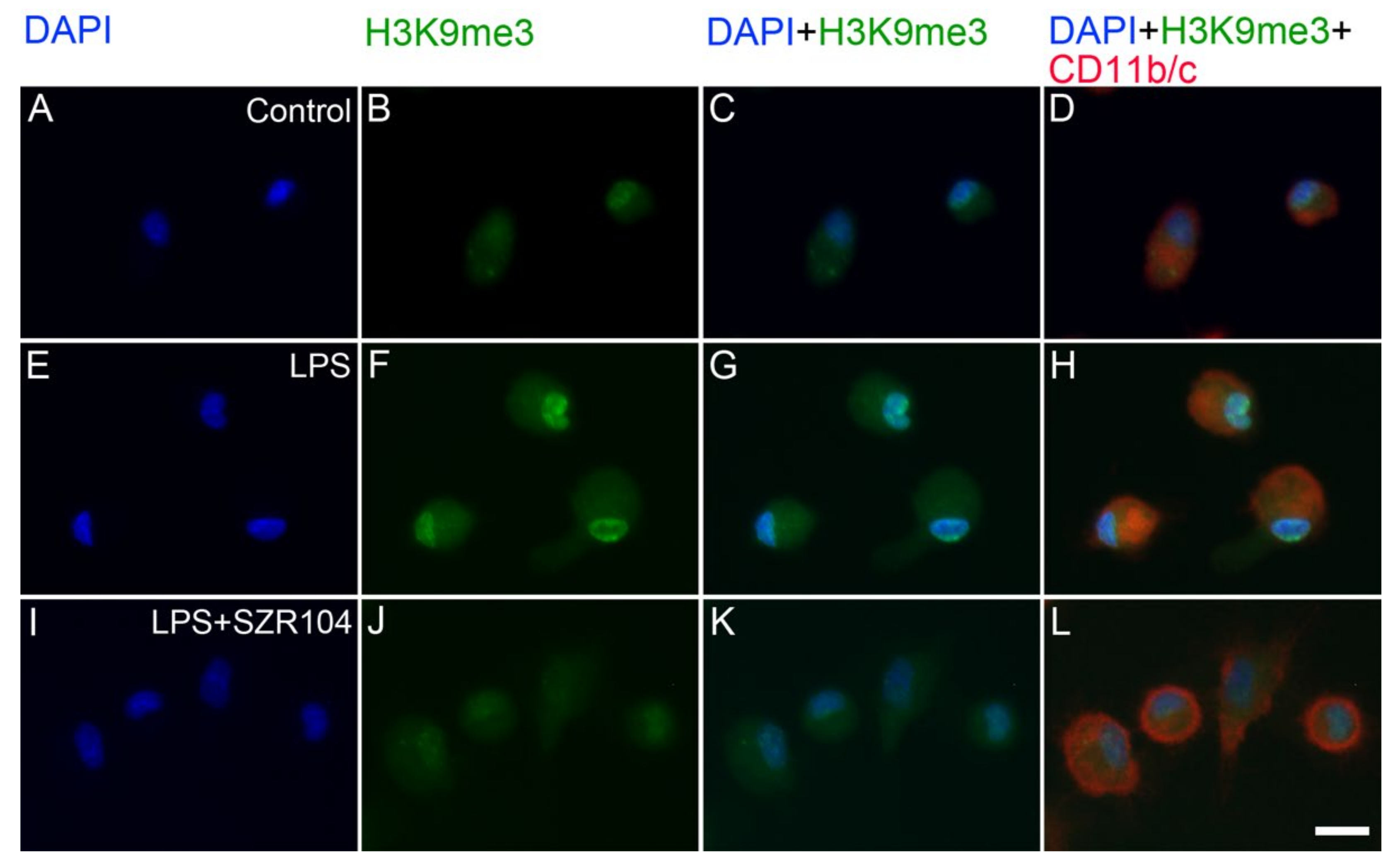
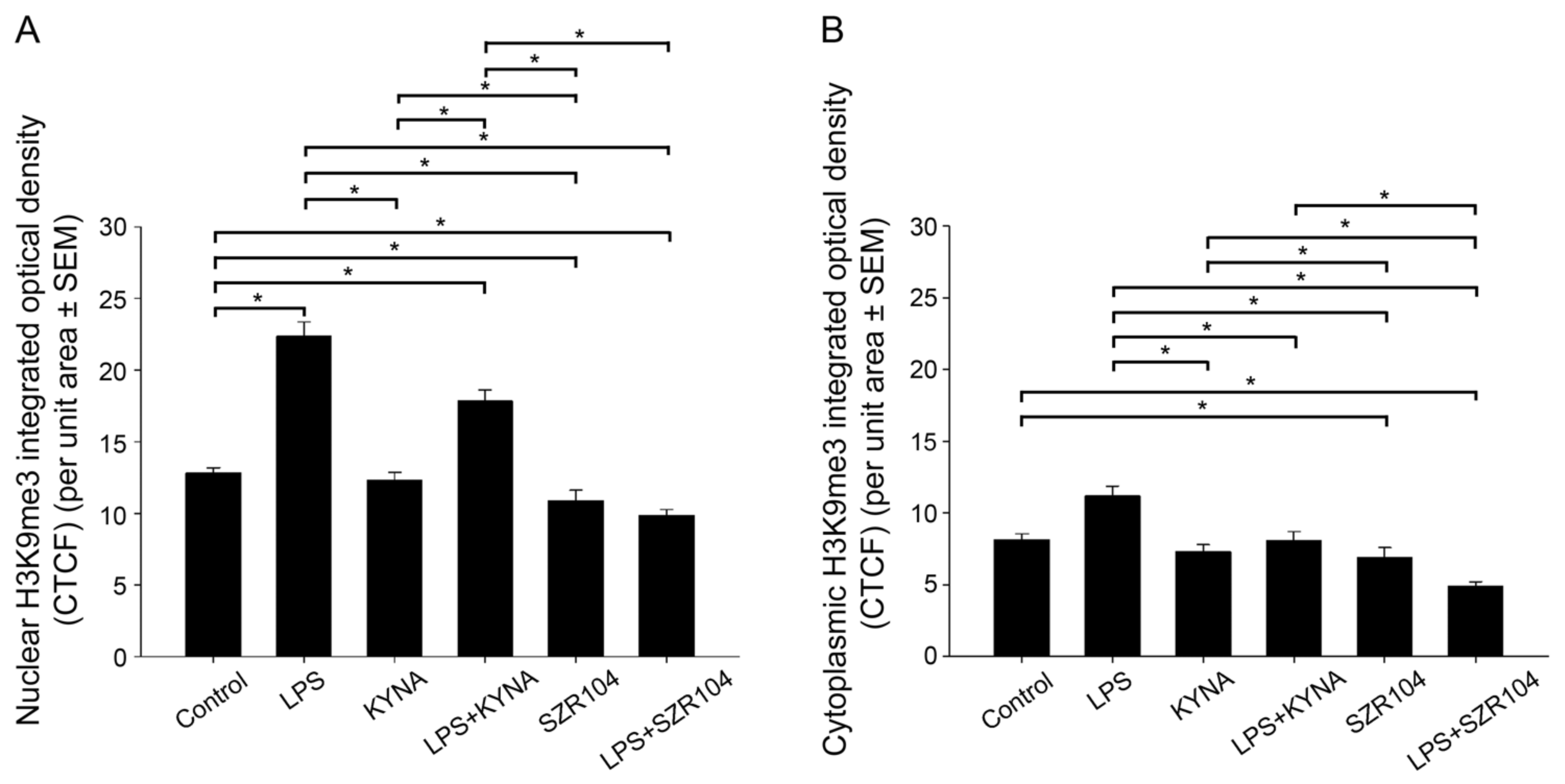
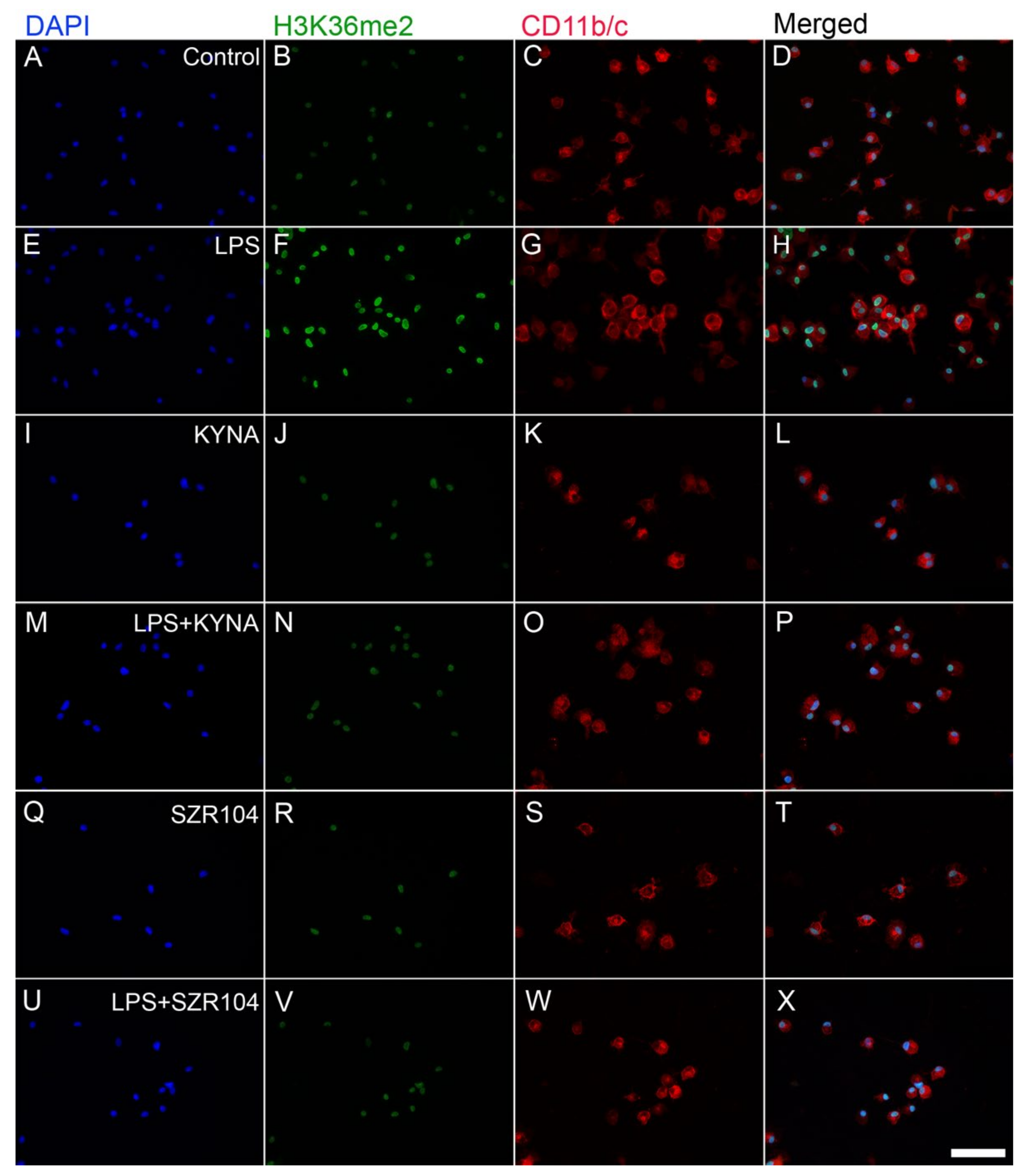
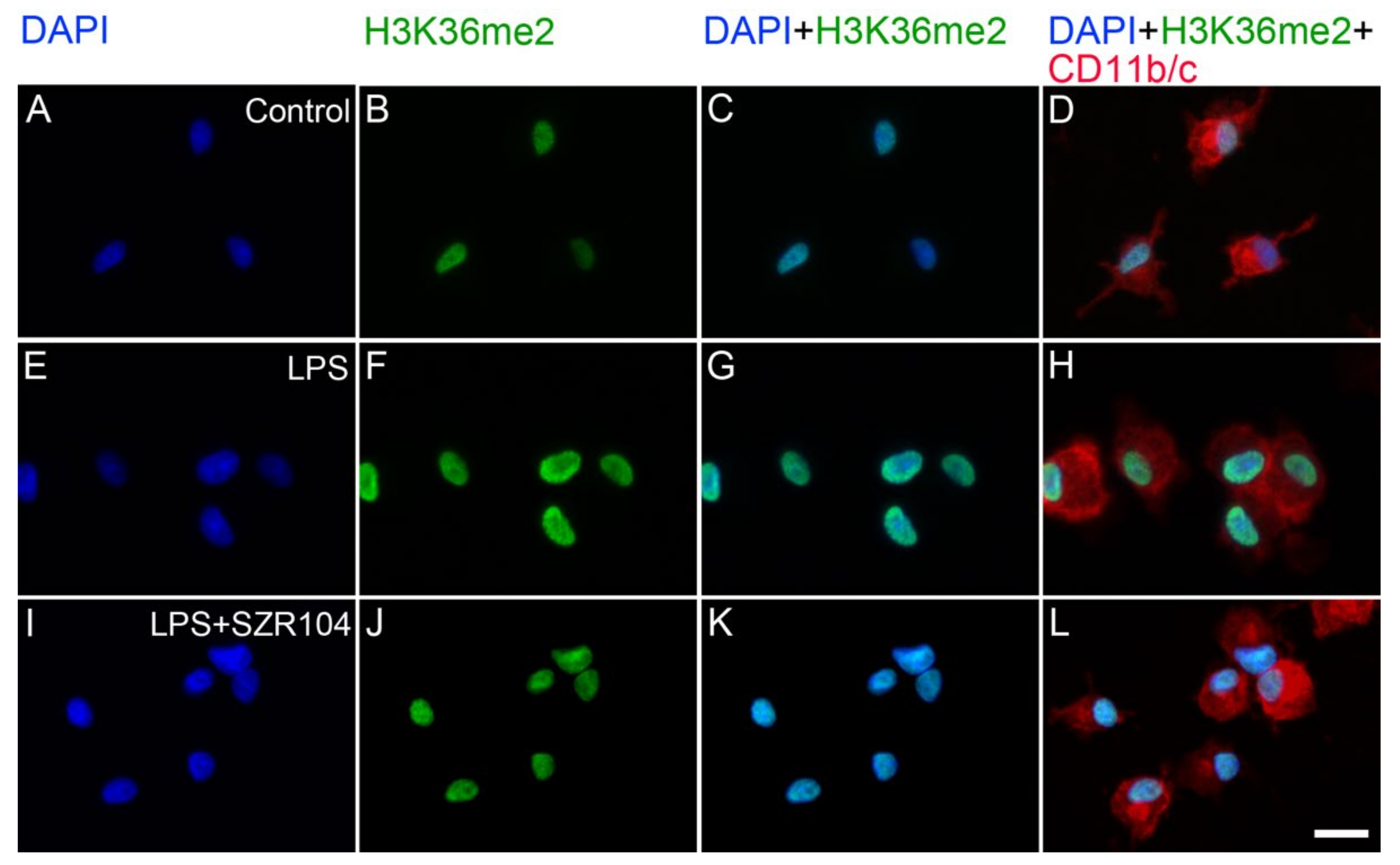
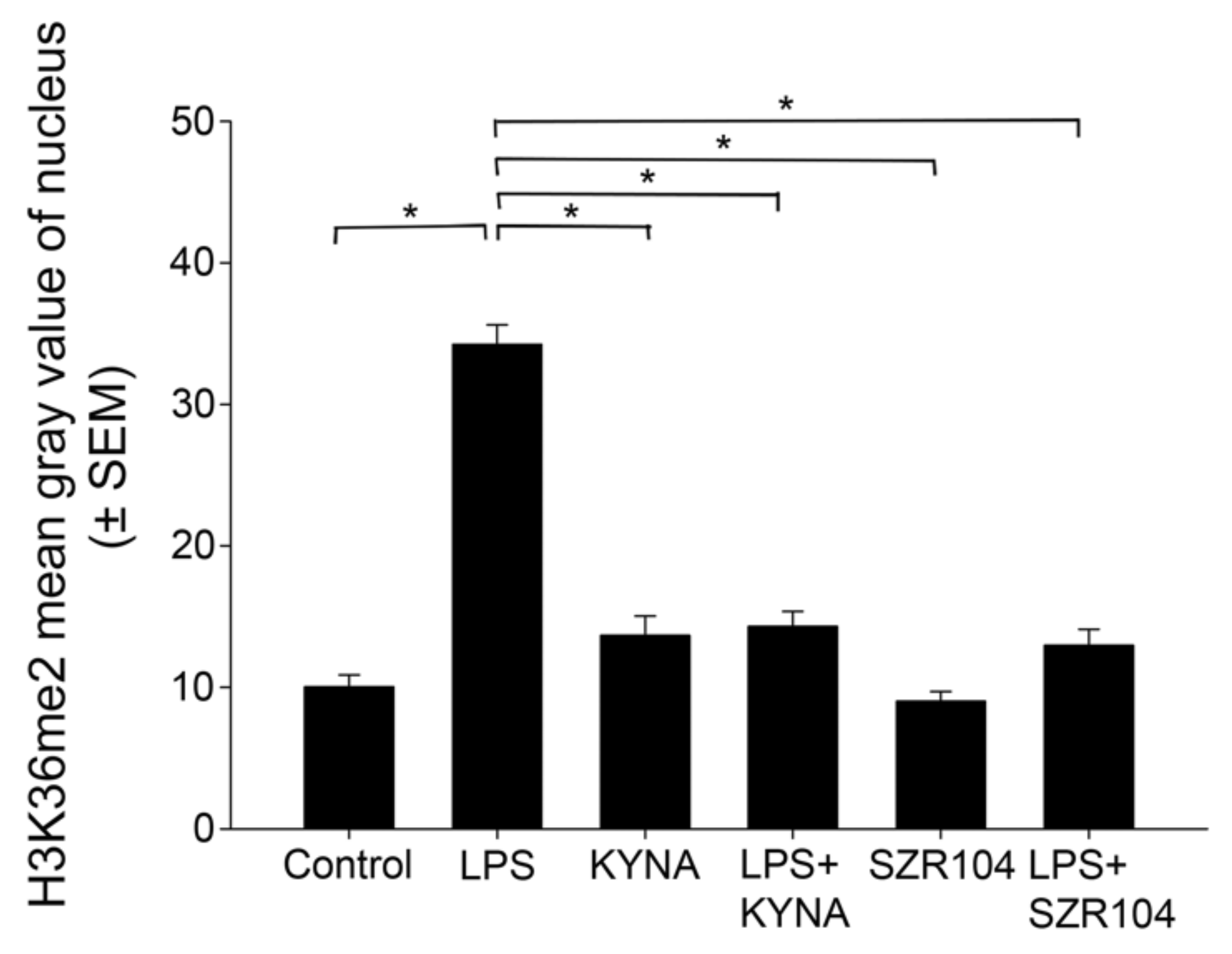
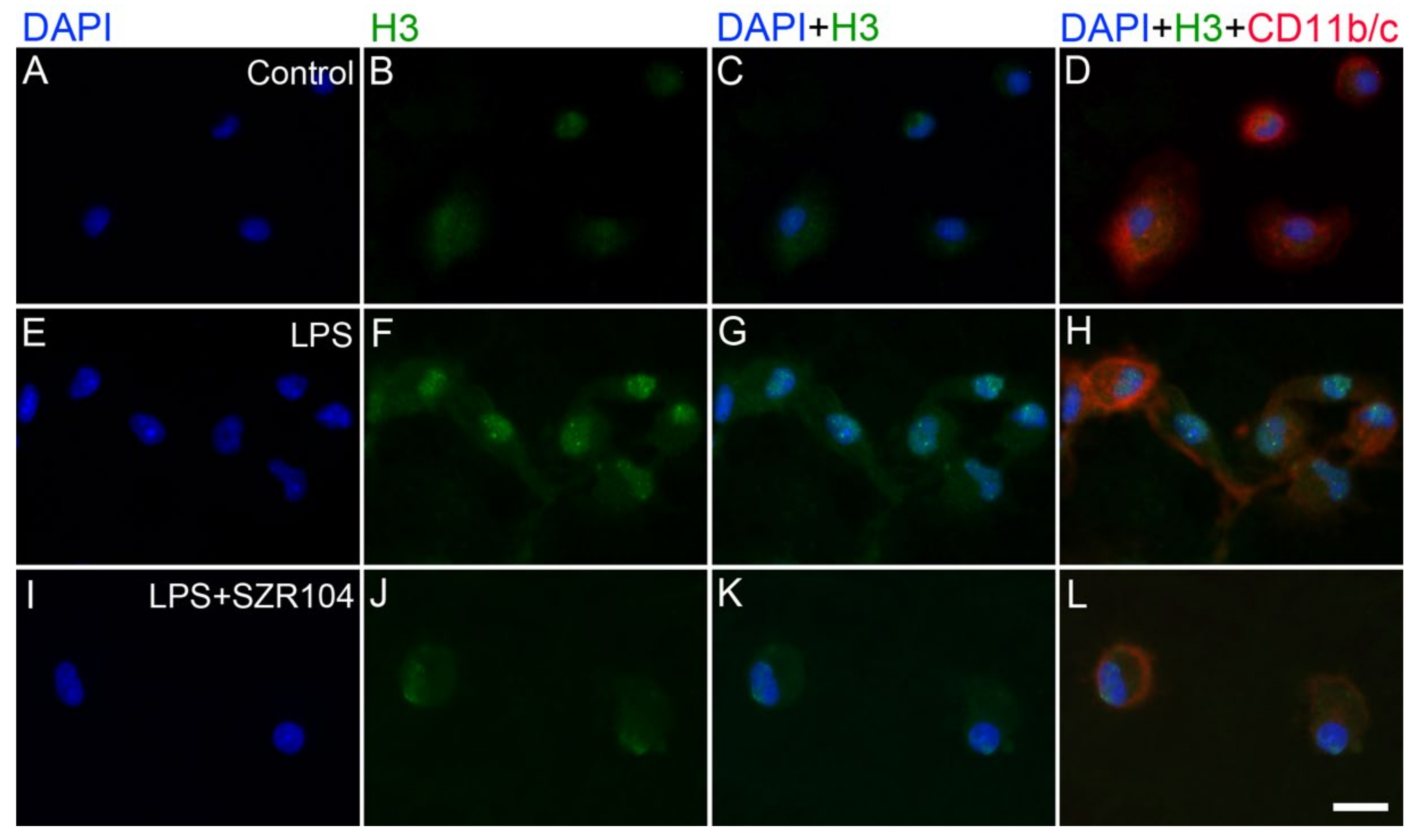
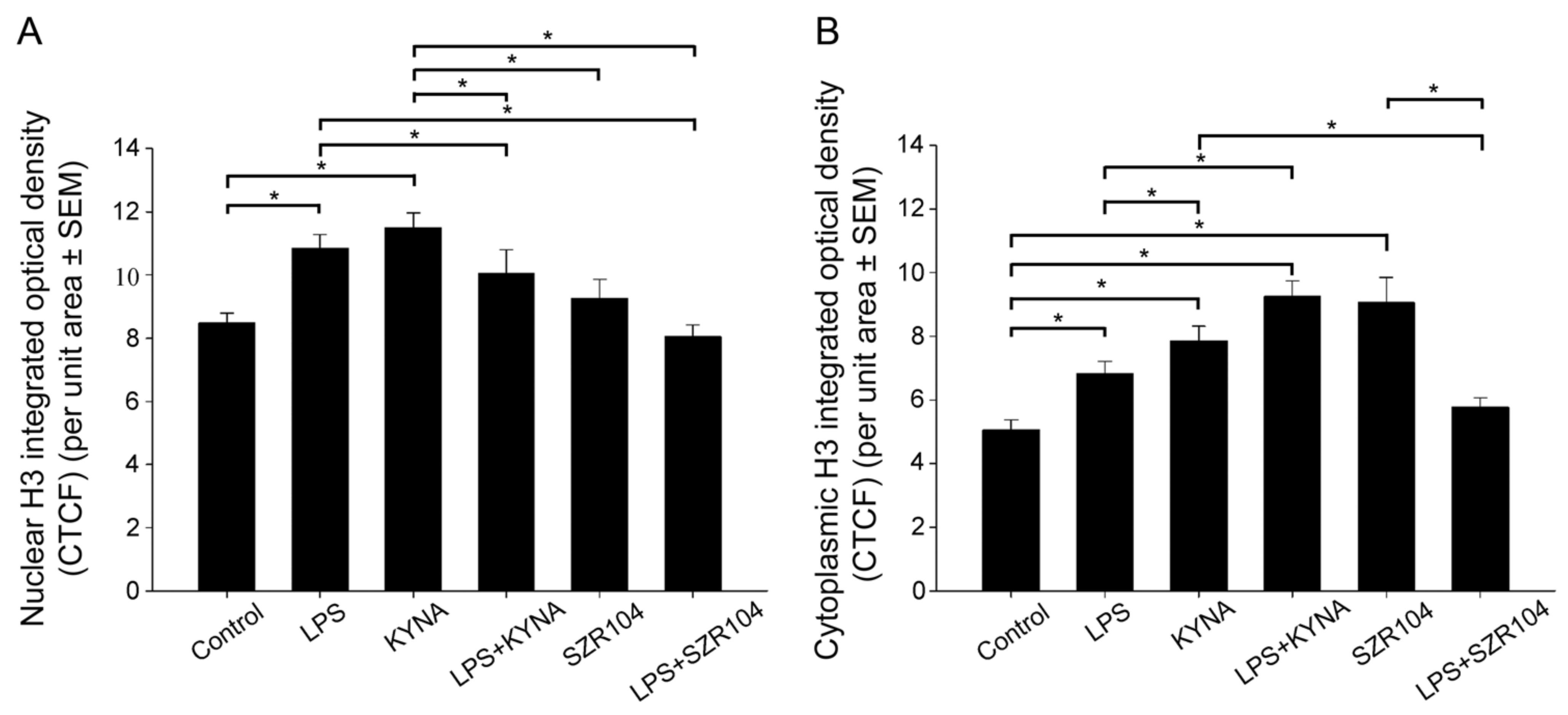
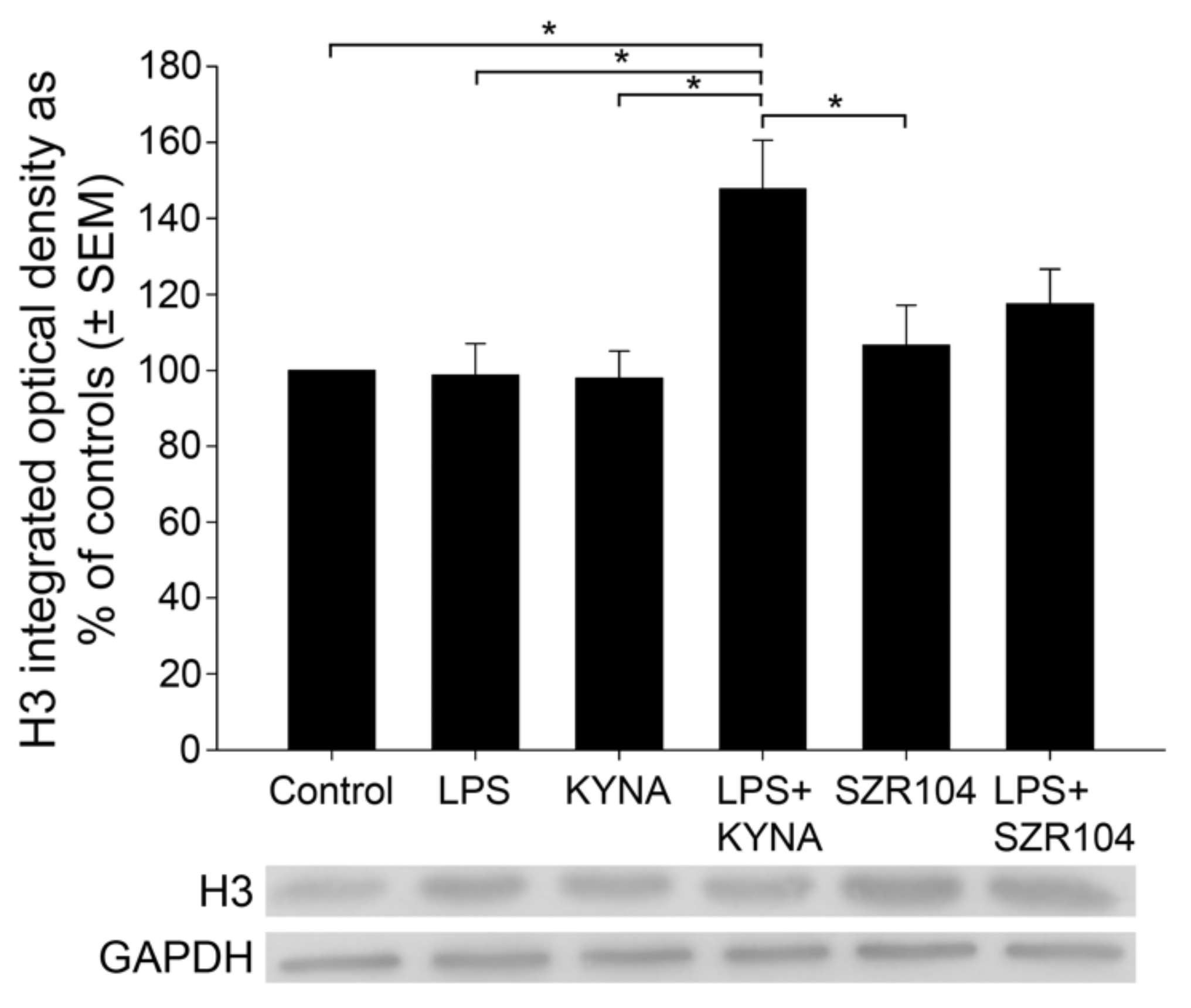
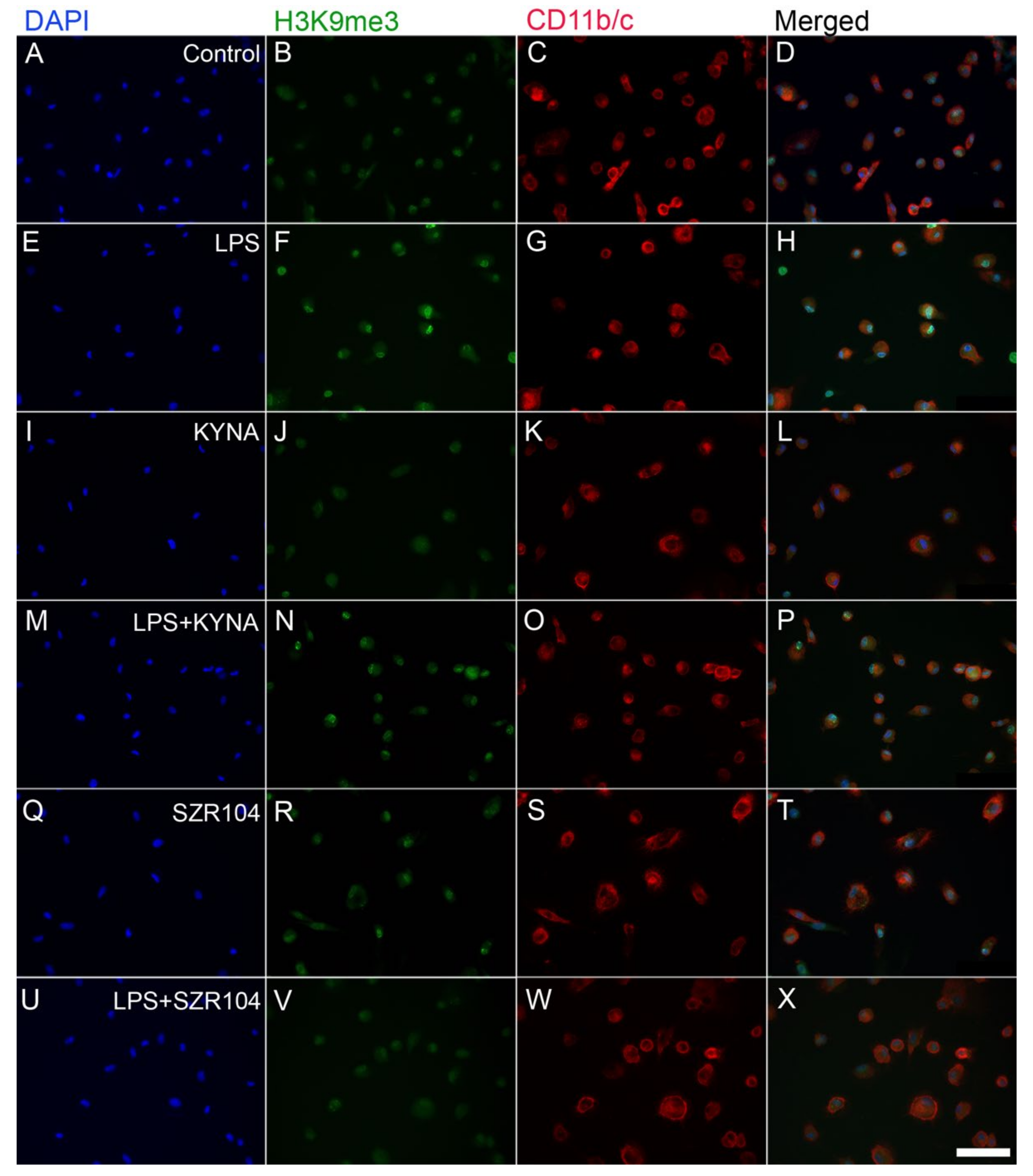
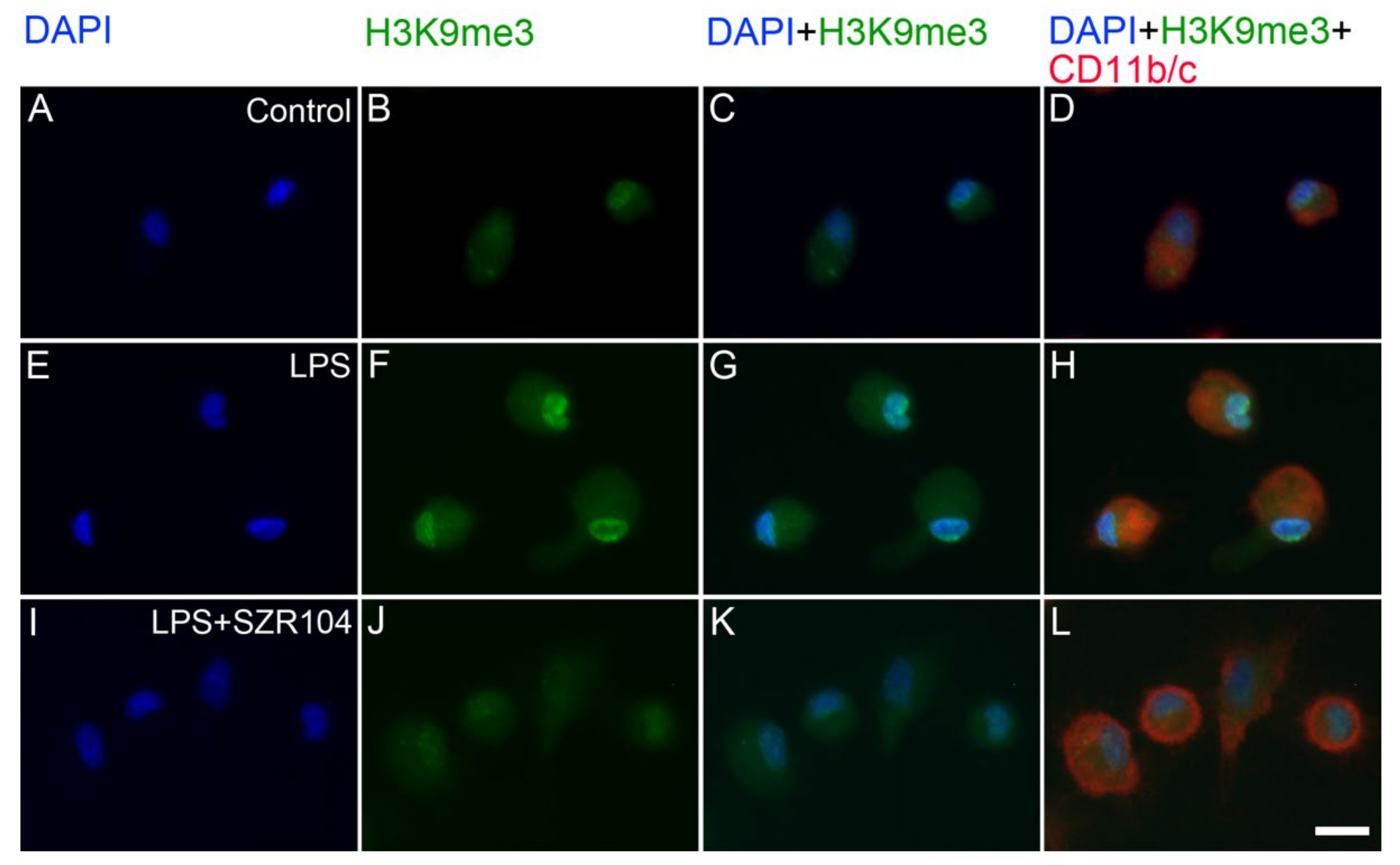
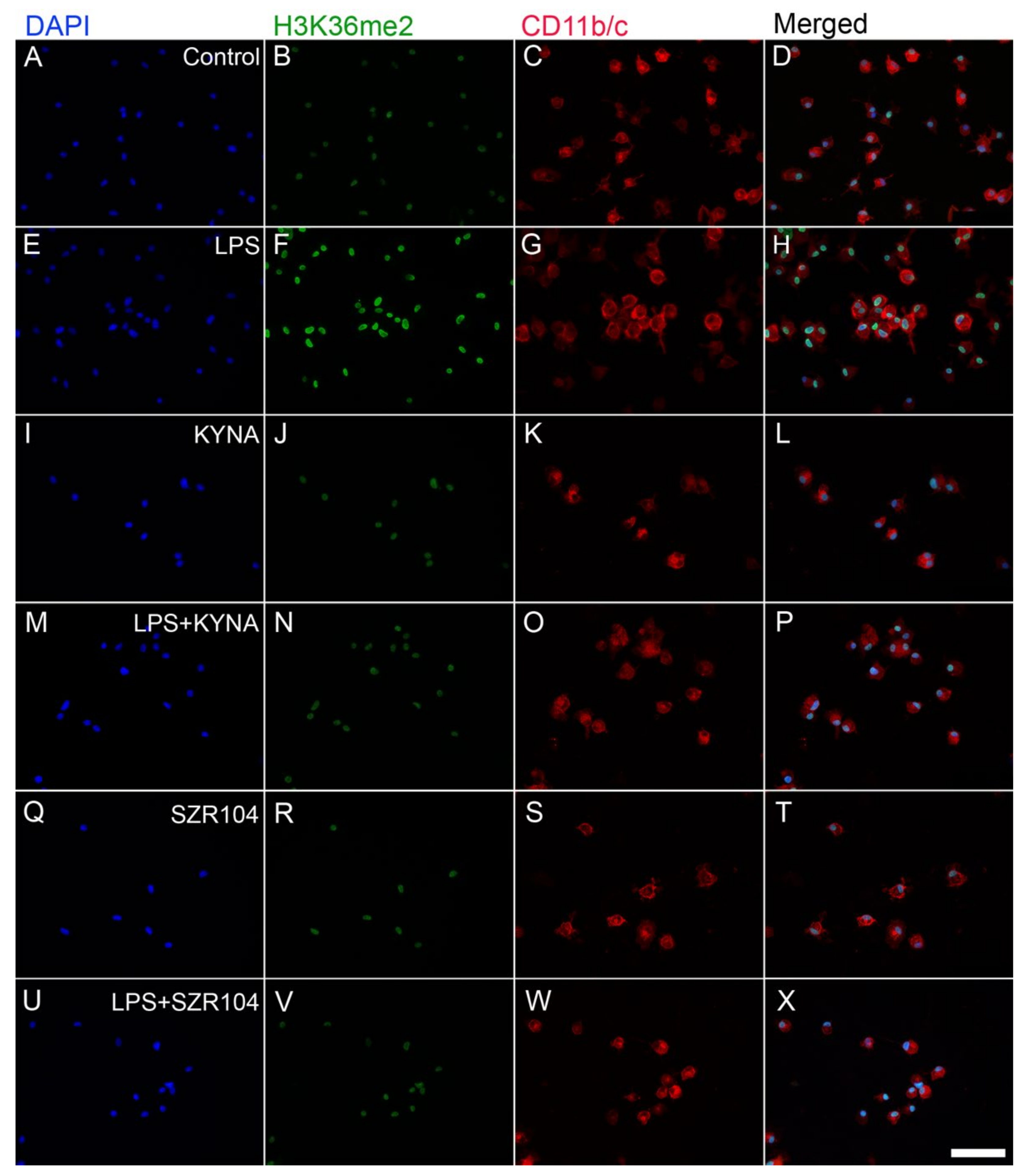
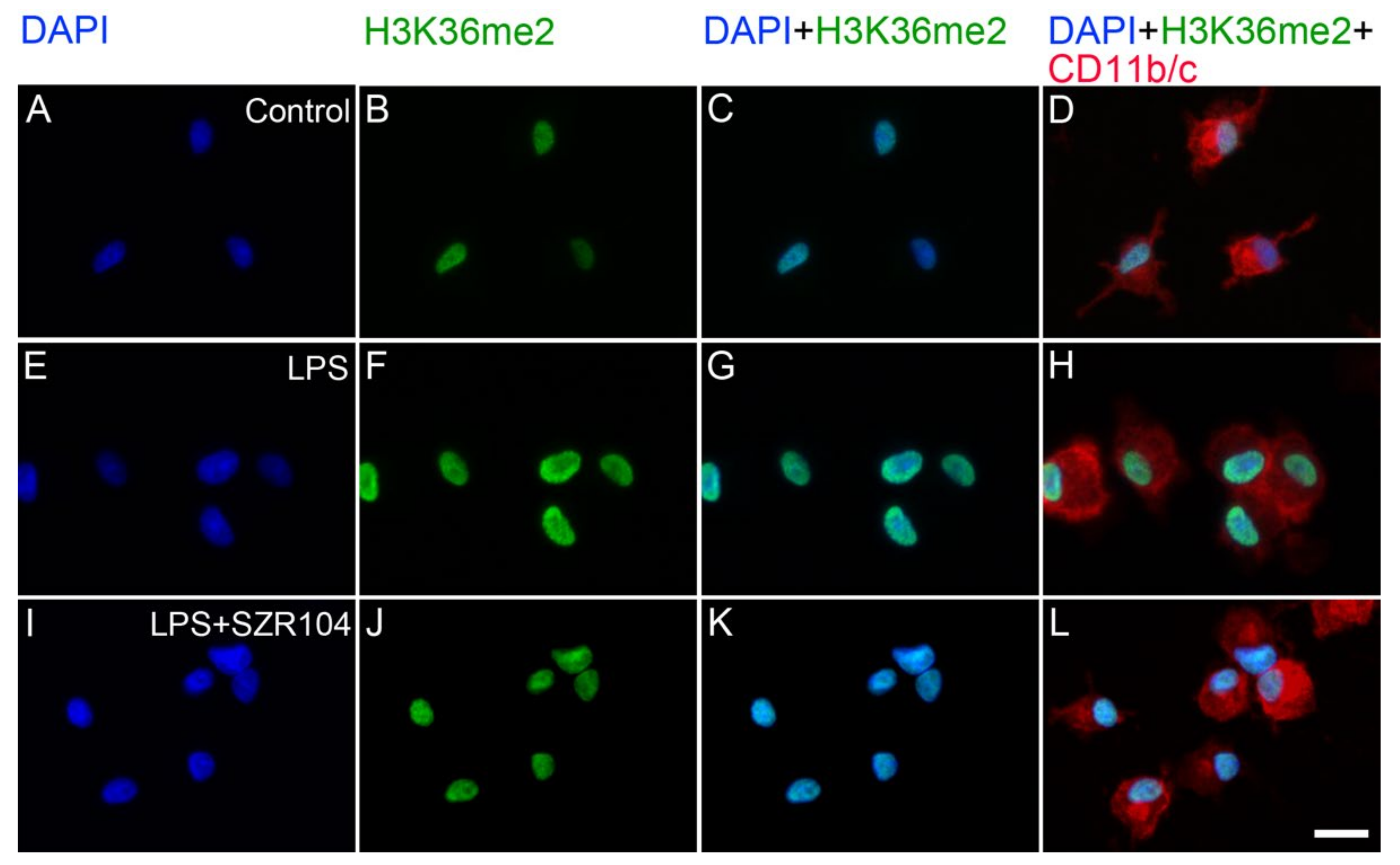
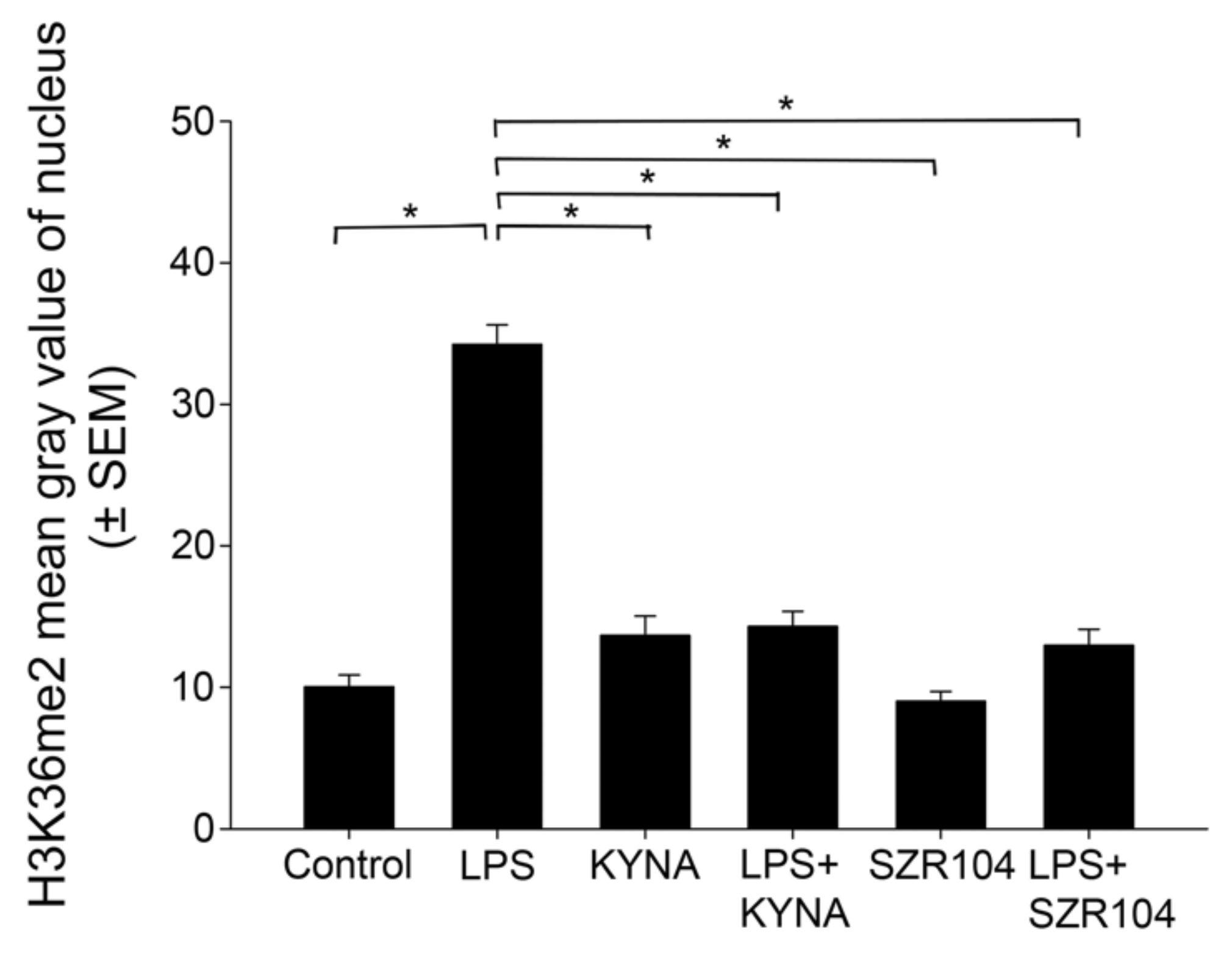
2.2. KYNA and SZR104 Alter the Intracellular Histone H3 Distribution and H3 lys Methylation Patterns
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Reagents and Antibodies
4.3. Cell Culture
4.4. Immunocytochemistry
4.5. Western Blot Analysis
4.6. Image Analysis and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ANOVA | one-way analysis of variance |
| CCR1 | C-C motif chemokine receptor 1 (also known as chemokine receptor 1) |
| CD11b/c | cluster of differentiation 11b/c, the rat CR3 complement receptor |
| CTCF | corrected total cell fluorescence, an area-dependent value |
| CXCL10 | C-X-C motif chemokine ligand 10 (also known as interferon-inducible cytokine IP-10) |
| DAPI | 2-[4-(aminoiminomethyl)phenyl]-1H-indole-6-carboximidamide hydrochloride |
| DIV | day(s) in vitro |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| FBS | fetal bovine serum |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase (EC 1.2.1.12) |
| H | histone |
| H3K | histone H3 lys modification |
| Iba1 | ionized calcium-binding adaptor molecule 1 |
| IgG | immunoglobulin G |
| ICC | immunocytochemistry |
| KYNA | kynurenic acid |
| PBS | phosphate-buffered saline |
| rpm | revolutions per minute |
| RT | room temperature |
| SEM | standard error of the mean |
| subDIV | subcloned day(s) in vitro |
| SZR104 | N-(2-(dimethylamino)ethyl)-3-(morpholinomethyl)-4-hydroxyquinoline-2-carboxamide |
| TBS | Tris-buffered saline |
| WB | western blot |
References
- Vazirinejad, R.; Ahmadi, Z.; Kazemi Arababadi, M.; Hassanshahi, G.; Kennedy, D. The biological functions, structure and sources of CXCL10 and its outstanding part in the pathophysiology of multiple sclerosis. Neuroimmunomodulation 2014, 21, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Vécsei, L.; Szalárdy, L.; Fülöp, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef]
- Wirthgen, E.; Hoeflich, A.; Rebl, A.; Günther, J. Kynurenic acid: The janus-faced role of an immunomodulatory tryptophan metabolite and its link to pathological conditions. Front. Immunol. 2017, 8, 1957. [Google Scholar] [CrossRef] [Green Version]
- Routy, J.P.; Routy, B.; Graziani, G.M.; Mehraj, V. The kynurenine pathway is a double-edged sword in immune-privileged sites and in cancer: Implications for immunotherapy. Int. J. Tryptophan Res. 2016, 9, 67–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lajkó, N.; Kata, D.; Szabó, M.; Mátyás, A.; Dulka, K.; Földesi, I.; Fülöp, F.; Gulya, K.; Vécsei, L.; Mihály, A. Sensitivity of rodent microglia to kynurenines in models of epilepsy and inflammation in vivo and in vitro: Microglia activation is inhibited by kynurenic acid and the synthetic analogue SZR104. Int. J. Mol. Sci. 2020, 21, 9333. [Google Scholar] [CrossRef] [PubMed]
- Molnár, K.; Lőrinczi, B.; Fazakas, C.; Szatmári, I.; Fülöp, F.; Kmetykó, N.; Berkecz, R.; Ilisz, I.; Krizbai, I.A.; Wilhelm, I.; et al. SZR-104, a Novel Kynurenic Acid Analogue with High Permeability through the Blood-Brain Barrier. Pharmaceutics 2021, 13, 61. [Google Scholar] [CrossRef]
- Biernacki, T.; Sandi, D.; Bencsik, K.; Vecsei, L. Kynurenines in the pathogenesis of multiple sclerosis: Therapeutic perspectives. Cells 2020, 9, 1564. [Google Scholar] [CrossRef]
- Mándi, Y.; Endrész, V.; Mosolygó, T.; Burián, K.; Lantos, I.; Fülöp, F.; Szatmári, I.; Lőrinczi, B.; Balog, A.; Vécsei, L. The opposite effects of kynurenic acid and different kynurenic acid analogs on tumor necrosis factor-α (TNF-α) production and tumor necrosis factor-stimulated gene-6 (TSG-6) expression. Front. Immunol. 2019, 10, 1406. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, F.; Szatmári, I.; Toldi, J.; Vécsei, L. Modifications on the carboxylic function of kynurenic acid. J. Neural Transm. 2012, 119, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, F.; Szatmári, I.; Toldi, J.; Vécsei, L. Novel Types of c-3 Substituted Kinurenic Acid Derivatives with Improved Neuroprotective Activity. Patent No: WO2017149333A1, 8 September 2017. Available online: https://patents.google.com/patent/WO2017149333A1/en (accessed on 2 June 2021).
- Demeter, I.; Nagy, K.; Gellért, L.; Vécsei, L.; Fülöp, F.; Toldi, J. A novel kynurenic acid analog (SZR104) inhibits pentylenetetrazole-induced epileptiform seizures. An electrophysiological study. J. Neural Transm. 2012, 119, 151–154. [Google Scholar] [CrossRef]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Ubogu, E.E.; Cossoy, M.B.; Ransohoff, R.M. The expression and function of chemokines involved in CNS inflammation. Trends Pharmacol. Sci. 2006, 27, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Cheeran, M.C.-J.; Hu, S.; Sheng, W.S.; Peterson, P.K.; Lokensgard, J.R. CXCL10 production from cytomegalovirus-stimulated microglia is regulated by both human and viral interleukin-10. J. Virol. 2003, 77, 4502–4515. [Google Scholar] [CrossRef] [Green Version]
- Nakamichi, K.; Saiki, M.; Sawada, M.; Takayama-Ito, M.; Yamamuro, Y.; Morimoto, K.; Kurane, I. Rabies virus-induced activation of mitogen-activated protein kinase and NF-kappaB signaling pathways regulates expression of CXC and CC chemokine ligands in microglia. J. Virol. 2005, 79, 11801–11812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyer, K.D.; Percopo, C.M.; Fischer, E.R.; Gabryszewski, S.J.; Rosenberg, H.F. Pneumoviruses infect eosinophils and elicit MyD88-dependent release of chemoattractant cytokines and interleukin-6. Blood 2009, 114, 2649–2656. [Google Scholar] [CrossRef]
- Lo, B.K.K.; Yu, M.; Zloty, D.; Cowan, B.; Shapiro, J.; McElwee, K.J. CXCR3/ligands are significantly involved in the tumorigenesis of basal cell carcinomas. Am. J. Pathol. 2010, 176, 2435–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, N.O.; Huang, M.B.; Anderson, W.; Bond, V.; Powell, M.; Thompson, W.E.; Armah, H.B.; Adjei, A.A.; Gyasi, R.; Tettey, Y.; et al. Soluble factors from Plasmodium falciparum-infected erythrocytes induce apoptosis in human brain vascular endothelial and neuroglia cells. Mol. Biochem. Parasitol. 2008, 162, 172–176. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Guo, S.; Hibbert, J.M.; Jain, V.; Singh, N.; Wilson, N.O.; Stiles, J.K. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 2011, 22, 121–130. [Google Scholar] [CrossRef]
- Klein, R.S. Regulation of neuroinflammation: The role of CXCL10 in lymphocyte infiltration during autoimmune encephalomyelitis. J. Cell Biochem. 2004, 92, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, R.; Matsui, M. Biomarkers for multiple sclerosis. Intern. Med. 2014, 53, 361–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczuciński, A.; Losy, J. Chemokines and chemokine receptors in multiple sclerosis. Potential targets for new therapies. Acta Neurol. Scand. 2007, 115, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral thinking: How histone modifications regulate gene expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlatanova, J.S.; Srebreva, L.N.; Banchev, T.B.; Tasheva, B.T.; Tsanev, R.G. Cytoplasmic pool of histone H1 in mammalian cells. J. Cell Sci. 1990, 96 Pt 3, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Parseghian, M.H.; Luhrs, K.A. Beyond the walls of the nucleus: The role of histones in cellular signaling and innate immunity. Biochem. Cell Biol. 2006, 84, 589–604. [Google Scholar] [CrossRef]
- Chen, R.; Kang, R.; Fan, X.G.; Tang, D. Release and activity of histone in diseases. Cell Death Dis. 2014, 5, e1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekaney, M.L.; Otto, G.P.; Sossdorf, M.; Sponholz, C.; Boehringer, M.; Loesche, W.; Rittirsch, D.; Wilharm, A.; Kurzai, O.; Bauer, M.; et al. Impact of plasma histones in human sepsis and their contribution to cellular injury and inflammation. Crit. Care 2014, 18, 543. [Google Scholar] [CrossRef] [Green Version]
- Frydman, G.H.; Tessier, S.N.; Wong, K.H.K.; Vanderburg, C.R.; Fox, J.G.; Toner, M.; Tompkins, R.G.; Irimia, D. Megakaryocytes contain extranuclear histones and may be a source of platelet-associated histones during sepsis. Sci. Rep. 2020, 10, 4621. [Google Scholar] [CrossRef]
- Gabler, C.; Blank, N.; Hieronymus, T.; Schiller, M.; Berden, J.H.M.; Kalden, J.R.; Lorenz, H.M. Extranuclear detection of histones and nucleosomes in activated human lymphoblasts as an early event in apoptosis. Ann. Rheum. Dis. 2004, 63, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- Gabler, C.; Kalden, J.R.; Lorenz, H.M. The putative role of apoptosis-modified histones for the induction of autoimmunity in systemic lupus erythematosus. Biochem. Pharmacol. 2003, 66, 1441–1446. [Google Scholar] [CrossRef]
- Gabler, C.; Blank, N.; Winkler, S.; Kalden, J.R.; Lorenz, H.M. Accumulation of histones in cell lysates precedes expression of apoptosis-related phagocytosis signals in human lymphoblasts. Ann. N. Y. Acad. Sci. 2003, 1010, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Konishi, A.; Shimizu, S.; Hirota, J.; Takao, T.; Fan, Y.; Matsuoka, Y.; Zhang, L.; Yoneda, Y.; Fujii, Y.; Skoultchi, A.I.; et al. Involvement of histone H1.2 in apoptosis induced by DNA double-strand breaks. Cell 2003, 114, 673–688. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, S.; Workman, J.L. Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [Green Version]
- Honma, K.; Machida, C.; Mochizuki, K.; Goda, T. Glucose and TNF enhance expression of TNF and IL1B, and histone H3 acetylation and K4/K36 methylation, in juvenile macrophage cells. Gene 2020, 763S, 100034. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Vogel, V. Spatial confinement downsizes the inflammatory response of macrophages. Nat. Mater. 2018, 17, 1134–1144. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Jang, Y.; Park, Y.K.; Lee, J.E.; Jain, S.; Froimchuk, E.; Broun, A.; Liu, C.; Gavrilova, O.; Ge, K. Depletion of Nsd2-mediated histone H3K36 methylation impairs adipose tissue development and function. Nat. Commun. 2018, 9, 1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saccani, S.; Natoli, G. Dynamic changes in histone H3 Lys 9 methylation occurring at tightly regulated inducible inflammatory genes. Genes Dev. 2002, 16, 2219–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, L.; Gold, M.; Mengel, D.; Dodel, R.; Bach, J.P. The endogenous α7 nicotinic acetylcholine receptor antagonist kynurenic acid modulates amyloid-β-induced inflammation in BV-2 microglial cells. J. Neurol. Sci. 2014, 344, 94–99. [Google Scholar] [CrossRef]
- Poles, M.Z.; Nászai, A.; Gulácsi, L.; Czakó, B.L.; Gál, K.G.; Glenz, R.J.; Dookhun, D.; Rutai, A.; Tallósy, S.P.; Szabó, A.; et al. Kynurenic acid and its synthetic derivatives protect against sepsis-associated neutrophil activation and brain mitochondrial dysfunction in rats. Front. Immunol. 2021, 12, 717157. [Google Scholar] [CrossRef]
- Balog, A.; Varga, B.; Fülöp, F.; Lantos, I.; Toldi, G.; Vécsei, L.; Mándi, Y. Kynurenic acid analog attenuates the production of tumor necrosis factor-α, calgranulins (S100A 8/9 and S100A 12), and the secretion of HNP1-3 and stimulates the production of tumor necrosis factor-stimulated gene-6 in whole blood cultures of patients with rheumatoid arthritis. Front. Immunol. 2021, 12, 632513. [Google Scholar] [CrossRef]
- Boros, F.A.; Vécsei, L. Immunomodulatory effects of genetic alterations affecting the kynurenine pathway. Front. Immunol. 2019, 10, 2570. [Google Scholar] [CrossRef] [PubMed]
- Hankittichai, P.; Lou, H.J.; Wikan, N.; Smith, D.R.; Potikanond, S.; Nimlamool, W. Oxyresveratrol inhibits IL-1β-induced inflammation via suppressing AKT and ERK1/2 activation in human microglia, HMC3. Int. J. Mol. Sci. 2020, 21, 6054. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Ye, J.; Yan, H. Investigation of key genes and pathways in inhibition of oxycodone on vincristine-induced microglia activation by using bioinformatics analysis. Dis. Markers 2019, 2019, 3521746. [Google Scholar] [CrossRef] [PubMed]
- Kata, D.; Földesi, I.; Feher, L.Z.; Hackler, L.; Puskas, L.G.; Gulya, K. A novel pleiotropic effect of aspirin: Beneficial regulation of pro- and anti-inflammatory mechanisms in microglial cells. Brain Res. Bull. 2017, 132, 61–74. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Ingram, A.; Lahti, J.H.; Mazza, B.; Grenet, J.; Kapoor, A.; Liu, L.; Kidd, V.J.; Tang, D. Apoptotic release of histones from nucleosomes. J. Biol. Chem. 2002, 277, 12001–12008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, Y.K.; Jaiswal, M.K.; Agrawal, V.; Chaturvedi, M.M. Bacterial endotoxin (LPS)-induced DNA damage in preimplanting embryonic and uterine cells inhibits implantation. Fertil. Steril. 2009, 91 (Suppl. 5), 2095–2103. [Google Scholar] [CrossRef] [PubMed]
- Kovalchuk, I.; Walz, P.; Thomas, J.; Kovalchuk, O. Genomic instability in liver cells caused by an LPS-induced bystander-like effect. PLoS ONE 2013, 8, e67342. [Google Scholar] [CrossRef] [Green Version]
- Kobiyama, K.; Kawashima, A.; Jounai, N.; Takeshita, F.; Ishii, K.J.; Ito, T.; Suzuki, K. Role of extrachromosomal histone H2B on recognition of DNA viruses and cell damage. Front. Genet. 2013, 4, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobiyama, K.; Takeshita, F.; Jounai, N.; Sakaue-Sawano, A.; Miyawaki, A.; Ishii, K.J.; Kawai, T.; Sasaki, S.; Hirano, H.; Ishii, N.; et al. Extrachromosomal histone H2B mediates innate antiviral immune responses induced by intracellular double-stranded DNA. J. Virol. 2010, 84, 822–832. [Google Scholar] [CrossRef] [Green Version]
- Talati, M.; Seeley, E.; Ihida-Stansbury, K.; Delisser, H.; McDonald, H.; Ye, F.; Zhang, X.; Shyr, Y.; Caprioli, R.; Meyrick, B. Altered expression of nuclear and cytoplasmic histone H1 in pulmonary artery and pulmonary artery smooth muscle cells in patients with IPAH. Pulm. Circ. 2012, 2, 340–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nánási, P., Jr.; Imre, L.; Firouzi Niaki, E.; Bosire, R.; Mocsár, G.; Türk-Mázló, A.; Ausio, J.; Szabó, G. Doxorubicin induces large-scale and differential H2A and H2B redistribution in live cells. PLoS ONE 2020, 15, e0231223. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.Q.; Lowman, X.H.; Kong, M. Molecular Pathways: Metabolic control of histone methylation and gene expression in cancer. Clin. Cancer Res. 2017, 23, 4004–4009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiles, E.T.; Selker, E.U. H3K27 methylation: A promiscuous repressive chromatin mark. Curr. Opin. Genet. Dev. 2017, 43, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Meng, M.; Liu, H.; Chen, S.; Zhao, H.; Gao, X.; Zhang, J.; Chen, D. Methylation of H3K27 and H3K4 in key gene promoter regions of thymus in RA mice is involved in the abnormal development and differentiation of iNKT cells. Immunogenetics 2019, 71, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kumari, N.; Sharma, U.; Ram, S.; Singh, S.K.; Kakkar, N.; Kaushal, K.; Prasad, R. Reduction in H3K4me patterns due to aberrant expression of methyltransferases and demethylases in renal cell carcinoma: Prognostic and therapeutic implications. Sci. Rep. 2019, 9, 8189. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Zhang, J.; Chen, R.; Wang, L.; Li, B.; Cheng, H.; Duan, X.; Zhu, H.; Wei, W.; Li, J.; et al. Dissecting the precise role of H3K9 methylation in crosstalk with DNA maintenance methylation in mammals. Nat. Commun. 2016, 7, 12464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, K.; Nishitani, K.; Tanaka, S. Kynurenine, 3-OH-kynurenine, and anthranilate are nutrient metabolites that alter H3K4 trimethylation and H2AS40 O-GlcNAcylation at hypothalamus-related loci. Sci. Rep. 2019, 9, 19768. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, J.A.; Wang, Z.; Schones, D.E.; Zhao, K.; DeSalle, R.; Zhang, M.Q. Determination of enriched histone modifications in non-genic portions of the human genome. BMC Genom. 2009, 10, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, T.C.; Schaefer, U.; Mecklenbrauker, I.; Stienen, A.; Dewell, S.; Chen, M.S.; Rioja, I.; Parravicini, V.; Prinjha, R.K.; Chandwani, R.; et al. Histone H3 lysine 9 di-methylation as an epigenetic signature of the interferon response. J. Exp. Med. 2012, 209, 661–669. [Google Scholar] [CrossRef] [Green Version]
- Harman, J.L.; Dobnikar, L.; Chappell, J.; Stokell, B.G.; Dalby, A.; Foote, K.; Finigan, A.; Freire-Pritchett, P.; Taylor, A.L.; Worssam, M.D.; et al. Epigenetic regulation of vascular smooth muscle cells by histone H3 lysine 9 dimethylation attenuates target gene-induction by inflammatory signaling. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2289–2302. [Google Scholar] [CrossRef]
- Torrano, J.; Al Emran, A.; Hammerlindl, H.; Schaider, H. Emerging roles of H3K9me3, SETDB1 and SETDB2 in therapy-induced cellular reprogramming. Clin. Epigenetics 2019, 11, 43. [Google Scholar] [CrossRef] [Green Version]
- Li, M.F.; Zhang, R.; Li, T.T.; Chen, M.Y.; Li, L.X.; Lu, J.X.; Jia, W.P. High glucose increases the expression of inflammatory cytokine genes in macrophages through H3K9 methyltransferase mechanism. J. Interferon Cytokine Res. 2016, 36, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, F.; Szatmári, I.; Vámos, E.; Zádori, D.; Toldi, J.; Vécsei, L. Syntheses, transformations and pharmaceutical applications of kynurenic acid derivatives. Curr. Med. Chem. 2009, 16, 4828–4842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, M.; Gulya, K. Development of the microglial phenotype in culture. Neuroscience 2013, 241, 280–295. [Google Scholar] [CrossRef]
- Neote, K.; DiGregorio, D.; Mak, J.Y.; Horuk, R.; Schall, T.J. Molecular cloning, functional expression, and signaling characteristics of a C-C chemokine receptor. Cell 1993, 72, 415–425. [Google Scholar] [CrossRef]
- Dulka, K.; Szabo, M.; Lajkó, N.; Belecz, I.; Hoyk, Z.; Gulya, K. Epigenetic consequences of in utero exposure to rosuvastatin: Alteration of histone methylation patterns in newborn rat brains. Int. J. Mol. Sci. 2021, 22, 3412. [Google Scholar] [CrossRef] [PubMed]
- Dulka, K.; Nacsa, K.; Lajkó, N.; Gulya, K. Quantitative morphometric and cell-type-specific population analysis of microglia-enriched cultures subcloned to high purity from newborn rat brains. IBRO Neurosci. Rep. 2021, 10, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szabo, M.; Lajkó, N.; Dulka, K.; Szatmári, I.; Fülöp, F.; Mihály, A.; Vécsei, L.; Gulya, K. Kynurenic Acid and Its Analog SZR104 Exhibit Strong Antiinflammatory Effects and Alter the Intracellular Distribution and Methylation Patterns of H3 Histones in Immunochallenged Microglia-Enriched Cultures of Newborn Rat Brains. Int. J. Mol. Sci. 2022, 23, 1079. https://doi.org/10.3390/ijms23031079
Szabo M, Lajkó N, Dulka K, Szatmári I, Fülöp F, Mihály A, Vécsei L, Gulya K. Kynurenic Acid and Its Analog SZR104 Exhibit Strong Antiinflammatory Effects and Alter the Intracellular Distribution and Methylation Patterns of H3 Histones in Immunochallenged Microglia-Enriched Cultures of Newborn Rat Brains. International Journal of Molecular Sciences. 2022; 23(3):1079. https://doi.org/10.3390/ijms23031079
Chicago/Turabian StyleSzabo, Melinda, Noémi Lajkó, Karolina Dulka, István Szatmári, Ferenc Fülöp, András Mihály, László Vécsei, and Karoly Gulya. 2022. "Kynurenic Acid and Its Analog SZR104 Exhibit Strong Antiinflammatory Effects and Alter the Intracellular Distribution and Methylation Patterns of H3 Histones in Immunochallenged Microglia-Enriched Cultures of Newborn Rat Brains" International Journal of Molecular Sciences 23, no. 3: 1079. https://doi.org/10.3390/ijms23031079
APA StyleSzabo, M., Lajkó, N., Dulka, K., Szatmári, I., Fülöp, F., Mihály, A., Vécsei, L., & Gulya, K. (2022). Kynurenic Acid and Its Analog SZR104 Exhibit Strong Antiinflammatory Effects and Alter the Intracellular Distribution and Methylation Patterns of H3 Histones in Immunochallenged Microglia-Enriched Cultures of Newborn Rat Brains. International Journal of Molecular Sciences, 23(3), 1079. https://doi.org/10.3390/ijms23031079