
Changes of Gut Microbiota by Natural mtDNA Variant Differences Augment Susceptibility to Metabolic Disease and Ageing


Abstract
:1. Introduction
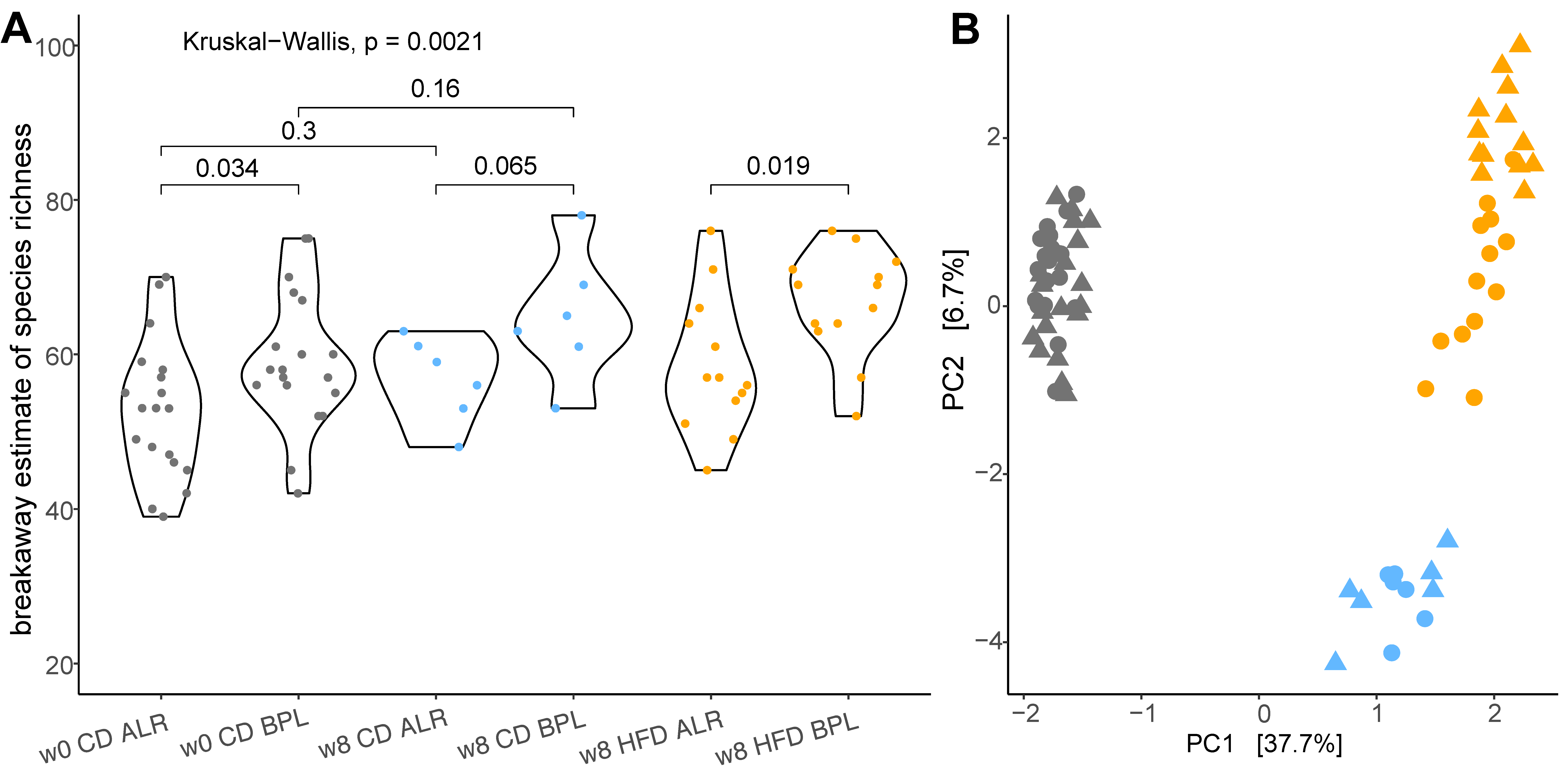
2. Results
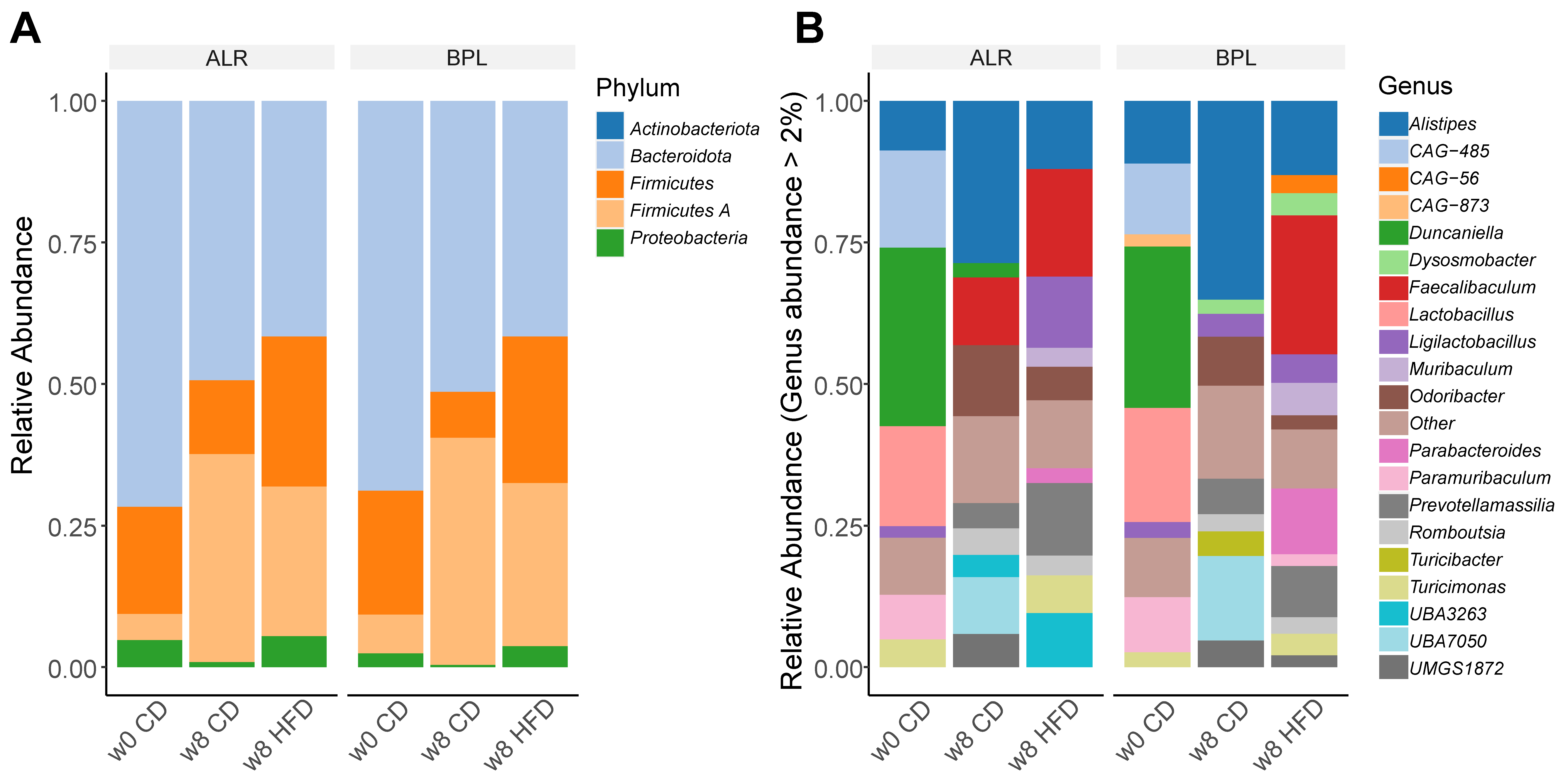
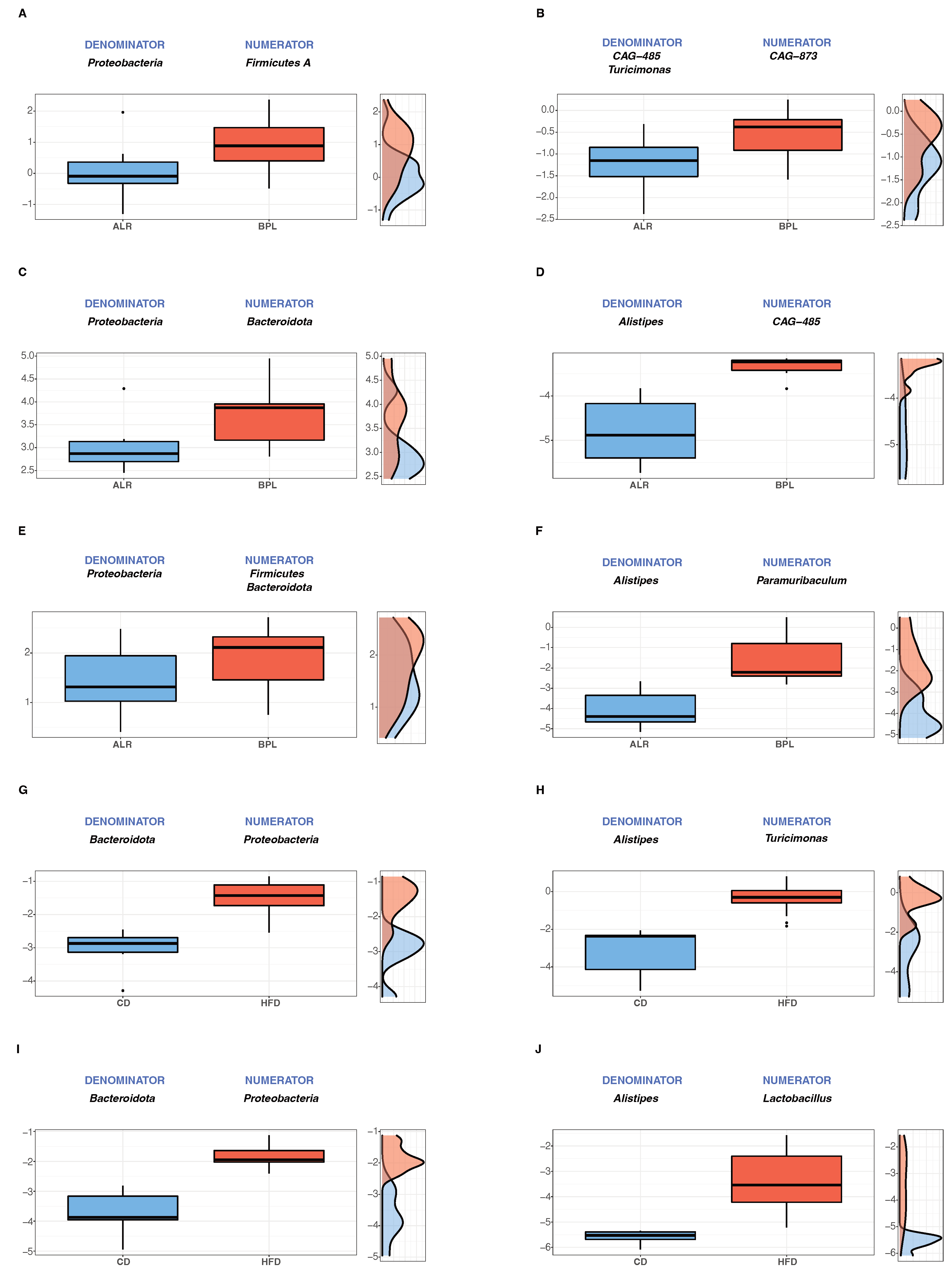
2.1. Proteobacteria, a Marker for Host Health, were Differently Abundant in B6-mtBPL Mice Compared with B6-mtALR Mice
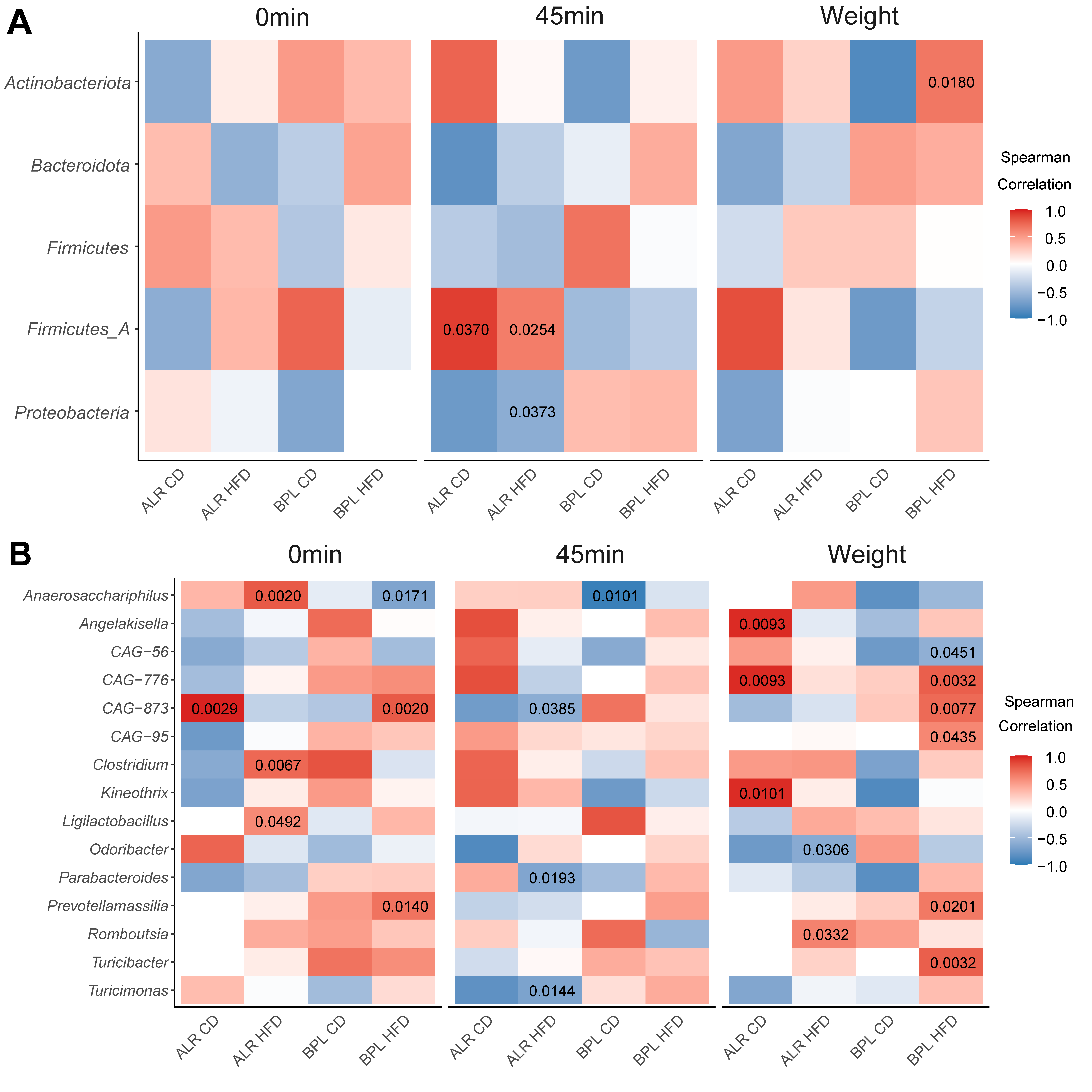
2.2. Correlation between the Abundance of Commensal Bacteria and Alteration in Metabolic Parameters upon the Dietary Metabolic Stress
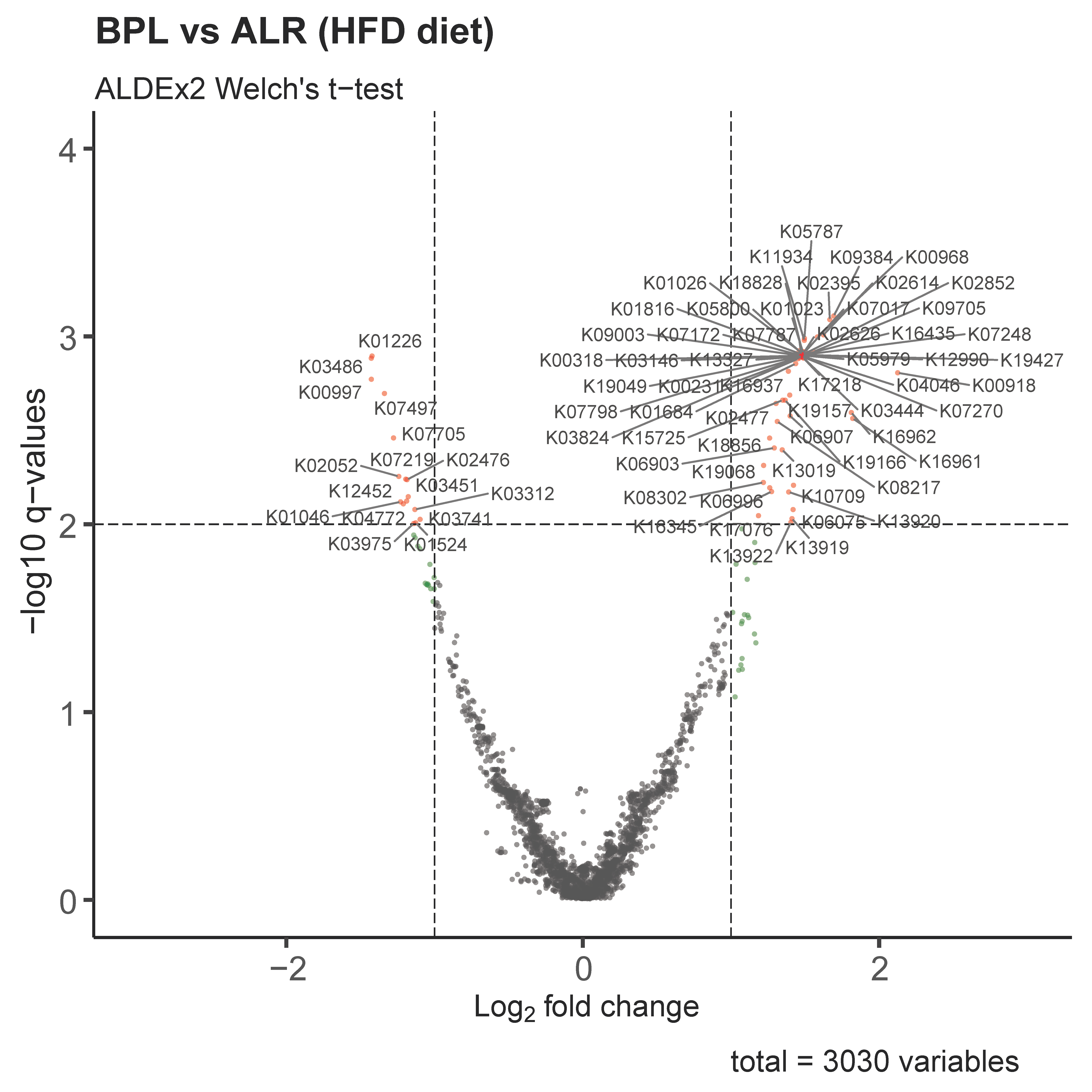
2.3. Functional Profiles of Differentially Abundant Gut Bacteria between HFD-fed B6-mtBPL and B6-mtALR Revealed Significant Involvement of Glucose Metabolism in Gut-Microbially Derived Pathways
3. Discussion
4. Materials and Methods
4.1. Mice, and Stool Sample Collection
4.2. Bacterial DNA Isolation and Library Preparation and Sequencing for the Bacterial 16S Ribosomal RNA Gene
4.3. Data Process and Analysis
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and Organization of the Human Mitochondrial Genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J.; Nikoskelainen, E.K. Mitochondrial DNA Mutation Associated with Leber’s Hereditary Optic Neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.D.; Trounce, I.A.; Jun, A.S.; Allen, J.C.; Wallace, D.C. Functional Analysis of Lymphoblast and Cybrid Mitochondria Containing the 3460, 11778, or 14484 Leber’s Hereditary Optic Neuropathy Mitochondrial DNA Mutation *. J. Biol. Chem. 2000, 275, 39831–39836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.S.; Sharpley, M.S.; Fan, W.; Waymire, K.G.; Sadun, A.A.; Carelli, V.; Ross-Cisneros, F.N.; Baciu, P.; Sung, E.; McManus, M.J.; et al. Mouse MtDNA Mutant Model of Leber Hereditary Optic Neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 20065–20070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indo, H.P.; Yen, H.-C.; Nakanishi, I.; Matsumoto, K.-I.; Tamura, M.; Nagano, Y.; Matsui, H.; Gusev, O.; Cornette, R.; Okuda, T.; et al. A Mitochondrial Superoxide Theory for Oxidative Stress Diseases and Aging. J. Clin. Biochem. Nutr. 2015, 56, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonova-Doing, E.; Calabrese, C.; Gomez-Duran, A.; Schon, K.; Wei, W.; Karthikeyan, S.; Chinnery, P.F.; Howson, J.M.M. An Atlas of Mitochondrial DNA Genotype-Phenotype Associations in the UK Biobank. Nat. Genet. 2021, 53, 982–993. [Google Scholar] [CrossRef]
- Latorre-Pellicer, A.; Moreno-Loshuertos, R.; Lechuga-Vieco, A.V.; Sánchez-Cabo, F.; Torroja, C.; Acín-Pérez, R.; Calvo, E.; Aix, E.; González-Guerra, A.; Logan, A.; et al. Mitochondrial and Nuclear DNA Matching Shapes Metabolism and Healthy Ageing. Nature 2016, 535, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Hirose, M.; Schilf, P.; Gupta, Y.; Zarse, K.; Künstner, A.; Fähnrich, A.; Busch, H.; Yin, J.; Wright, M.N.; Ziegler, A.; et al. Low-Level Mitochondrial Heteroplasmy Modulates DNA Replication, Glucose Metabolism and Lifespan in Mice. Sci. Rep. 2018, 8, 5872. [Google Scholar] [CrossRef] [Green Version]
- Hirose, M.; Künstner, A.; Schilf, P.; Tietjen, A.K.; Jöhren, O.; Huebbe, P.; Rimbach, G.; Rupp, J.; Schwaninger, M.; Busch, H.; et al. A Natural MtDNA Polymorphism in Complex III Is a Modifier of Healthspan in Mice. Int. J. Mol. Sci. 2019, 20, 2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McManus, M.J.; Picard, M.; Chen, H.-W.; de Haas, H.J.; Potluri, P.; Leipzig, J.; Towheed, A.; Angelin, A.; Sengupta, P.; Morrow, R.M.; et al. Mitochondrial DNA Variation Dictates Expressivity and Progression of Nuclear DNA Mutations Causing Cardiomyopathy. Cell. Metab. 2019, 29, 78–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schilf, P.; Künstner, A.; Olbrich, M.; Waschina, S.; Fuchs, B.; Galuska, C.E.; Braun, A.; Neuschütz, K.; Seutter, M.; Bieber, K.; et al. A Mitochondrial Polymorphism Alters Immune Cell Metabolism and Protects Mice from Skin Inflammation. Int. J. Mol. Sci. 2021, 22, 1006. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, P.W.; Jeffery, I.B. Gut Microbiota and Aging. Science 2015, 350, 1214–1215. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Hirose, M.; Künstner, A.; Schilf, P.; Sünderhauf, A.; Rupp, J.; Jöhren, O.; Schwaninger, M.; Sina, C.; Baines, J.F.; Ibrahim, S.M. Mitochondrial Gene Polymorphism Is Associated with Gut Microbial Communities in Mice. Sci. Rep. 2017, 7, 15293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yardeni, T.; Tanes, C.E.; Bittinger, K.; Mattei, L.M.; Schaefer, P.M.; Singh, L.N.; Wu, G.D.; Murdock, D.G.; Wallace, D.C. Host Mitochondria Influence Gut Microbiome Diversity: A Role for ROS. Sci. Signal 2019, 12, 588. [Google Scholar] [CrossRef]
- Houghton, D.; Stewart, C.J.; Stamp, C.; Nelson, A.; Aj Ami, N.J.; Petrosino, J.F.; Wipat, A.; Trenell, M.I.; Turnbull, D.M.; Greaves, L.C. Impact of Age-Related Mitochondrial Dysfunction and Exercise on Intestinal Microbiota Composition. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 571–578. [Google Scholar] [CrossRef] [Green Version]
- Hirose, M.; Schilf, P.; Zarse, K.; Busch, H.; Fuellen, G.; Jöhren, O.; Köhling, R.; König, I.R.; Richer, B.; Rupp, J.; et al. Maternally Inherited Differences within Mitochondrial Complex I Control Murine Healthspan. Genes 2019, 10, 532. [Google Scholar] [CrossRef] [Green Version]
- Hirose, M.; Schilf, P.; Gupta, Y.; Wright, M.N.; Wright, M.N.; Jöhren, O.; Wagner, A.E.; Sina, C.; Ziegler, A.; Ristow, M.; et al. Lifespan Effects of Mitochondrial Mutations. Nature 2016, 540, E13–E14. [Google Scholar] [CrossRef]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial Signature of Dysbiosis in Gut Microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Badal, V.D.; Vaccariello, E.D.; Murray, E.R.; Yu, K.E.; Knight, R.; Jeste, D.V.; Nguyen, T.T. The Gut Microbiome, Aging, and Longevity: A Systematic Review. Nutrients 2020, 12, E3759. [Google Scholar] [CrossRef]
- Kaur, H.; Bose, C.; Mande, S.S. Tryptophan Metabolism by Gut Microbiome and Gut-Brain-Axis: An in Silico Analysis. Front. Neurosci. 2019, 13, 1365. [Google Scholar] [CrossRef]
- Zhou, L.; Xiao, X.; Zhang, Q.; Zheng, J.; Li, M.; Yu, M.; Wang, X.; Deng, M.; Zhai, X.; Li, R. Improved Glucose and Lipid Metabolism in the Early Life of Female Offspring by Maternal Dietary Genistein Is Associated With Alterations in the Gut Microbiota. Front. Endocrinol. 2018, 9, 516. [Google Scholar] [CrossRef]
- Yu, X.; Gimsa, U.; Wester-Rosenlöf, L.; Kanitz, E.; Otten, W.; Kunz, M.; Ibrahim, S.M. Dissecting the Effects of MtDNA Variations on Complex Traits Using Mouse Conplastic Strains. Genome Res. 2009, 19, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murali, A.; Bhargava, A.; Wright, E.S. IDTAXA: A Novel Approach for Accurate Taxonomic Classification of Microbiome Sequences. Microbiome 2018, 6, 140. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Chuvochina, M.; Rinke, C.; Mussig, A.J.; Chaumeil, P.-A.; Hugenholtz, P. GTDB: An Ongoing Census of Bacterial and Archaeal Diversity through a Phylogenetically Consistent, Rank Normalized and Complete Genome-Based Taxonomy. Nucleic Acids Res. 2021, 50, D785–D794. [Google Scholar] [CrossRef]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple Statistical Identification and Removal of Contaminant Sequences in Marker-Gene and Metagenomics Data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef] [Green Version]
- Willis, A.; Bunge, J. Estimating Diversity via Frequency Ratios. Biometrics 2015, 71, 1042–1049. [Google Scholar] [CrossRef] [Green Version]
- Willis, A.D.; Martin, B.D. Estimating Diversity in Networked Ecological Communities. Biostatistics 2020, 23, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Aitchison, J. The Statistical Analysis of Compositional Data; Chapman and Hall Ltd.: London, UK, 1986. [Google Scholar]
- Martin, B.D.; Witten, D.; Willis, A.D. Modeling Microbial Abundances and Dysbiosis with Beta-Binomial Regression. Ann. Appl. Stat. 2020, 14, 94–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera-Pinto, J.; Egozcue, J.J.; Pawlowsky-Glahn, V.; Paredes, R.; Noguera-Julian, M.; Calle, M.L. Balances: A New Perspective for Microbiome Analysis. mSystems 2018, 3, e00053-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S. Ppcor: An R Package for a Fast Calculation to Semi-Partial Correlation Coefficients. Commun. Stat. Appl. Methods 2015, 22, 665–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.D.; Reid, J.N.; Macklaim, J.M.; McMurrough, T.A.; Edgell, D.R.; Gloor, G.B. Unifying the Analysis of High-Throughput Sequencing Datasets: Characterizing RNA-Seq, 16S RRNA Gene Sequencing and Selective Growth Experiments by Compositional Data Analysis. Microbiome 2014, 2, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison | HFD vs. CD in ALR | HFD vs. CD in BPL | BPL vs. ALR in CD | BPL vs. ALR in HFD | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Abundance | Taxa | Fdr | Effect | Taxa | Fdr | Effect | Taxa | Fdr | Effect | Taxa | Fdr | Effect |
| Decreased | CAG-269 | 0.00005 | −4.72158 | Clostridium | 0.01590 | −3.29675 | UBA3263 | 0.00000 | −1.18785 | UBA3263 | 0.00000 | −2.02792 |
| Duncaniella | 0.00018 | −3.28685 | Duncaniella | 0.00160 | −3.22098 | Duncaniella | 0.00384 | −1.02234 | Emergencia | 0.00076 | −1.14646 | |
| CAG-873 | 0.00120 | −2.87259 | Acutalibacter | 0.00000 | −2.29577 | Romboutsia | 0.00021 | −0.75258 | Ligilactobacillus | 0.00133 | −0.89182 | |
| Schaedlerella | 0.01477 | −2.23013 | Alistipes | 0.00000 | −1.65379 | Turicimonas | 0.00317 | −0.61016 | ||||
| UMGS1872 | 0.00119 | −1.88033 | CAG-873 | 0.00000 | −1.28945 | |||||||
| Acutalibacter | 0.00003 | −1.44531 | Odoribacter | 0.01590 | −1.23079 | |||||||
| Alistipes | 0.00000 | −1.27705 | Anaerosacchariphilus | 0.00001 | −1.20352 | |||||||
| Odoribacter | 0.02196 | −0.91225 | UMGS1872 | 0.00019 | −1.12353 | |||||||
| Romboutsia | 0.00000 | −0.53529 | ||||||||||
| Increased | UBA3263 | 0.00000 | 1.58432 | Muribaculum | 0.02202 | 1.00961 | Acutalibacter | 0.04802 | 0.43005 | Muribaculum | 0.03175 | 0.82913 |
| Ligilactobacillus | 0.00000 | 2.28024 | Parabacteroides | 0.00000 | 1.97159 | Bacteroides | 0.00000 | 0.58530 | Dysosmobacter | 0.00133 | 1.43358 | |
| Emergencia | 0.04810 | 3.84358 | Turicimonas | 0.00000 | 2.75714 | Turicibacter | 0.00073 | 0.81148 | Parabacteroides | 0.00000 | 1.59726 | |
| Faecalibaculum | 0.00001 | 3.14495 | Ligilactobacillus | 0.04507 | 1.08915 | CAG-873 | 0.04156 | 1.98442 | ||||
| CAG-56 | 0.00118 | 5.71160 | Faecalibaculum | 0.01479 | 1.12808 | Paramuribaculum | 0.00533 | 3.61420 | ||||
| CAG-485 | 0.01479 | 1.69376 | ||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Künstner, A.; Schilf, P.; Busch, H.; Ibrahim, S.M.; Hirose, M. Changes of Gut Microbiota by Natural mtDNA Variant Differences Augment Susceptibility to Metabolic Disease and Ageing. Int. J. Mol. Sci. 2022, 23, 1056. https://doi.org/10.3390/ijms23031056
Künstner A, Schilf P, Busch H, Ibrahim SM, Hirose M. Changes of Gut Microbiota by Natural mtDNA Variant Differences Augment Susceptibility to Metabolic Disease and Ageing. International Journal of Molecular Sciences. 2022; 23(3):1056. https://doi.org/10.3390/ijms23031056
Chicago/Turabian StyleKünstner, Axel, Paul Schilf, Hauke Busch, Saleh M. Ibrahim, and Misa Hirose. 2022. "Changes of Gut Microbiota by Natural mtDNA Variant Differences Augment Susceptibility to Metabolic Disease and Ageing" International Journal of Molecular Sciences 23, no. 3: 1056. https://doi.org/10.3390/ijms23031056
APA StyleKünstner, A., Schilf, P., Busch, H., Ibrahim, S. M., & Hirose, M. (2022). Changes of Gut Microbiota by Natural mtDNA Variant Differences Augment Susceptibility to Metabolic Disease and Ageing. International Journal of Molecular Sciences, 23(3), 1056. https://doi.org/10.3390/ijms23031056