
PS1 Affects the Pathology of Alzheimer’s Disease by Regulating BACE1 Distribution in the ER and BACE1 Maturation in the Golgi Apparatus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
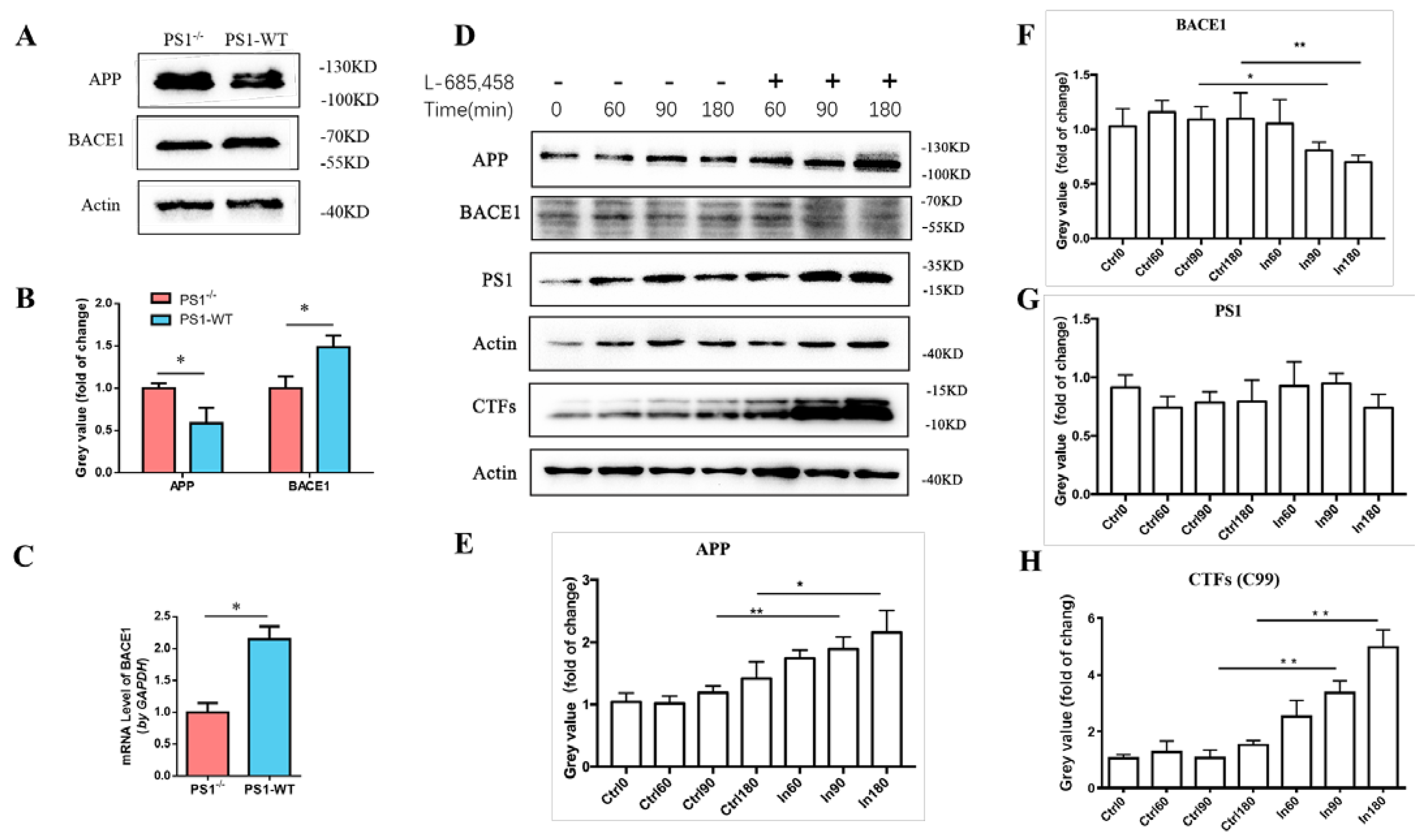
2.1. PS1 Overexpression Upregulated the Expression Levels of Endogenous BACE1 In Vitro
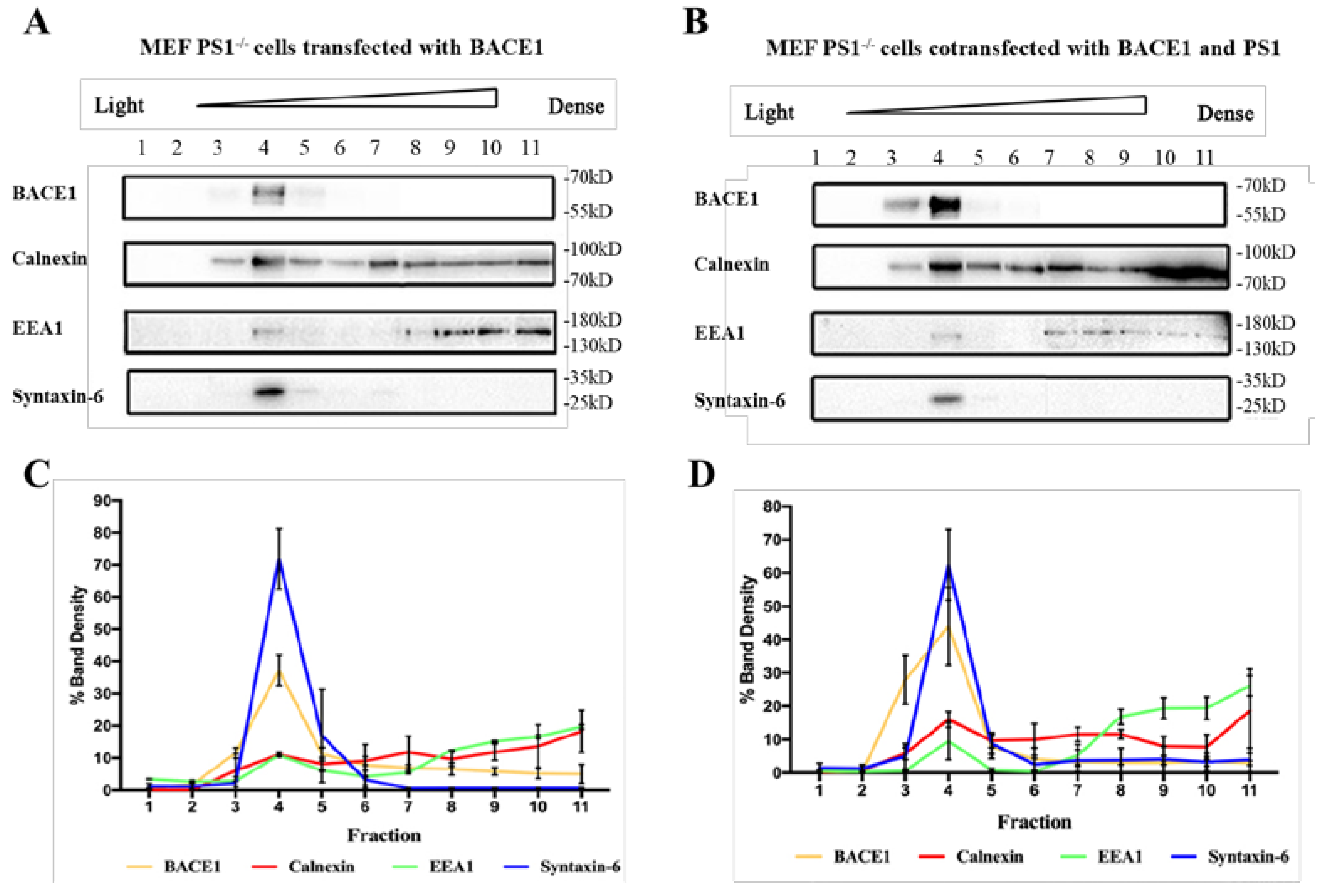
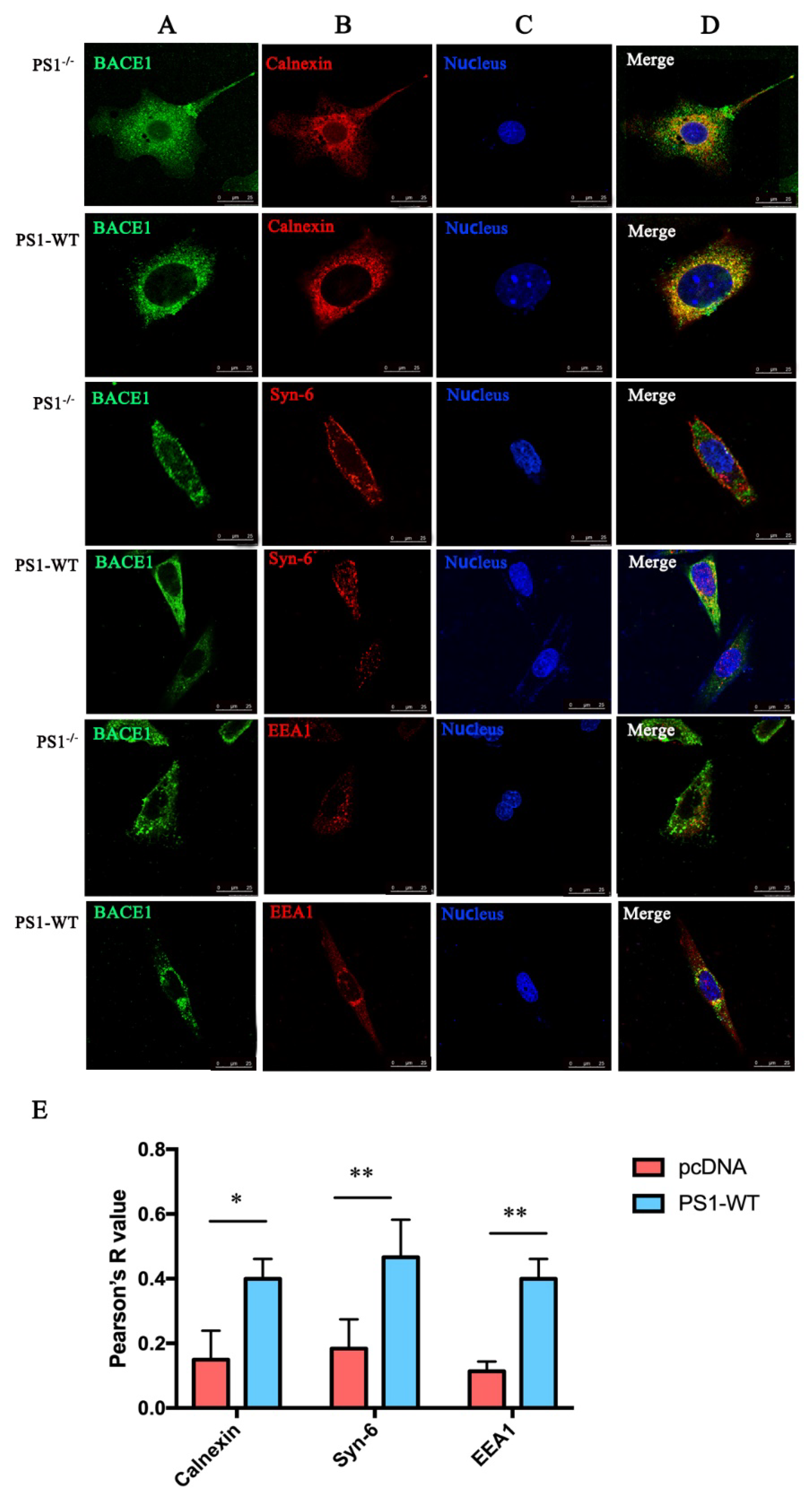
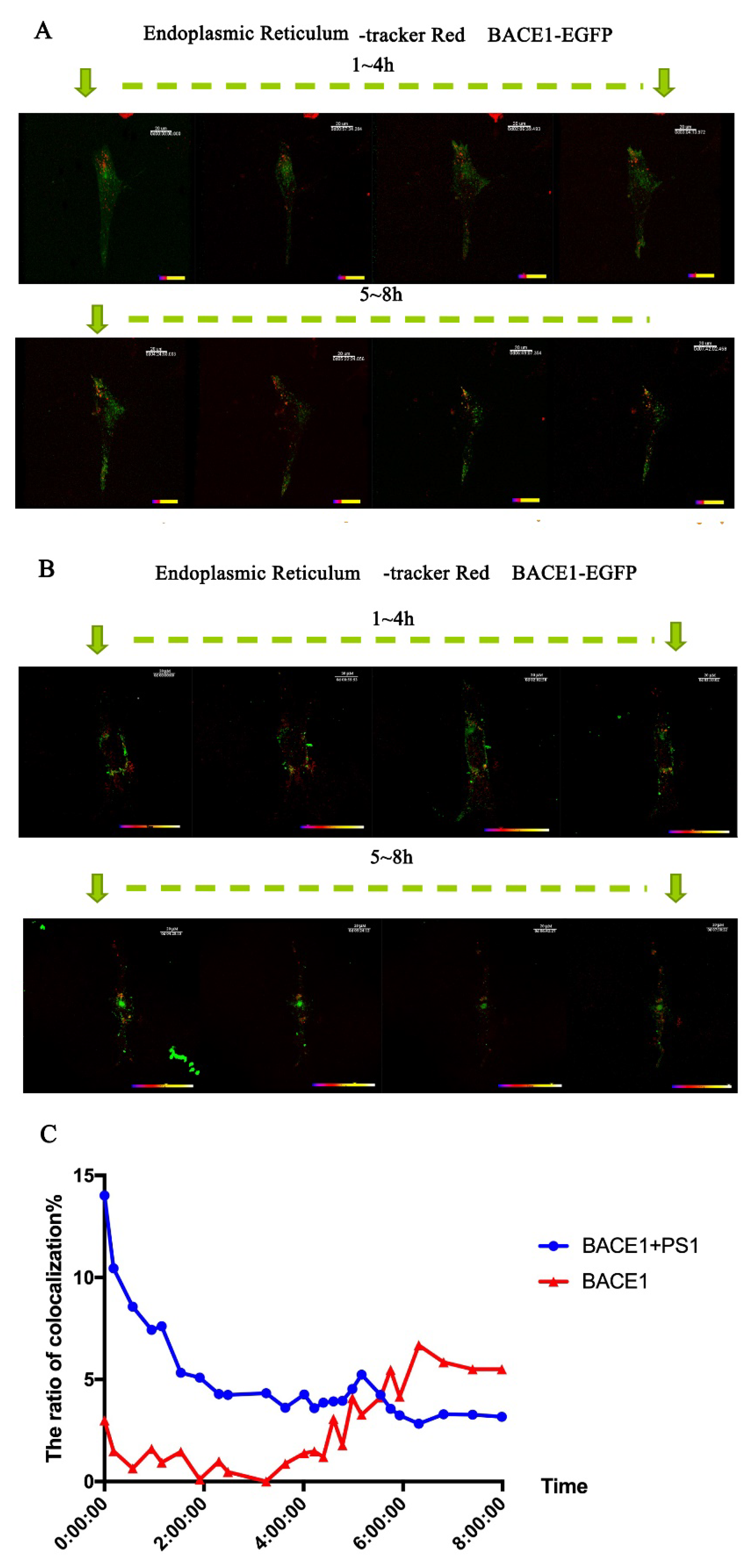
2.2. PS1 Overexpression Upregulated BACE1 Colocalization with ER, Golgi/TGN, and Early Endosome
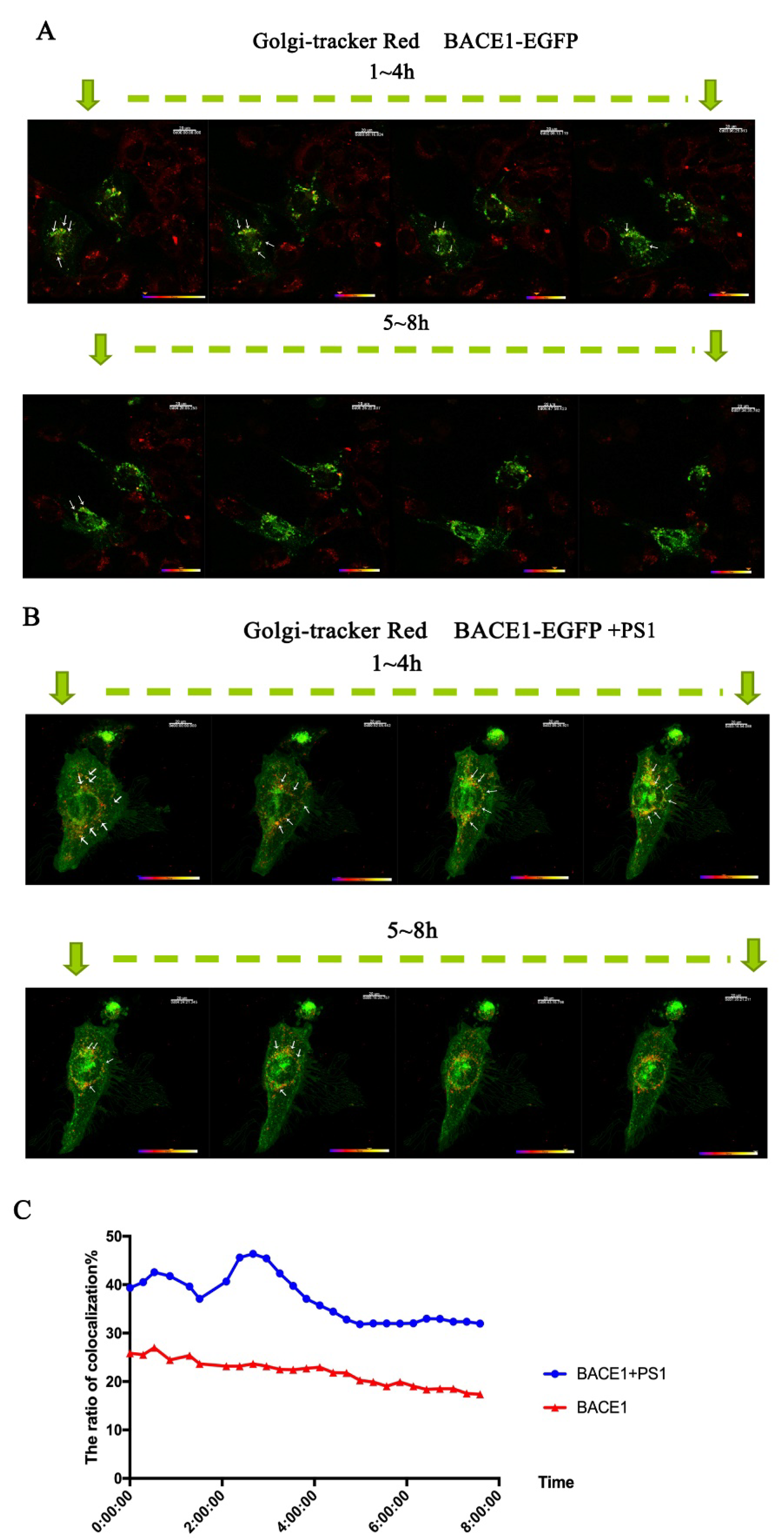
2.3. PS1 Overexpression Upregulated BACE1 Transportation to the ER and Golgi/TGN
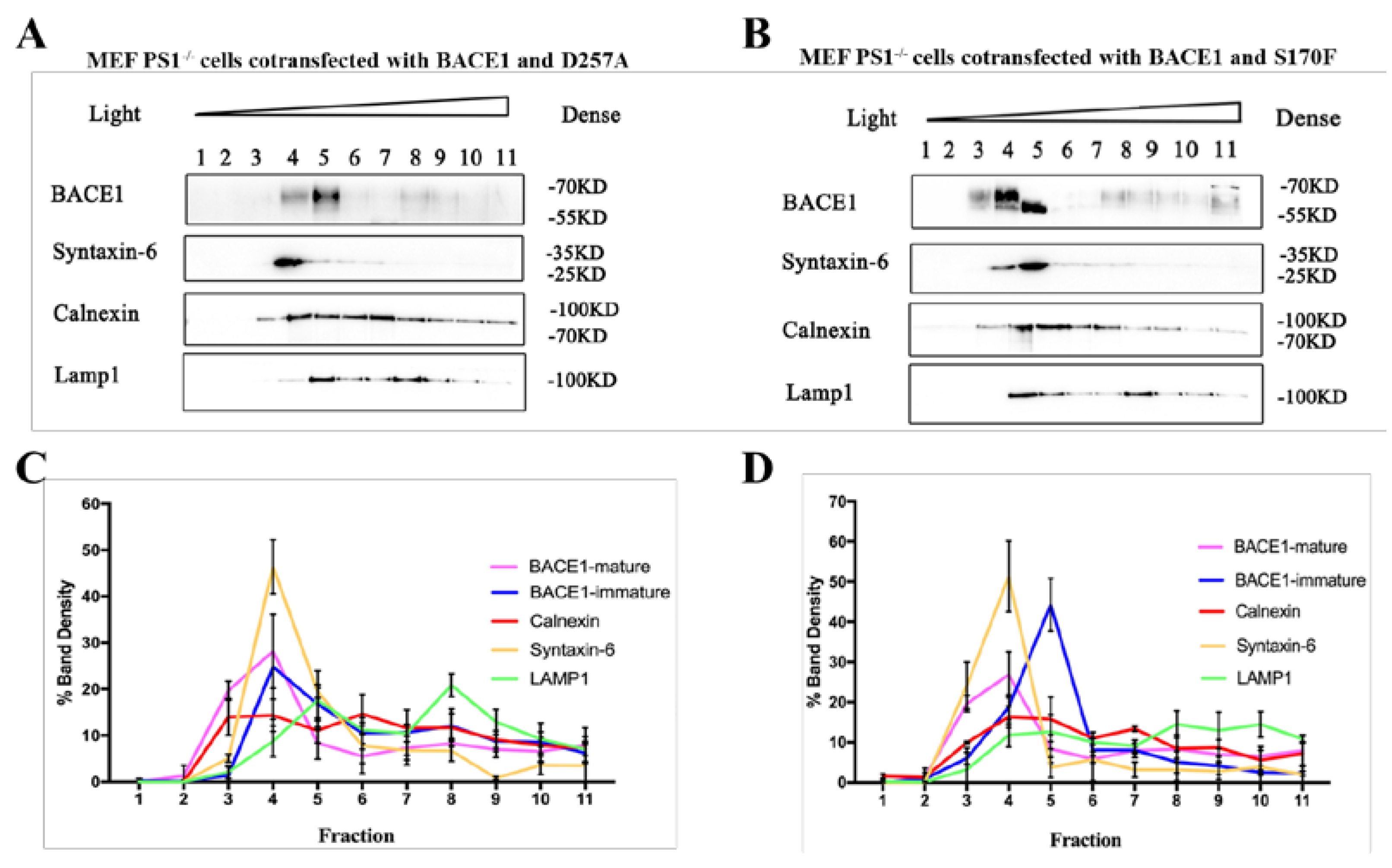
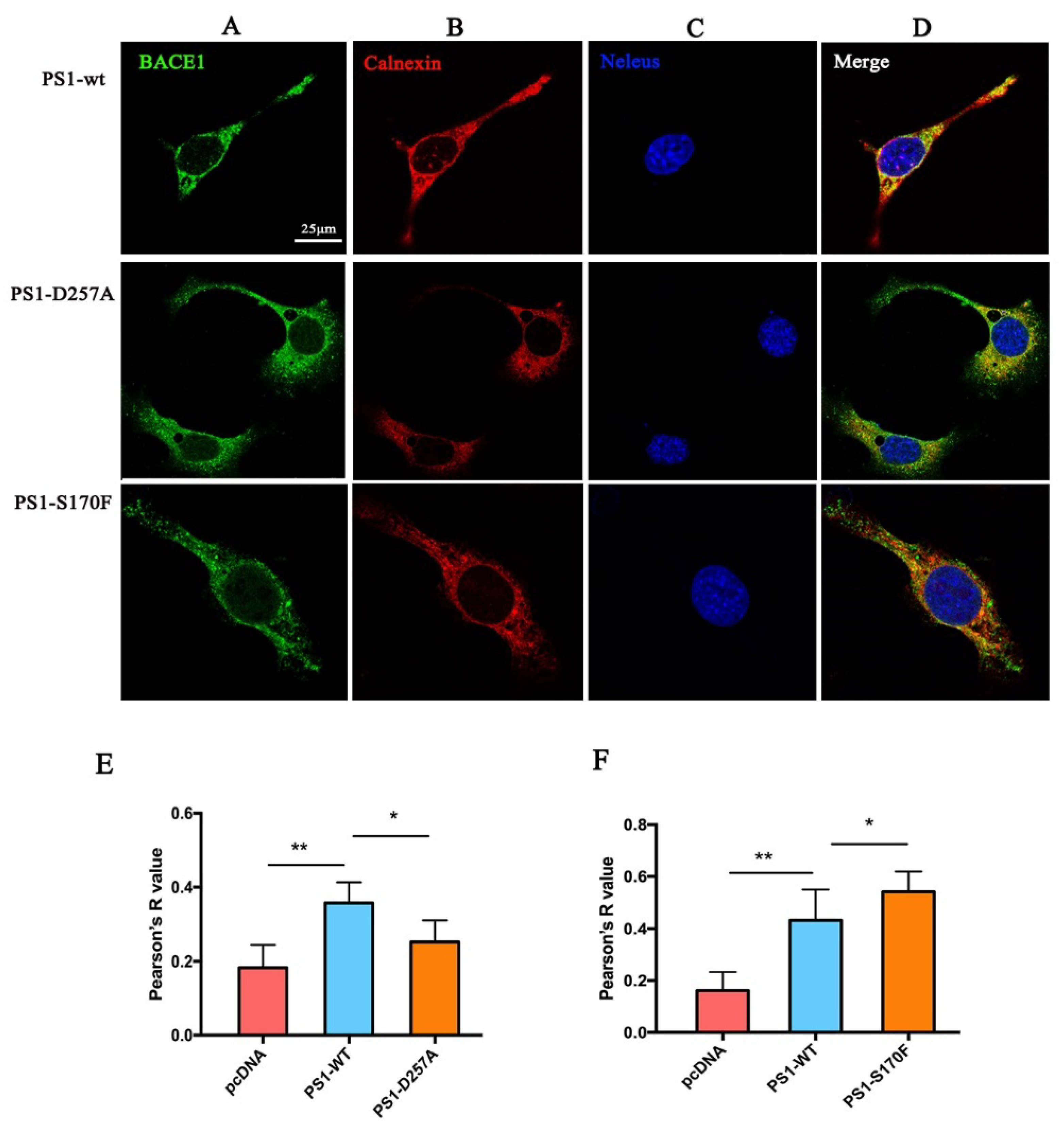
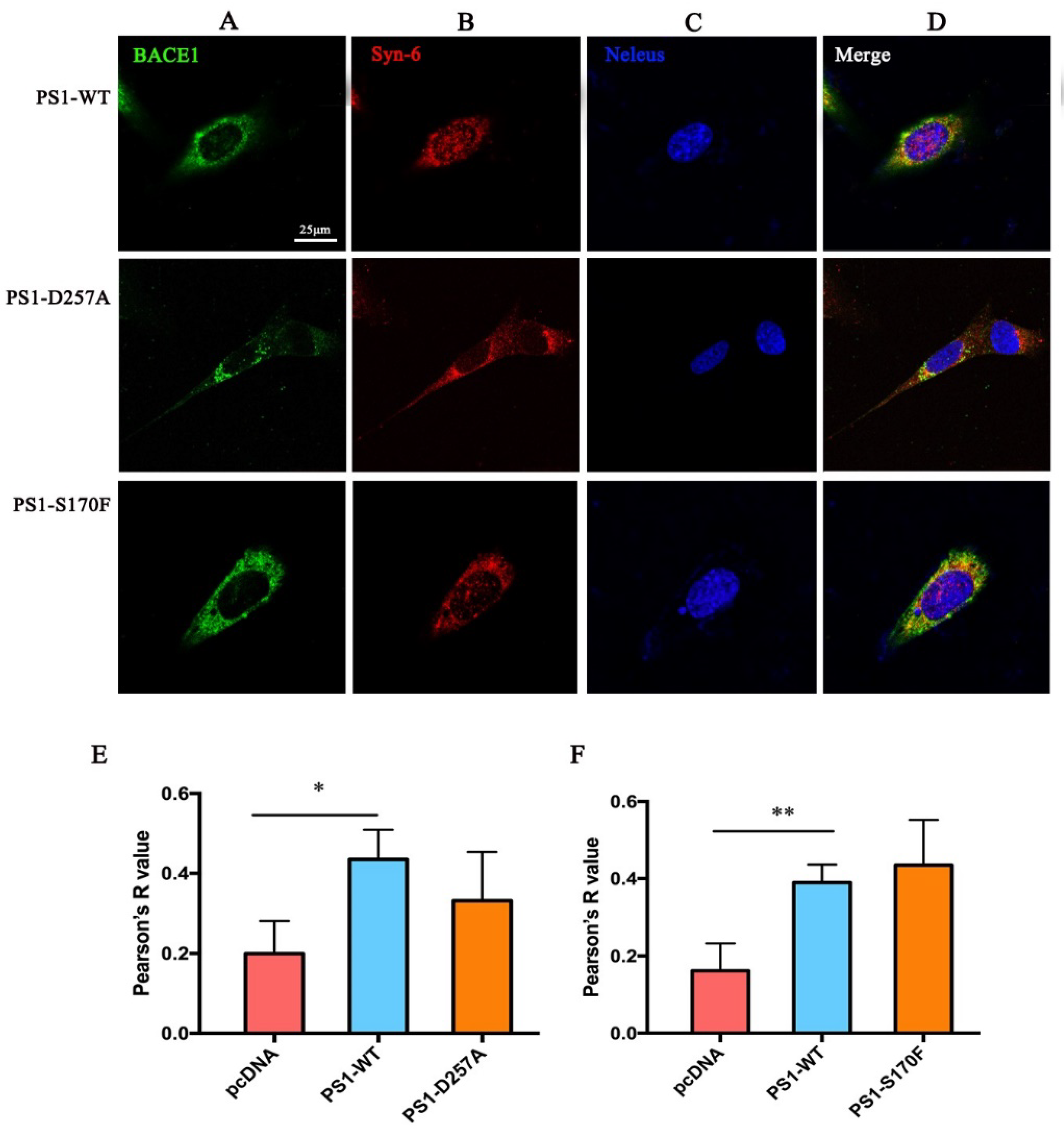
2.4. PS1 Mutants Affect the Distribution and Maturation of BACE1 In Vitro
3. Discussion
4. Materials and Methods
4.1. Plasmid Construction
4.2. Cell Culture and Transfection
4.3. Subcellular Fractionation
4.4. Immunocytochemistry
4.5. Time-Lapse
4.6. Immunoblotting
4.7. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, X.; Song, W. The role of APP and BACE1 trafficking in APP processing and amyloid-β generation. Alzheimer’s Res. Ther. 2013, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Zohar, O.; Cavallaro, S.; D’Agata, V.; Alkon, D.L. Quantification and distribution of beta-secretase alternative splice variants in the rat and human brain. Mol. Brain Res. 2003, 115, 63–68. [Google Scholar] [CrossRef]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Huse, J.T.; Liu, K.; Pijak, D.S.; Carlin, D.; Lee, V.M.; Doms, R.W. Beta-secretase processing in the trans-Golgi network preferentially generates truncated amyloid species that accumulate in Alzheimer’s disease brain. J. Biol. Chem. 2002, 277, 16278–16284. [Google Scholar] [CrossRef]
- Cole, S.L.; Vassar, R. The role of amyloid precursor protein processing by BACE1, the beta-secretase, in Alzheimer disease pathophysiology. J. Biol. Chem. 2008, 283, 29621–29625. [Google Scholar] [CrossRef]
- Small, D.H.; Klaver, D.W.; Beckman, M. Regulation of proBACE1 by glycosaminoglycans. Neurodegener. Dis. 2008, 5, 206–208. [Google Scholar] [CrossRef]
- Murayama, K.S.; Kametani, F.; Saito, S.; Kume, H.; Akiyama, H.; Araki, W. Reticulons RTN3 and RTN4-B/C interact with BACE1 and inhibit its ability to produce amyloid beta-protein. Eur. J. Neurosci. 2006, 24, 1237–1244. [Google Scholar] [CrossRef]
- Lu, P.; Bai, X.C.; Ma, D.; Xie, T.; Yan, C.; Sun, L.; Yang, G.; Zhao, Y.; Zhou, R.; Scheres, S.H.W.; et al. Three-dimensional structure of human γ-secretase. Nature 2014, 512, 166–170. [Google Scholar] [CrossRef]
- Chelban, V.; Breza, M.; Szaruga, M.; Vandrovcova, J.; Murphy, D.; Lee, C.J.; Alikhwan, S.; Bourinaris, T.; Vavougios, G.; Ilyas, M.; et al. Spastic paraplegia preceding PSEN1-related familial Alzheimer’s disease. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2021, 13, e12186. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.M.; Kim, G.; Seekins, S.; Yager, D.; et al. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron 1996, 17, 1005–1013. [Google Scholar] [CrossRef]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef]
- Sun, L.; Zhou, R.; Yang, G.; Shi, Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase. Proc. Natl. Acad. Sci. USA 2017, 114, E476–E485. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, K.; Qiu, Y.; Ren, Z.; Dai, R.; Deng, Y.; Qing, H. Effect of Presenilin Mutations on APP Cleavage; Insights into the Pathogenesis of FAD. Front. Aging Neurosci. 2016, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Leem, J.Y.; Greenfield, J.P.; Wang, P.; Kim, B.S.; Wang, R.; Lopes, K.O.; Kim, S.H.; Zheng, H.; Greengard, P.; et al. Presenilin-1 regulates intracellular trafficking and cell surface delivery of beta-amyloid precursor protein. J. Biol. Chem. 2003, 278, 3446–3454. [Google Scholar] [CrossRef] [PubMed]
- Leem, J.Y.; Vijayan, S.; Han, P.; Cai, D.; Machura, M.; Lopes, K.O.; Veselits, M.L.; Xu, H.; Thinakaran, G. Presenilin 1 is required for maturation and cell surface accumulation of nicastrin. J. Biol. Chem. 2002, 277, 19236–19240. [Google Scholar] [CrossRef] [PubMed]
- Zou, K.; Hosono, T.; Nakamura, T.; Shiraishi, H.; Maeda, T.; Komano, H.; Yanagisawa, K.; Michikawa, M. Novel role of presenilins in maturation and transport of integrin beta 1. Biochemistry 2008, 47, 3370–3378. [Google Scholar] [CrossRef] [PubMed]
- Giliberto, L.; Borghi, R.; Piccini, A.; Mangerini, R.; Sorbi, S.; Cirmena, G.; Garuti, A.; Ghetti, B.; Tagliavini, F.; Mughal, M.R.; et al. Mutant presenilin 1 increases the expression and activity of BACE1. J. Biol. Chem. 2009, 284, 9027–9038. [Google Scholar] [CrossRef]
- Kuzuya, A.; Uemura, K.; Kitagawa, N.; Aoyagi, N.; Kihara, T.; Ninomiya, H.; Ishiura, S.; Takahashi, R.; Shimohama, S. Presenilin 1 is involved in the maturation of beta-site amyloid precursor protein-cleaving enzyme 1 (BACE1). J. Neurosci. Res. 2007, 85, 153–165. [Google Scholar] [CrossRef]
- Kim, S.H.; Lah, J.J.; Thinakaran, G.; Levey, A.; Sisodia, S.S. Subcellular localization of presenilins: Association with a unique membrane pool in cultured cells. Neurobiol. Dis. 2000, 7, 99–117. [Google Scholar] [CrossRef]
- Pasternak, S.H.; Bagshaw, R.D.; Guiral, M.; Zhang, S.; Ackerley, C.A.; Pak, B.J.; Callahan, J.W.; Mahuran, D.J. Presenilin-1, nicastrin, amyloid precursor protein, and gamma-secretase activity are co-localized in the lysosomal membrane. J. Biol. Chem. 2003, 278, 26687–26694. [Google Scholar] [CrossRef]
- Teng, L.; Zhao, J.; Wang, F.; Ma, L.; Pei, G. A GPCR/secretase complex regulates beta- and gamma-secretase specificity for Abeta production and contributes to AD pathogenesis. Cell Res. 2010, 20, 138–153. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Wang, X.; Li, X.; Wang, X.; Zhang, C.; Li, W.; Zhang, Y.; Gu, H.; Xie, X.; Nan, F.; et al. Targeting the γ-/β-secretase interaction reduces β-amyloid generation and ameliorates Alzheimer’s disease-related pathogenesis. Cell Discov. 2015, 1, 15021. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Leem, J.Y.; Lah, J.J.; Slunt, H.H.; Levey, A.I.; Thinakaran, G.; Sisodia, S.S. Multiple effects of aspartate mutant presenilin 1 on the processing and trafficking of amyloid precursor protein. J. Biol. Chem. 2001, 276, 43343–43350. [Google Scholar] [CrossRef] [PubMed]
- Golan, M.P.; Styczynska, M.; Jozwiak, K.; Walecki, J.; Maruszak, A.; Pniewski, J.; Lugiewicz, R.; Filipek, S.; Zekanowski, C.; Barcikowska, M. Early-onset Alzheimer’s disease with a de novo mutation in the presenilin 1 gene. Exp. Neurol. 2007, 208, 264–268. [Google Scholar] [CrossRef]
- Piccini, A.; Zanusso, G.; Borghi, R.; Noviello, C.; Monaco, S.; Russo, R.; Damonte, G.; Armirotti, A.; Gelati, M.; Giordano, R.; et al. Association of a presenilin 1 S170F mutation with a novel Alzheimer disease molecular phenotype. Arch. Neurol. 2007, 64, 738–745. [Google Scholar] [CrossRef]
- Capell, A.; Steiner, H.; Willem, M.; Kaiser, H.; Meyer, C.; Walter, J.; Lammich, S.; Multhaup, G.; Haass, C. Maturation and pro-peptide cleavage of beta-secretase. J. Biol. Chem. 2000, 275, 30849–30854. [Google Scholar] [CrossRef]
- Haniu, M.; Denis, P.; Young, Y.; Mendiaz, E.A.; Fuller, J.; Hui, J.O.; Bennett, B.D.; Kahn, S.; Ross, S.; Burgess, T.; et al. Characterization of Alzheimer’s beta -secretase protein BACE. A pepsin family member with unusual properties. J. Biol. Chem. 2000, 275, 21099–21106. [Google Scholar] [CrossRef]
- Creemers, J.W.; Ines Dominguez, D.; Plets, E.; Serneels, L.; Taylor, N.A.; Multhaup, G.; Craessaerts, K.; Annaert, W.; De Strooper, B. Processing of beta-secretase by furin and other members of the proprotein convertase family. J. Biol. Chem. 2001, 276, 4211–4217. [Google Scholar] [CrossRef]
- Soldano, A.; Hassan, B.A. Beyond pathology: APP, brain development and Alzheimer’s disease. Curr. Opin. Neurobiol. 2014, 27, 61–67. [Google Scholar] [CrossRef]
- Jiang, Y.; Mullaney, K.A.; Peterhoff, C.M.; Che, S.; Schmidt, S.D.; Boyer-Boiteau, A.; Ginsberg, S.D.; Cataldo, A.M.; Mathews, P.M.; Nixon, R.A. Alzheimer’s-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc. Natl. Acad. Sci. USA 2010, 107, 1630–1635. [Google Scholar] [CrossRef]
- Qing, H.; Zhou, W.; Christensen, M.A.; Sun, X.; Tong, Y.; Song, W. Degradation of BACE by the ubiquitin-proteasome pathway. FASEB J. 2004, 18, 1571–1573. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.M.; Cho, S.J.; Song, J.C.; Song, S.Y.; Jo, S.A.; Jo, C.; Yoon, K.; Tanzi, R.E.; Choi, E.J.; Koh, Y.H. SUMO1 modulates Abeta generation via BACE1 accumulation. Neurobiol. Aging 2013, 34, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Lemere, C.A.; Capell, A.; Citron, M.; Seubert, P.; Schenk, D.; Lannfelt, L.; Selkoe, D.J. The Swedish mutation causes early-onset Alzheimer’s disease by beta-secretase cleavage within the secretory pathway. Nat. Med. 1995, 1, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef] [PubMed]
- Sisodia, S.S.; Kim, S.H.; Thinakaran, G. Function and dysfunction of the presenilins. Am. J. Hum. Genet. 1999, 65, 7–12. [Google Scholar] [CrossRef][Green Version]
- Wolfe, M.S. When loss is gain: Reduced presenilin proteolytic function leads to increased Abeta42/Abeta40. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007, 8, 136–140. [Google Scholar] [CrossRef]
- Katayama, T.; Imaizumi, K.; Honda, A.; Yoneda, T.; Kudo, T.; Takeda, M.; Mori, K.; Rozmahel, R.; Fraser, P.; George-Hyslop, P.S.; et al. Disturbed activation of endoplasmic reticulum stress transducers by familial Alzheimer’s disease-linked presenilin-1 mutations. J. Biol. Chem. 2001, 276, 43446–43454. [Google Scholar] [CrossRef]
- Yasuda, Y.; Kudo, T.; Katayama, T.; Imaizumi, K.; Yatera, M.; Okochi, M.; Yamamori, H.; Matsumoto, N.; Kida, T.; Fukumori, A.; et al. FAD-linked presenilin-1 mutants impede translation regulation under ER stress. Biochem. Biophys. Res. Commun. 2002, 296, 313–318. [Google Scholar] [CrossRef]
- Hashimoto, S.; Ishii, A.; Kamano, N.; Watamura, N.; Saito, T.; Ohshima, T.; Yokosuka, M.; Saido, T.C. Endoplasmic reticulum stress responses in mouse models of Alzheimer’s disease: Overexpression paradigm versus knockin paradigm. J. Biol. Chem. 2018, 293, 3118–3125. [Google Scholar] [CrossRef]
- Rojas-Charry, L.; Calero-Martinez, S.; Morganti, C.; Morciano, G.; Park, K.; Hagel, C.; Marciniak, S.J.; Glatzel, M.; Pinton, P.; Sepulveda-Falla, D. Susceptibility to cellular stress in PS1 mutant N2a cells is associated with mitochondrial defects and altered calcium homeostasis. Sci. Rep. 2020, 10, 6455. [Google Scholar] [CrossRef]
- Chia, P.Z.; Gleeson, P.A. Intracellular trafficking of the β-secretase and processing of amyloid precursor protein. IUBMB Life 2011, 63, 721–729. [Google Scholar] [PubMed]
- Waugh, M.G. Raft-like membranes from the trans-Golgi network and endosomal compartments. Nat. Protoc. 2013, 8, 2429–2439. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.A.; Klutkowski, J.A.; Freret, T.; Wolfe, M.S. Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid beta-peptides (Abeta) by gamma-secretase to increase 42-to-40-residue Abeta. J. Biol. Chem. 2014, 289, 31043–31052. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, N.; Qiu, Y.; Wang, H.; Zhao, J.; Qing, H. PS1 Affects the Pathology of Alzheimer’s Disease by Regulating BACE1 Distribution in the ER and BACE1 Maturation in the Golgi Apparatus. Int. J. Mol. Sci. 2022, 23, 16151. https://doi.org/10.3390/ijms232416151
Li N, Qiu Y, Wang H, Zhao J, Qing H. PS1 Affects the Pathology of Alzheimer’s Disease by Regulating BACE1 Distribution in the ER and BACE1 Maturation in the Golgi Apparatus. International Journal of Molecular Sciences. 2022; 23(24):16151. https://doi.org/10.3390/ijms232416151
Chicago/Turabian StyleLi, Nuomin, Yunjie Qiu, Hao Wang, Juan Zhao, and Hong Qing. 2022. "PS1 Affects the Pathology of Alzheimer’s Disease by Regulating BACE1 Distribution in the ER and BACE1 Maturation in the Golgi Apparatus" International Journal of Molecular Sciences 23, no. 24: 16151. https://doi.org/10.3390/ijms232416151
APA StyleLi, N., Qiu, Y., Wang, H., Zhao, J., & Qing, H. (2022). PS1 Affects the Pathology of Alzheimer’s Disease by Regulating BACE1 Distribution in the ER and BACE1 Maturation in the Golgi Apparatus. International Journal of Molecular Sciences, 23(24), 16151. https://doi.org/10.3390/ijms232416151