
Oral Therapy for the Treatment of Transthyretin-Related Amyloid Cardiomyopathy


Abstract
1. Introduction
2. Transthyretin Structure and Function
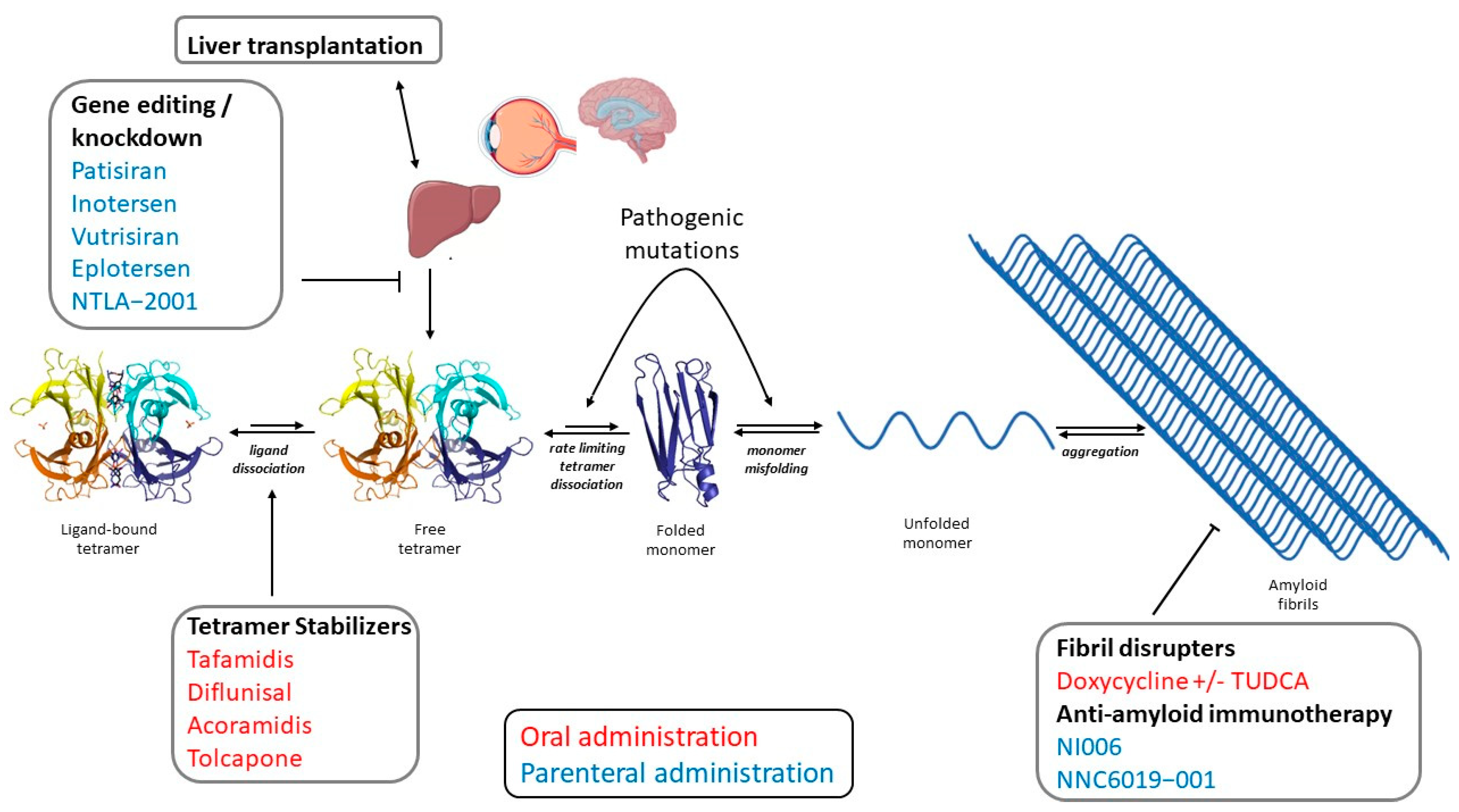
3. Molecular Mechanisms of ATTR Amyloidosis
4. TTR-Related Cardiac Amyloidosis
5. Principles of Therapy
6. TTR Tetramer Stabilizers
6.1. Tafamidis
6.2. Diflunisal
6.3. AG10/Acoramidis
6.4. Tolcapone
7. Amyloid Fibril Disruptors: Doxycycline
8. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Merlini, G.; Dispenzieri, A.; Sanchorawala, V.; Schonland, S.O.; Palladini, G.; Hawkins, P.N.; Gertz, M.A. Systemic immunoglobulin light chain amyloidosis. Nat. Rev. Dis. Prim. 2018, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Nuvolone, M.; Merlini, G. Systemic amyloidosis: Novel therapies and role of biomarkers. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transplant. Assoc. Eur. Ren. Assoc. 2017, 32, 770–780. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ruberg, F.L.; Grogan, M.; Hanna, M.; Kelly, J.W.; Maurer, M.S. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 2872–2891. [Google Scholar] [CrossRef]
- Maurer, M.S.; Bokhari, S.; Damy, T.; Dorbala, S.; Drachman, B.M.; Fontana, M.; Grogan, M.; Kristen, A.V.; Lousada, I.; Nativi-Nicolau, J.; et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ. Heart Fail. 2019, 12, e006075. [Google Scholar] [CrossRef] [PubMed]
- Mohamed-Salem, L.; Santos-Mateo, J.J.; Sanchez-Serna, J.; Hernandez-Vicente, A.; Reyes-Marle, R.; Castellon Sanchez, M.I.; Claver-Valderas, M.A.; Gonzalez-Vioque, E.; Haro-Del Moral, F.J.; Garcia-Pavia, P.; et al. Prevalence of wild type ATTR assessed as myocardial uptake in bone scan in the elderly population. Int. J. Cardiol. 2018, 270, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Nakamura, K.; Ito, H. Molecular Mechanisms of Cardiac Amyloidosis. Int. J. Mol. Sci. 2021, 23, 25. [Google Scholar] [CrossRef]
- Vieira, M.; Saraiva, M.J. Transthyretin: A multifaceted protein. Biomol. Concepts 2014, 5, 45–54. [Google Scholar] [CrossRef]
- Vahlquist, A.; Peterson, P.A.; Wibell, L. Metabolism of the vitamin A transporting protein complex. I. Turnover studies in normal persons and in patients with chronic renal failure. Eur. J. Clin. Investig. 1973, 3, 352–362. [Google Scholar] [CrossRef]
- Benson, M.D.; Kincaid, J.C. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve 2007, 36, 411–423. [Google Scholar] [CrossRef]
- Myron Johnson, A.; Merlini, G.; Sheldon, J.; Ichihara, K. Clinical indications for plasma protein assays: Transthyretin (prealbumin) in inflammation and malnutrition: International Federation of Clinical Chemistry and Laboratory Medicine (IFCC): IFCC Scientific Division Committee on Plasma Proteins (C-PP). Clin. Chem. Lab. Med. 2007, 45, 419–426. [Google Scholar] [CrossRef]
- Kanda, Y.; Goodman, D.S.; Canfield, R.E.; Morgan, F.J. The amino acid sequence of human plasma prealbumin. J. Biol. Chem. 1974, 249, 6796–6805. [Google Scholar] [CrossRef] [PubMed]
- Blake, C.C.; Geisow, M.J.; Swan, I.D.; Rerat, C.; Rerat, B. Structure of human plasma prealbumin at 2-5 A resolution. A preliminary report on the polypeptide chain conformation, quaternary structure and thyroxine binding. J. Mol. Biol. 1974, 88, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Blake, C.C.; Geisow, M.J.; Oatley, S.J.; Rérat, B.; Rérat, C. Structure of prealbumin: Secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 A. J. Mol. Biol. 1978, 121, 339–356. [Google Scholar] [CrossRef] [PubMed]
- Monaco, H.L.; Rizzi, M.; Coda, A. Structure of a complex of two plasma proteins: Transthyretin and retinol-binding protein. Science 1995, 268, 1039–1041. [Google Scholar] [CrossRef] [PubMed]
- Bartalena, L.; Robbins, J. Thyroid hormone transport proteins. Clin. Lab. Med. 1993, 13, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Nuvolone, M.; Obici, L.; Merlini, G. Transthyretin-associated Familial Amyloid Polyneuropathy—Current and Emerging Therapies. Eur. J. Neurol. 2012, 7, 14–21. [Google Scholar]
- Aldred, A.R.; Brack, C.M.; Schreiber, G. The cerebral expression of plasma protein genes in different species. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 1995, 111, 1–15. [Google Scholar] [CrossRef]
- Hagen, G.A.; Elliott, W.J. Transport of thyroid hormones in serum and cerebrospinal fluid. J. Clin. Endocrinol. Metab. 1973, 37, 415–422. [Google Scholar] [CrossRef]
- Gião, T.; Saavedra, J.; Cotrina, E.; Quintana, J.; Llop, J.; Arsequell, G.; Cardoso, I. Undiscovered Roles for Transthyretin: From a Transporter Protein to a New Therapeutic Target for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 2075. [Google Scholar] [CrossRef]
- Li, X.; Buxbaum, J.N. Transthyretin and the brain re-visited: Is neuronal synthesis of transthyretin protective in Alzheimer’s disease? Mol. Neurodegener. 2011, 6, 79. [Google Scholar] [CrossRef]
- Buxbaum, J.N.; Ye, Z.; Reixach, N.; Friske, L.; Levy, C.; Das, P.; Golde, T.; Masliah, E.; Roberts, A.R.; Bartfai, T. Transthyretin protects Alzheimer’s mice from the behavioral and biochemical effects of Abeta toxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 2681–2686. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Anderson, D.H.; Liang, W.Y.; Chou, J.A.; Saelices, L. The inhibition of cellular toxicity of amyloid-beta by dissociated transthyretin. J. Biol. Chem. 2020, 295, 14015–14024. [Google Scholar] [CrossRef] [PubMed]
- Cascella, R.; Conti, S.; Mannini, B.; Li, X.; Buxbaum, J.N.; Tiribilli, B.; Chiti, F.; Cecchi, C. Transthyretin suppresses the toxicity of oligomers formed by misfolded proteins in vitro. Biochim. Biophys. Acta 2013, 1832, 2302–2314. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Leight, S.N.; Lee, V.M.; Li, T.; Wong, P.C.; Johnson, J.A.; Saraiva, M.J.; Sisodia, S.S. Accelerated Abeta deposition in APPswe/PS1deltaE9 mice with hemizygous deletions of TTR (transthyretin). J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 7006–7010. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.; Gonçalves, A.; Saraiva, M.J.; Cardoso, I. Transthyretin binding to A-Beta peptide--impact on A-Beta fibrillogenesis and toxicity. FEBS Lett. 2008, 582, 936–942. [Google Scholar] [CrossRef]
- Garai, K.; Posey, A.E.; Li, X.; Buxbaum, J.N.; Pappu, R.V. Inhibition of amyloid beta fibril formation by monomeric human transthyretin. Protein Sci. A Publ. Protein Soc. 2018, 27, 1252–1261. [Google Scholar] [CrossRef]
- Schwarzman, A.L.; Gregori, L.; Vitek, M.P.; Lyubski, S.; Strittmatter, W.J.; Enghilde, J.J.; Bhasin, R.; Silverman, J.; Weisgraber, K.H.; Coyle, P.K.; et al. Transthyretin sequesters amyloid beta protein and prevents amyloid formation. Proc. Natl. Acad. Sci. USA 1994, 91, 8368–8372. [Google Scholar] [CrossRef]
- Stein, T.D.; Anders, N.J.; DeCarli, C.; Chan, S.L.; Mattson, M.P.; Johnson, J.A. Neutralization of transthyretin reverses the neuroprotective effects of secreted amyloid precursor protein (APP) in APPSW mice resulting in tau phosphorylation and loss of hippocampal neurons: Support for the amyloid hypothesis. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 7707–7717. [Google Scholar] [CrossRef]
- Hebert, D.N.; Molinari, M. In and out of the ER: Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007, 87, 1377–1408. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef]
- Cornwell, G.G., 3rd; Murdoch, W.L.; Kyle, R.A.; Westermark, P.; Pitkänen, P. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am. J. Med. 1983, 75, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Tanskanen, M.; Peuralinna, T.; Polvikoski, T.; Notkola, I.L.; Sulkava, R.; Hardy, J.; Singleton, A.; Kiuru-Enari, S.; Paetau, A.; Tienari, P.J.; et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: A population-based autopsy study. Ann. Med. 2008, 40, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Marcoux, J.; Mangione, P.P.; Porcari, R.; Degiacomi, M.T.; Verona, G.; Taylor, G.W.; Giorgetti, S.; Raimondi, S.; Sanglier-Cianférani, S.; Benesch, J.L.; et al. A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol. Med. 2015, 7, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, S.; Mangione, P.P.; Verona, G.; Canetti, D.; Nocerino, P.; Marchese, L.; Piccarducci, R.; Mondani, V.; Faravelli, G.; Taylor, G.W.; et al. Comparative study of the stabilities of synthetic in vitro and natural ex vivo transthyretin amyloid fibrils. J. Biol. Chem. 2020, 295, 11379–11387. [Google Scholar] [CrossRef]
- Schneider, F.; Hammarström, P.; Kelly, J.W. Transthyretin slowly exchanges subunits under physiological conditions: A convenient chromatographic method to study subunit exchange in oligomeric proteins. Protein Sci. A Publ. Protein Soc. 2001, 10, 1606–1613. [Google Scholar] [CrossRef]
- Hurshman Babbes, A.R.; Powers, E.T.; Kelly, J.W. Quantification of the thermodynamically linked quaternary and tertiary structural stabilities of transthyretin and its disease-associated variants: The relationship between stability and amyloidosis. Biochemistry 2008, 47, 6969–6984. [Google Scholar] [CrossRef]
- Jiang, X.; Buxbaum, J.N.; Kelly, J.W. The V122I cardiomyopathy variant of transthyretin increases the velocity of rate-limiting tetramer dissociation, resulting in accelerated amyloidosis. Proc. Natl. Acad. Sci. USA 2001, 98, 14943–14948. [Google Scholar] [CrossRef]
- Johnson, S.M.; Connelly, S.; Fearns, C.; Powers, E.T.; Kelly, J.W. The transthyretin amyloidoses: From delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J. Mol. Biol. 2012, 421, 185–203. [Google Scholar] [CrossRef]
- Jacobson, D.R.; Pastore, R.; Pool, S.; Malendowicz, S.; Kane, I.; Shivji, A.; Embury, S.H.; Ballas, S.K.; Buxbaum, J.N. Revised transthyretin Ile 122 allele frequency in African-Americans. Hum. Genet. 1996, 98, 236–238. [Google Scholar] [CrossRef]
- Quarta, C.C.; Buxbaum, J.N.; Shah, A.M.; Falk, R.H.; Claggett, B.; Kitzman, D.W.; Mosley, T.H.; Butler, K.R.; Boerwinkle, E.; Solomon, S.D. The amyloidogenic V122I transthyretin variant in elderly black Americans. N. Engl. J. Med. 2015, 372, 21–29. [Google Scholar] [CrossRef]
- Alves, I.L.; Altland, K.; Almeida, M.R.; Winter, P.; Saraiva, M.J. Screening and biochemical characterization of transthyretin variants in the Portuguese population. Hum. Mutat 1997, 9, 226–233. [Google Scholar] [CrossRef]
- Andersson, R. Familial amyloidosis with polyneuropathy. A clinical study based on patients living in northern Sweden. Acta Med. Scand. Suppl. 1976, 590, 1–64. [Google Scholar] [PubMed]
- Araki, S. Type I familial amyloidotic polyneuropathy (Japanese type). Brain Dev. 1984, 6, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Munar-Ques, M.; Saraiva, M.J.; Viader-Farre, C.; Zabay-Becerril, J.M.; Mulet-Ferrer, J. Genetic epidemiology of familial amyloid polyneuropathy in the Balearic Islands (Spain). Amyloid 2005, 12, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Yazaki, M.; Tokuda, T.; Nakamura, A.; Higashikata, T.; Koyama, J.; Higuchi, K.; Harihara, Y.; Baba, S.; Kametani, F.; Ikeda, S. Cardiac amyloid in patients with familial amyloid polyneuropathy consists of abundant wild-type transthyretin. Biochem. Biophys. Res. Commun. 2000, 274, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, A.; Yazaki, M.; Kametani, F.; Takei, Y.; Ikeda, S. Marked regression of abdominal fat amyloid in patients with familial amyloid polyneuropathy during long-term follow-up after liver transplantation. Liver Transpl. 2008, 14, 563–570. [Google Scholar] [CrossRef]
- Reixach, N.; Deechongkit, S.; Jiang, X.; Kelly, J.W.; Buxbaum, J.N. Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc. Natl. Acad. Sci. USA 2004, 101, 2817–2822. [Google Scholar] [CrossRef]
- Pepys, M.B. Amyloidosis. Annu. Rev. Med. 2006, 57, 223–241. [Google Scholar] [CrossRef]
- Connors, L.H.; Sam, F.; Skinner, M.; Salinaro, F.; Sun, F.; Ruberg, F.L.; Berk, J.L.; Seldin, D.C. Heart Failure Resulting From Age-Related Cardiac Amyloid Disease Associated With Wild-Type Transthyretin: A Prospective, Observational Cohort Study. Circulation 2016, 133, 282–290. [Google Scholar] [CrossRef]
- Geller, H.I.; Singh, A.; Alexander, K.M.; Mirto, T.M.; Falk, R.H. Association Between Ruptured Distal Biceps Tendon and Wild-Type Transthyretin Cardiac Amyloidosis. JAMA 2017, 318, 962–963. [Google Scholar] [CrossRef]
- Grogan, M.; Scott, C.G.; Kyle, R.A.; Zeldenrust, S.R.; Gertz, M.A.; Lin, G.; Klarich, K.W.; Miller, W.L.; Maleszewski, J.J.; Dispenzieri, A. Natural History of Wild-Type Transthyretin Cardiac Amyloidosis and Risk Stratification Using a Novel Staging System. J. Am. Coll. Cardiol. 2016, 68, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Pinney, J.H.; Whelan, C.J.; Petrie, A.; Dungu, J.; Banypersad, S.M.; Sattianayagam, P.; Wechalekar, A.; Gibbs, S.D.; Venner, C.P.; Wassef, N.; et al. Senile systemic amyloidosis: Clinical features at presentation and outcome. J. Am. Heart Assoc. 2013, 2, e000098. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Westermark, G.T.; Suhr, O.B.; Berg, S. Transthyretin-derived amyloidosis: Probably a common cause of lumbar spinal stenosis. Upsala J. Med. Sci. 2014, 119, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Sperry, B.W.; Reyes, B.A.; Ikram, A.; Donnelly, J.P.; Phelan, D.; Jaber, W.A.; Shapiro, D.; Evans, P.J.; Maschke, S.; Kilpatrick, S.E.; et al. Tenosynovial and Cardiac Amyloidosis in Patients Undergoing Carpal Tunnel Release. J. Am. Coll. Cardiol. 2018, 72, 2040–2050. [Google Scholar] [CrossRef] [PubMed]
- Sueyoshi, T.; Ueda, M.; Jono, H.; Irie, H.; Sei, A.; Ide, J.; Ando, Y.; Mizuta, H. Wild-type transthyretin-derived amyloidosis in various ligaments and tendons. Hum. Pathol. 2011, 42, 1259–1264. [Google Scholar] [CrossRef]
- Rubin, J.; Alvarez, J.; Teruya, S.; Castano, A.; Lehman, R.A.; Weidenbaum, M.; Geller, J.A.; Helmke, S.; Maurer, M.S. Hip and knee arthroplasty are common among patients with transthyretin cardiac amyloidosis, occurring years before cardiac amyloid diagnosis: Can we identify affected patients earlier? Amyloid 2017, 24, 226–230. [Google Scholar] [CrossRef]
- Pinto, M.V.; Milone, M.; Mauermann, M.L.; Dyck, P.J.B.; Alhammad, R.; McPhail, E.D.; Grogan, M.; Liewluck, T. Transthyretin amyloidosis: Putting myopathy on the map. Muscle Nerve 2020, 61, 95–100. [Google Scholar] [CrossRef]
- Wajnsztajn Yungher, F.; Kim, A.; Boehme, A.; Kleyman, I.; Weimer, L.H.; Maurer, M.S.; Brannagan, T.H., 3rd. Peripheral neuropathy symptoms in wild type transthyretin amyloidosis. J. Peripher. Nerv. Syst. JPNS 2020, 25, 265–272. [Google Scholar] [CrossRef]
- Zeldenrust, S.R.; Cooper, L.T. Getting to the heart of the matter: Cardiac involvement in transthyretin-related amyloidosis. Eur. Heart J. 2013, 34, 483–485. [Google Scholar] [CrossRef]
- Ando, E.; Ando, Y.; Okamura, R.; Uchino, M.; Ando, M.; Negi, A. Ocular manifestations of familial amyloidotic polyneuropathy type I: Long-term follow up. Br. J. Ophthalmol. 1997, 81, 295–298. [Google Scholar] [CrossRef]
- Benson, M.D. Leptomeningeal amyloid and variant transthyretins. Am. J. Pathol. 1996, 148, 351–354. [Google Scholar] [PubMed]
- Ihse, E.; Rapezzi, C.; Merlini, G.; Benson, M.D.; Ando, Y.; Suhr, O.B.; Ikeda, S.; Lavatelli, F.; Obici, L.; Quarta, C.C.; et al. Amyloid fibrils containing fragmented ATTR may be the standard fibril composition in ATTR amyloidosis. Amyloid 2013, 20, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Lane, T.; Fontana, M.; Martinez-Naharro, A.; Quarta, C.C.; Whelan, C.J.; Petrie, A.; Rowczenio, D.M.; Gilbertson, J.A.; Hutt, D.F.; Rezk, T.; et al. Natural History, Quality of Life, and Outcome in Cardiac Transthyretin Amyloidosis. Circulation 2019, 140, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Hanna, M.; Grogan, M.; Dispenzieri, A.; Witteles, R.; Drachman, B.; Judge, D.P.; Lenihan, D.J.; Gottlieb, S.S.; Shah, S.J.; et al. Genotype and Phenotype of Transthyretin Cardiac Amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J. Am. Coll. Cardiol. 2016, 68, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Gillmore, J.D.; Damy, T.; Fontana, M.; Hutchinson, M.; Lachmann, H.J.; Martinez-Naharro, A.; Quarta, C.C.; Rezk, T.; Whelan, C.J.; Gonzalez-Lopez, E.; et al. A new staging system for cardiac transthyretin amyloidosis. Eur. Heart J. 2018, 39, 2799–2806. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Maurer, M.S.; Falk, R.H.; Merlini, G.; Damy, T.; Dispenzieri, A.; Wechalekar, A.D.; Berk, J.L.; Quarta, C.C.; Grogan, M.; et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation 2016, 133, 2404–2412. [Google Scholar] [CrossRef] [PubMed]
- Ericzon, B.G.; Wilczek, H.E.; Larsson, M.; Wijayatunga, P.; Stangou, A.; Pena, J.R.; Furtado, E.; Barroso, E.; Daniel, J.; Samuel, D.; et al. Liver Transplantation for Hereditary Transthyretin Amyloidosis: After 20 Years Still the Best Therapeutic Alternative? Transplantation 2015, 99, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Bulawa, C.E.; Connelly, S.; Devit, M.; Wang, L.; Weigel, C.; Fleming, J.A.; Packman, J.; Powers, E.T.; Wiseman, R.L.; Foss, T.R.; et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. USA 2012, 109, 9629–9634. [Google Scholar] [CrossRef]
- Coelho, T.; Maia, L.F.; Martins da Silva, A.; Waddington Cruz, M.; Planté-Bordeneuve, V.; Lozeron, P.; Suhr, O.B.; Campistol, J.M.; Conceição, I.M.; Schmidt, H.H.; et al. Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology 2012, 79, 785–792. [Google Scholar] [CrossRef]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef]
- Sekijima, Y.; Dendle, M.A.; Kelly, J.W. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid 2006, 13, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing diflunisal for familial amyloid polyneuropathy: A randomized clinical trial. JAMA 2013, 310, 2658–2667. [Google Scholar] [CrossRef] [PubMed]
- Sekijima, Y.; Tojo, K.; Morita, H.; Koyama, J.; Ikeda, S. Safety and efficacy of long-term diflunisal administration in hereditary transthyretin (ATTR) amyloidosis. Amyloid 2015, 22, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Castaño, A.; Helmke, S.; Alvarez, J.; Delisle, S.; Maurer, M.S. Diflunisal for ATTR cardiac amyloidosis. Congest. Heart Fail. (Greenwich Conn.) 2012, 18, 315–319. [Google Scholar] [CrossRef]
- Ikram, A.; Donnelly, J.P.; Sperry, B.W.; Samaras, C.; Valent, J.; Hanna, M. Diflunisal tolerability in transthyretin cardiac amyloidosis: A single center’s experience. Amyloid 2018, 25, 197–202. [Google Scholar] [CrossRef]
- Koyama, J.; Minamisawa, M.; Sekijima, Y.; Ikeda, S.I.; Kozuka, A.; Ebisawa, S.; Miura, T.; Motoki, H.; Okada, A.; Izawa, A.; et al. Left ventricular deformation and torsion assessed by speckle-tracking echocardiography in patients with mutated transthyretin-associated cardiac amyloidosis and the effect of diflunisal on myocardial function. Int. J. Cardiol. Heart Vasc. 2015, 9, 1–10. [Google Scholar] [CrossRef][Green Version]
- Lohrmann, G.; Pipilas, A.; Mussinelli, R.; Gopal, D.M.; Berk, J.L.; Connors, L.H.; Vellanki, N.; Hellawell, J.; Siddiqi, O.K.; Fox, J.; et al. Stabilization of Cardiac Function With Diflunisal in Transthyretin (ATTR) Cardiac Amyloidosis. J. Card. Fail. 2019, 26, 753–759. [Google Scholar] [CrossRef]
- Rosenblum, H.; Castano, A.; Alvarez, J.; Goldsmith, J.; Helmke, S.; Maurer, M.S. TTR (Transthyretin) Stabilizers Are Associated With Improved Survival in Patients With TTR Cardiac Amyloidosis. Circ. Heart Fail. 2018, 11, e004769. [Google Scholar] [CrossRef]
- Fox, J.C.; Hellawell, J.L.; Rao, S.; O’Reilly, T.; Lumpkin, R.; Jernelius, J.; Gretler, D.; Sinha, U. First-in-Human Study of AG10, a Novel, Oral, Specific, Selective, and Potent Transthyretin Stabilizer for the Treatment of Transthyretin Amyloidosis: A Phase 1 Safety, Tolerability, Pharmacokinetic, and Pharmacodynamic Study in Healthy Adult Volunteers. Clin. Pharmacol. Drug Dev. 2020, 9, 115–129. [Google Scholar] [CrossRef]
- Alhamadsheh, M.M.; Connelly, S.; Cho, A.; Reixach, N.; Powers, E.T.; Pan, D.W.; Wilson, I.A.; Kelly, J.W.; Graef, I.A. Potent kinetic stabilizers that prevent transthyretin-mediated cardiomyocyte proteotoxicity. Sci. Transl. Med. 2011, 3, 97ra81. [Google Scholar] [CrossRef]
- Penchala, S.C.; Connelly, S.; Wang, Y.; Park, M.S.; Zhao, L.; Baranczak, A.; Rappley, I.; Vogel, H.; Liedtke, M.; Witteles, R.M.; et al. AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc. Natl. Acad. Sci. USA 2013, 110, 9992–9997. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Pal, A.; Albusairi, W.; Joo, H.; Pappas, B.; Haque Tuhin, M.T.; Liang, D.; Jampala, R.; Liu, F.; Khan, J.; et al. Enthalpy-Driven Stabilization of Transthyretin by AG10 Mimics a Naturally Occurring Genetic Variant That Protects from Transthyretin Amyloidosis. J. Med. Chem. 2018, 61, 7862–7876. [Google Scholar] [CrossRef] [PubMed]
- Nelson, L.T.; Paxman, R.J.; Xu, J.; Webb, B.; Powers, E.T.; Kelly, J.W. Blinded potency comparison of transthyretin kinetic stabilisers by subunit exchange in human plasma. Amyloid 2020, 28, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Judge, D.P.; Heitner, S.B.; Falk, R.H.; Maurer, M.S.; Shah, S.J.; Witteles, R.M.; Grogan, M.; Selby, V.N.; Jacoby, D.; Hanna, M.; et al. Transthyretin Stabilization by AG10 in Symptomatic Transthyretin Amyloid Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 285–295. [Google Scholar] [CrossRef] [PubMed]
- BridgeBio. Available online: https://bridgebio.com/news/bridgebio-pharma-reports-month-12-topline-results-from-phase-3-attribute-cm-study/ (accessed on 30 November 2022).
- Sant’Anna, R.; Gallego, P.; Robinson, L.Z.; Pereira-Henriques, A.; Ferreira, N.; Pinheiro, F.; Esperante, S.; Pallares, I.; Huertas, O.; Almeida, M.R.; et al. Repositioning tolcapone as a potent inhibitor of transthyretin amyloidogenesis and associated cellular toxicity. Nat. Commun. 2016, 7, 10787. [Google Scholar] [CrossRef]
- Gamez, J.; Salvadó, M.; Reig, N.; Suñé, P.; Casasnovas, C.; Rojas-Garcia, R.; Insa, R. Transthyretin stabilization activity of the catechol-O-methyltransferase inhibitor tolcapone (SOM0226) in hereditary ATTR amyloidosis patients and asymptomatic carriers: Proof-of-concept study. Amyloid 2019, 26, 74–84. [Google Scholar] [CrossRef]
- Pinheiro, F.; Varejão, N.; Esperante, S.; Santos, J.; Velázquez-Campoy, A.; Reverter, D.; Pallarès, I.; Ventura, S. Tolcapone, a potent aggregation inhibitor for the treatment of familial leptomeningeal amyloidosis. FEBS J. 2020, 288, 310–324. [Google Scholar] [CrossRef]
- Cardoso, I.; Merlini, G.; Saraiva, M.J. 4′-iodo-4′-deoxydoxorubicin and tetracyclines disrupt transthyretin amyloid fibrils in vitro producing noncytotoxic species: Screening for TTR fibril disrupters. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 803–809. [Google Scholar] [CrossRef]
- Cardoso, I.; Saraiva, M.J. Doxycycline disrupts transthyretin amyloid: Evidence from studies in a FAP transgenic mice model. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 234–239. [Google Scholar] [CrossRef]
- Obici, L.; Cortese, A.; Lozza, A.; Lucchetti, J.; Gobbi, M.; Palladini, G.; Perlini, S.; Saraiva, M.J.; Merlini, G. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: A phase II study. Amyloid 2012, 19 (Suppl. 1), 34–36. [Google Scholar] [CrossRef]
- Wixner, J.; Pilebro, B.; Lundgren, H.E.; Olsson, M.; Anan, I. Effect of doxycycline and ursodeoxycholic acid on transthyretin amyloidosis. Amyloid 2017, 24 (Suppl. 1), 78–79. [Google Scholar] [CrossRef] [PubMed]
- Karlstedt, E.; Jimenez-Zepeda, V.; Howlett, J.G.; White, J.A.; Fine, N.M. Clinical Experience With the Use of Doxycycline and Ursodeoxycholic Acid for the Treatment of Transthyretin Cardiac Amyloidosis. J. Card. Fail. 2019, 25, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Carvalho, M.; Saraiva, M.J.; Alves, I.; Almeida, M.R.; Costa, P.P. A strikingly benign evolution of FAP in an individual found to be a compund heterozygote for two mutations: TTR MET 30 and TTR MET MET 119. J. Rheumatol. 1993, 20, 179. [Google Scholar]
- Coelho, T.; Chorao, R.; Sausa, A.; Alves, I.; Torres, M.F.; Saraiva, M.J. Compund heterozygotes of transthyretin Met30 and transtyretin Met119 are protected from the devastating effects of familial amyloid polyneuropathy. Neuromusc. Disord. 1996, 6. [Google Scholar] [CrossRef]
- Hammarström, P.; Schneider, F.; Kelly, J.W. Trans-suppression of misfolding in an amyloid disease. Science 2001, 293, 2459–2462. [Google Scholar] [CrossRef]
- Hammarström, P.; Wiseman, R.L.; Powers, E.T.; Kelly, J.W. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science 2003, 299, 713–716. [Google Scholar] [CrossRef] [PubMed]
- McCutchen, S.L.; Lai, Z.; Miroy, G.J.; Kelly, J.W.; Colón, W. Comparison of lethal and nonlethal transthyretin variants and their relationship to amyloid disease. Biochemistry 1995, 34, 13527–13536. [Google Scholar] [CrossRef] [PubMed]
- Miroy, G.J.; Lai, Z.; Lashuel, H.A.; Peterson, S.A.; Strang, C.; Kelly, J.W. Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc. Natl. Acad. Sci. USA 1996, 93, 15051–15056. [Google Scholar] [CrossRef]
- Razavi, H.; Palaninathan, S.K.; Powers, E.T.; Wiseman, R.L.; Purkey, H.E.; Mohamedmohaideen, N.N.; Deechongkit, S.; Chiang, K.P.; Dendle, M.T.; Sacchettini, J.C.; et al. Benzoxazoles as transthyretin amyloid fibril inhibitors: Synthesis, evaluation, and mechanism of action. Angew. Chem. (Int. Ed. Engl.) 2003, 42, 2758–2761. [Google Scholar] [CrossRef]
- Wiseman, R.L.; Johnson, S.M.; Kelker, M.S.; Foss, T.; Wilson, I.A.; Kelly, J.W. Kinetic stabilization of an oligomeric protein by a single ligand binding event. J. Am. Chem. Soc. 2005, 127, 5540–5551. [Google Scholar] [CrossRef]
- Tess, D.A.; Maurer, T.S.; Li, Z.; Bulawa, C.; Fleming, J.; Moody, A.T. Relationship of binding-site occupancy, transthyretin stabilisation and disease modification in patients with tafamidis-treated transthyretin amyloid cardiomyopathy. Amyloid 2022, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Merlini, G.; Planté-Bordeneuve, V.; Judge, D.P.; Schmidt, H.; Obici, L.; Perlini, S.; Packman, J.; Tripp, T.; Grogan, D.R. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J. Cardiovasc. Transl. Res. 2013, 6, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Lozeron, P.; Théaudin, M.; Mincheva, Z.; Ducot, B.; Lacroix, C.; Adams, D. Effect on disability and safety of Tafamidis in late onset of Met30 transthyretin familial amyloid polyneuropathy. Eur. J. Neurol. 2013, 20, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Merlini, G.; Coelho, T.; Waddington Cruz, M.; Li, H.; Stewart, M.; Ebede, B. Evaluation of Mortality During Long-Term Treatment with Tafamidis for Transthyretin Amyloidosis with Polyneuropathy: Clinical Trial Results up to 8.5 Years. Neurol. Ther. 2020, 9, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Klamerus, K.J.; Watsky, E.; Moller, R.; Wang, R.; Riley, S. The effect of tafamidis on the QTc interval in healthy subjects. Br. J. Clin. Pharmacol. 2015, 79, 918–925. [Google Scholar] [CrossRef]
- Maurer, M.S.; Grogan, D.R.; Judge, D.P.; Mundayat, R.; Packman, J.; Lombardo, I.; Quyyumi, A.A.; Aarts, J.; Falk, R.H. Tafamidis in transthyretin amyloid cardiomyopathy: Effects on transthyretin stabilization and clinical outcomes. Circ. Heart Fail. 2015, 8, 519–526. [Google Scholar] [CrossRef]
- Li, H.; Rozenbaum, M.; Casey, M.; Sultan, M.B. Estimating the effect of tafamidis on cardiovascular-related hospitalization in NYHA class III patients with transthyretin amyloid cardiomyopathy in the presence of death. Cardiology 2022, 147, 398–405. [Google Scholar] [CrossRef]
- Vong, C.; Boucher, M.; Riley, S.; Harnisch, L.O. Modeling of Survival and Frequency of Cardiovascular-Related Hospitalization in Patients with Transthyretin Amyloid Cardiomyopathy Treated with Tafamidis. Am. J. Cardiovasc. Drugs 2021, 21, 535–543. [Google Scholar] [CrossRef]
- Basset, M.; Nuvolone, M.; Palladini, G.; Merlini, G. Novel challenges in the management of immunoglobulin light chain amyloidosis: From the bench to the bedside. Expert Rev. Hematol. 2020, 13, 1003–1015. [Google Scholar] [CrossRef]
- Hanna, M.; Damy, T.; Grogan, M.; Stewart, M.; Gundapaneni, B.; Patterson, T.A.; Schwartz, J.H.; Sultan, M.B.; Maurer, M.S. Impact of Tafamidis on Health-Related Quality of Life in Patients With Transthyretin Amyloid Cardiomyopathy (from the Tafamidis in Transthyretin Cardiomyopathy Clinical Trial). Am. J. Cardiol. 2021, 141, 98–105. [Google Scholar] [CrossRef]
- Rapezzi, C.; Elliott, P.; Damy, T.; Nativi-Nicolau, J.; Berk, J.L.; Velazquez, E.J.; Boman, K.; Gundapaneni, B.; Patterson, T.A.; Schwartz, J.H.; et al. Efficacy of Tafamidis in Patients With Hereditary and Wild-Type Transthyretin Amyloid Cardiomyopathy: Further Analyses From ATTR-ACT. JACC Heart Fail. 2021, 9, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Lockwood, P.A.; Le, V.H.; O’Gorman, M.T.; Patterson, T.A.; Sultan, M.B.; Tankisheva, E.; Wang, Q.; Riley, S. The Bioequivalence of Tafamidis 61-mg Free Acid Capsules and Tafamidis Meglumine 4 × 20-mg Capsules in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2020, 9, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Alvir, J.; Stewart, M. Extrapolation of Survival Benefits in Patients with Transthyretin Amyloid Cardiomyopathy Receiving Tafamidis: Analysis of the Tafamidis in Transthyretin Cardiomyopathy Clinical Trial. Cardiol. Ther. 2020, 9, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; Drachman, B.M.; Gottlieb, S.S.; Hoffman, J.E.; Hummel, S.L.; Lenihan, D.J.; Ebede, B.; Gundapaneni, B.; Li, B.; Sultan, M.B.; et al. Long-Term Survival With Tafamidis in Patients With Transthyretin Amyloid Cardiomyopathy. Circ. Heart Fail. 2022, 15, e008193. [Google Scholar] [CrossRef] [PubMed]
- Damy, T.; Garcia-Pavia, P.; Hanna, M.; Judge, D.P.; Merlini, G.; Gundapaneni, B.; Patterson, T.A.; Riley, S.; Schwartz, J.H.; Sultan, M.B.; et al. Efficacy and safety of tafamidis doses in the Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR-ACT) and long-term extension study. Eur. J. Heart Fail. 2021, 23, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Miranda, D.; Sleiman, J.; Liang, H.; Ives, L.; Asher Craig, R.; Hanna Mazen, A.; Wolinsky David, G. DOES TAFAMIDIS BENEFIT OCTOGENARIANS WITH TRANSTHYRETIN AMYLOID CARDIOMYOPATHY? ANALYSIS OF THE CLEVELAND CLINIC AMYLOID REGISTRY. J. Am. Coll. Cardiol. 2022, 79, 300. [Google Scholar] [CrossRef]
- Elliott, P.; Gundapaneni, B.; Sultan, M.B. Response by Elliott et al to Letter Regarding Article, “Long-Term Survival With Tafamidis in Patients With Transthyretin Amyloid Cardiomyopathy”. Circ. Heart Fail. 2022, 15, e009637. [Google Scholar] [CrossRef]
- Bezard, M.; Kharoubi, M.; Galat, A.; Poullot, E.; Guendouz, S.; Fanen, P.; Funalot, B.; Moktefi, A.; Lefaucheur, J.P.; Abulizi, M.; et al. Natural history and impact of treatment with tafamidis on major cardiovascular outcome-free survival time in a cohort of patients with transthyretin amyloidosis. Eur. J. Heart Fail. 2021, 23, 264–274. [Google Scholar] [CrossRef]
- Oghina, S.; Josse, C.; Bezard, M.; Kharoubi, M.; Delbarre, M.A.; Eyharts, D.; Zaroui, A.; Guendouz, S.; Galat, A.; Hittinger, L.; et al. Prognostic Value of N-Terminal Pro-Brain Natriuretic Peptide and High-Sensitivity Troponin T Levels in the Natural History of Transthyretin Amyloid Cardiomyopathy and Their Evolution after Tafamidis Treatment. J. Clin. Med. 2021, 10, 4868. [Google Scholar] [CrossRef]
- Giblin, G.T.; Cuddy, S.A.M.; Gonzalez-Lopez, E.; Sewell, A.; Murphy, A.; Dorbala, S.; Falk, R.H. Effect of tafamidis on global longitudinal strain and myocardial work in transthyretin cardiac amyloidosis. Eur. Heart J. Cardiovasc Imaging 2022, 23, 1029–1039. [Google Scholar] [CrossRef]
- Rettl, R.; Mann, C.; Duca, F.; Dachs, T.M.; Binder, C.; Ligios, L.C.; Schrutka, L.; Dalos, D.; Koschutnik, M.; Dona, C.; et al. Tafamidis treatment delays structural and functional changes of the left ventricle in patients with transthyretin amyloid cardiomyopathy. Eur. Heart J. Cardiovasc Imaging 2022, 23, 767–780. [Google Scholar] [CrossRef]
- Munro, S.L.; Lim, C.F.; Hall, J.G.; Barlow, J.W.; Craik, D.J.; Topliss, D.J.; Stockigt, J.R. Drug competition for thyroxine binding to transthyretin (prealbumin): Comparison with effects on thyroxine-binding globulin. J. Clin. Endocrinol. Metab. 1989, 68, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Baures, P.W.; Peterson, S.A.; Kelly, J.W. Discovering transthyretin amyloid fibril inhibitors by limited screening. Bioorganic Med. Chem. 1998, 6, 1389–1401. [Google Scholar] [CrossRef] [PubMed]
- Baures, P.W.; Oza, V.B.; Peterson, S.A.; Kelly, J.W. Synthesis and evaluation of inhibitors of transthyretin amyloid formation based on the non-steroidal anti-inflammatory drug, flufenamic acid. Bioorganic Med. Chem. 1999, 7, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.R.; Sekijima, Y.; Kelly, J.W. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab. Investig. A J. Tech. Methods Pathol. 2004, 84, 545–552. [Google Scholar] [CrossRef]
- Klabunde, T.; Petrassi, H.M.; Oza, V.B.; Raman, P.; Kelly, J.W.; Sacchettini, J.C. Rational design of potent human transthyretin amyloid disease inhibitors. Nat. Struct. Biol. 2000, 7, 312–321. [Google Scholar]
- Tsai, F.J.; Nelson, L.T.; Kline, G.M.; Jager, M.; Berk, J.L.; Sekijima, Y.; Powers, E.T.; Kelly, J.W. Characterising diflunisal as a transthyretin kinetic stabilizer at relevant concentrations in human plasma using subunit exchange. Amyloid 2022, 1–5. [Google Scholar] [CrossRef]
- Adamski-Werner, S.L.; Palaninathan, S.K.; Sacchettini, J.C.; Kelly, J.W. Diflunisal analogues stabilize the native state of transthyretin. Potent inhibition of amyloidogenesis. J. Med. Chem. 2004, 47, 355–374. [Google Scholar] [CrossRef]
- Ibrahim, M.; Saint Croix, G.R.; Lacy, S.; Fattouh, M.; Barillas-Lara, M.I.; Behrooz, L.; Mechanic, O. The use of diflunisal for transthyretin cardiac amyloidosis: A review. Heart Fail. Rev. 2022, 27, 517–524. [Google Scholar] [CrossRef]
- Merlini, G.; Ascari, E.; Amboldi, N.; Bellotti, V.; Arbustini, E.; Perfetti, V.; Ferrari, M.; Zorzoli, I.; Marinone, M.G.; Garini, P.; et al. Interaction of the anthracycline 4′-iodo-4′-deoxydoxorubicin with amyloid fibrils: Inhibition of amyloidogenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 2959–2963. [Google Scholar] [CrossRef]
- Riond, J.L.; Riviere, J.E. Pharmacology and toxicology of doxycycline. Vet. Hum. Toxicol. 1988, 30, 431–443. [Google Scholar] [PubMed]
- Sloan, B.; Scheinfeld, N. The use and safety of doxycycline hyclate and other second-generation tetracyclines. Expert Opin. Drug Saf. 2008, 7, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.; Leyden, J.J. Safety of doxycycline and minocycline: A systematic review. Clin. Ther. 2005, 27, 1329–1342. [Google Scholar] [CrossRef] [PubMed]
- Westfall, L.K.; Mintzer, D.L.; Wiser, T.H. Potentiation of warfarin by tetracycline. Am. J. Hosp. Pharm. 1980, 37, 1620–1625. [Google Scholar] [CrossRef]
- Baciewicz, A.M.; Bal, B.S. Bleeding associated with doxycycline and warfarin treatment. Arch. Intern. Med. 2001, 161, 1231. [Google Scholar] [CrossRef]
- Nuvolone, M.; Merlini, G. Emerging therapeutic targets currently under investigation for the treatment of systemic amyloidosis. Expert Opin. Ther. Targets 2017, 21, 1095–1110. [Google Scholar] [CrossRef]
- Aimo, A.; Castiglione, V.; Rapezzi, C.; Franzini, M.; Panichella, G.; Vergaro, G.; Gillmore, J.; Fontana, M.; Passino, C.; Emdin, M. RNA-targeting and gene editing therapies for transthyretin amyloidosis. Nat. Rev. Cardiol. 2022, 19, 655–667. [Google Scholar] [CrossRef]
- Nevone, A.; Merlini, G.; Nuvolone, M. Treating Protein Misfolding Diseases: Therapeutic Successes Against Systemic Amyloidoses. Front. Pharmacol. 2020, 11, 1024. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Solomon, S.D.; Adams, D.; Kristen, A.; Grogan, M.; González-Duarte, A.; Maurer, M.S.; Merlini, G.; Damy, T.; Slama, M.S.; Brannagan, T.H., 3rd; et al. Effects of Patisiran, an RNA Interference Therapeutic, on Cardiac Parameters in Patients With Hereditary Transthyretin-Mediated Amyloidosis. Circulation 2019, 139, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, N.R.; Benson, M.D. Potential reversal of transthyretin amyloid cardiomyopathy with ttr specific antisense oligonucleotide therapy. J. Clin. Med. 2018, 71, A660. [Google Scholar] [CrossRef]
- Habtemariam, B.A.; Karsten, V.; Attarwala, H.; Goel, V.; Melch, M.; Clausen, V.A.; Garg, P.; Vaishnaw, A.K.; Sweetser, M.T.; Robbie, G.J.; et al. Single-Dose Pharmacokinetics and Pharmacodynamics of Transthyretin Targeting N-acetylgalactosamine-Small Interfering Ribonucleic Acid Conjugate, Vutrisiran, in Healthy Subjects. Clin. Pharm. 2021, 109, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Viney, N.J.; Guo, S.; Tai, L.J.; Baker, B.F.; Aghajan, M.; Jung, S.W.; Yu, R.Z.; Booten, S.; Murray, H.; Machemer, T.; et al. Ligand conjugated antisense oligonucleotide for the treatment of transthyretin amyloidosis: Preclinical and phase 1 data. ESC Heart Fail. 2021, 8, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Alnylam. Available online: https://investors.alnylam.com/press-release?id=26851 (accessed on 30 November 2022).
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef]
- Maurer, M.S. Gene Editing—A Cure for Transthyretin Amyloidosis? N. Engl. J. Med. 2021, 385, 558–559. [Google Scholar] [CrossRef]
- Buning, H.; Schambach, A. A first step toward in vivo gene editing in patients. Nat. Med. 2021, 27, 1515–1517. [Google Scholar] [CrossRef]
- Batista, A.R.; Flotte, T.R. In vivo gene editing works in humans: Results of a phase 1 clinical trial for TTR amyloidosis. Mol. Ther. 2021, 29, 2633–2634. [Google Scholar] [CrossRef]
- Shen, K.N.; Fu, W.J.; Wu, Y.; Dong, Y.J.; Huang, Z.X.; Wei, Y.Q.; Li, C.R.; Sun, C.Y.; Chen, Y.; Miao, H.L.; et al. Doxycycline Combined With Bortezomib-Cyclophosphamide-Dexamethasone Chemotherapy for Newly Diagnosed Cardiac Light-Chain Amyloidosis: A Multicenter Randomized Controlled Trial. Circulation 2022, 145, 8–17. [Google Scholar] [CrossRef]
- Nuvolone, M.; Nevone, A.; Merlini, G. Targeting Amyloid Fibrils by Passive Immunotherapy in Systemic Amyloidosis. BioDrugs 2022, 36, 591–608. [Google Scholar] [CrossRef]
- Canepa, M.; Tini, G.; Musumeci, B.; Cappelli, F.; Milandri, A.; Mussinelli, R.; Autore, C.; Perfetto, F.; Rapezzi, C.; Perlini, S. Real-world versus trial patients with transthyretin amyloid cardiomyopathy. Eur. J. Heart Fail. 2019, 21, 1479–1481. [Google Scholar] [CrossRef] [PubMed]
- Luzzatto, L.; Hyry, H.I.; Schieppati, A.; Costa, E.; Simoens, S.; Schaefer, F.; Roos, J.C.P.; Merlini, G.; Kääriäinen, H.; Garattini, S.; et al. Outrageous prices of orphan drugs: A call for collaboration. Lancet (Lond. Engl.) 2018, 392, 791–794. [Google Scholar] [CrossRef]
- Gurwitz, J.H.; Maurer, M.S. Tafamidis-A Pricey Therapy for a Not-So-Rare Condition. JAMA Cardiol. 2020, 5, 247. [Google Scholar] [CrossRef] [PubMed]
- Kazi, D.S.; Bellows, B.K.; Baron, S.J.; Shen, C.; Cohen, D.J.; Spertus, J.A.; Yeh, R.W.; Arnold, S.V.; Sperry, B.W.; Maurer, M.S.; et al. Cost-Effectiveness of Tafamidis Therapy for Transthyretin Amyloid Cardiomyopathy. Circulation 2020, 141, 1214–1224. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Drug [References] | Absorption | Volume of Distribution | Protein Binding | Metabolism | Route of Elimination | Half-life | Clearance | Dosing * |
|---|---|---|---|---|---|---|---|---|
| Tafamidis and tafamidis meglumine [68,69,70] | Peak plasma concentration within 4 h following oral administration | 18.5 L | 99.9% protein bound in plasma, mostly to transthyretin | Largely not subject to first pass or oxidative metabolism, being 90% unchanged after in vitro experiments. Mainly metabolized through glucuronidation and excreted in bile. | 20 mg oral dose: 59% recovered in the feces, largely as unchanged drug; 22% recovered in the urine, mostly as the glucuronide metabolite | 49 h | Oral clearance: 0.263 L/h. Apparent total clearance: 0.44 L/h. | 20 mg or 80 mg (61 mg of tafamidis) QD |
| Diflunisal [71,72,73,74,75,76,77,78] | Bioavailability of 80–90%. Peak plasma concentration within 2–3 h following oral administration | Not available | 98 to 99% protein bound in plasma | Hepatic metabolism, primarily via glucuronide conjugation (90% of the administered dose). | Excreted in the urine as two soluble glucuronide conjugates accounting for about 90% of the administered dose. Little or no excretion in the feces. | 8–12 h | Not available. | 250 mg BID |
| Acoramidis [79,80,81,82,83,84,85] | Peak plasma concentration within 1 h following oral administration | Not available | High binding selectivity for TTR. No data on binding to albumin or plasma proteins other than TTR | Predominantly acyl glucuronidation based on in vitro studies. | Up to 9.5% excreted as intact AG10 in urine. 19.5–23.5% extreted as AG10 acylglucuronide. No data on fecal elimination. | 25 h | 1.58–5.98 L/h. | 800 mg BID |
| Tolcapone [86,87,88] | Absolute bioavailability of about 65% | 9 L | >99.9% (to serum albumin) | Mainly metabolized through glucuronidation. | Almost completely metabolized before excretion, with only a very small amount (0.5% of dose) found unchanged in the urine. The glucuronide conjugate is mainly excreted in the urine but is also excreted in the bile. | 2–3.5 h | 7 L/h | NA |
| Doxycycline [89,90,91,92,93] | Peak plasma concentration within 2 hours following oral administration | 0.7 L/kg | >90% | Metabolized in the liver and gastrointestinal tract and concentrated in bile. Major metabolic pathways have not been identified. | Mainly the urine-(40–60%) and feces (30%) as active and unchanged drug. | 18–22 h | Excretion by the kidney is about 40% over 72 h in individuals with normal kidney function. | 100 mg BID |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nuvolone, M.; Girelli, M.; Merlini, G. Oral Therapy for the Treatment of Transthyretin-Related Amyloid Cardiomyopathy. Int. J. Mol. Sci. 2022, 23, 16145. https://doi.org/10.3390/ijms232416145
Nuvolone M, Girelli M, Merlini G. Oral Therapy for the Treatment of Transthyretin-Related Amyloid Cardiomyopathy. International Journal of Molecular Sciences. 2022; 23(24):16145. https://doi.org/10.3390/ijms232416145
Chicago/Turabian StyleNuvolone, Mario, Maria Girelli, and Giampaolo Merlini. 2022. "Oral Therapy for the Treatment of Transthyretin-Related Amyloid Cardiomyopathy" International Journal of Molecular Sciences 23, no. 24: 16145. https://doi.org/10.3390/ijms232416145
APA StyleNuvolone, M., Girelli, M., & Merlini, G. (2022). Oral Therapy for the Treatment of Transthyretin-Related Amyloid Cardiomyopathy. International Journal of Molecular Sciences, 23(24), 16145. https://doi.org/10.3390/ijms232416145