

Targeting TLR2/Rac1/cdc42/JNK Pathway to Reveal That Ruxolitinib Promotes Thrombocytopoiesis
 ,
,
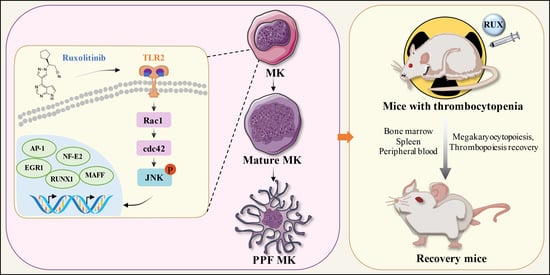
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Safe Concentration of Ruxolitinib for the Treatment of K562 and Meg-01 Cells
2.2. Ruxolitinib Induces Typical MK Differentiation
2.3. Ruxolitinib Promotes Platelet Recovery in Irradiated Mice
2.4. Ruxolitinib Rescues Bone Marrow MKs after Radiation Injury
2.5. Ruxolitinib Restores Splenic Hematopoiesis
2.6. Ruxolitinib Promotes the Number and Functional Recovery of Peripheral Blood Platlets
2.7. Gene Expression Profiling and Functional Analysis of Ruxolitinib-Induced Differentially Expressed Genes
2.8. Ruxolitinib Directly Bound to TLR2 to Stimulate MK Differentiation and Platelet Formation
2.9. Ruxolitinib Promotes MK Differentiation by Activating Rac1/cdc42/JNK
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Proliferation Assay
4.3. Lactate Dehydrogenase (LDH) Assay
4.4. Morphological Observations
4.5. Giemsa Staining
4.6. Analysis of Cell Differentiation
4.7. Phalloidin Staining
4.8. RIT Establishment in Mice and Ruxolitinib Treatment
4.9. Measurement of Hematologic Parameters
4.10. Flow Cytometry Analysis of BM, Spleen and Blood Cells
4.11. Platelet Isolation and Sample Preparation
4.12. Platelet Activation Analysis
4.13. Tail Bleeding Assay
4.14. Histology Analysis
4.15. Acquisition of Candidate Targets of Ruxolitinib against Thrombocytopenia
4.16. Protein–Protein Interaction (PPI) Network Construction and Screening of Core Targets
4.17. Molecular Docking
4.18. Drug Affinity Responsive Target Stability Assay (DARTS)
4.19. Cell Sample Preparation
4.20. RNA-seq and Data Analysis
4.21. Gene Ontology and KEGG Pathway Analysis
4.22. Western Blotting
4.23. Immunofluorescence Assay
4.24. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hur, W.; Yoon, S.K. Molecular Pathogenesis of Radiation-Induced Cell Toxicity in Stem Cells. Int. J. Mol. Sci. 2017, 18, 2749. [Google Scholar] [CrossRef] [PubMed]
- Taliaferro, L.P.; Cassatt, D.R.; Horta, Z.P.; Satyamitra, M.M. Meeting Report: A Poly-Pharmacy Approach to Mitigate Acute Radiation Syndrome. Radiat. Res. 2021, 196, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Bunin, D.I.; Bakke, J.; Green, C.E.; Javitz, H.S.; Fielden, M.; Chang, P.Y. Romiplostim (Nplate((R))) as an effective radiation countermeasure to improve survival and platelet recovery in mice. Int. J. Radiat. Biol. 2020, 96, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Tkaczynski, E.; Arulselvan, A.; Tkaczynski, J.; Avery, S.; Xiao, L.; Torok-Storb, B.; Abrams, K.; Rao, N.V.; Johnson, G.; Kennedy, T.P.; et al. 2-O, 3-O desulfated heparin mitigates murine chemotherapy- and radiation-induced thrombocytopenia. Blood. Adv. 2018, 2, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Che, F.; Zhao, J.; Zhao, Y.; Wang, Z.; Zhang, L.; Yang, Y. A Novel Heterozygous Pathogenic Variation in CYCS Gene Cause Autosomal Dominant Non-Syndromic Thrombocytopenia 4 in a Large Chinese Family. Front. Genet. 2022, 12, 783455. [Google Scholar] [CrossRef]
- Constantinescu-Bercu, A.; Wang, Y.A.; Woollard, K.J.; Mangin, P.; Vanhoorelbeke, K.; Crawley, J.T.B.; Salles, C., II. The GPIbalpha intracellular tail—Role in transducing VWF- and collagen/GPVI-mediated signaling. Haematologica 2022, 107, 933–946. [Google Scholar] [CrossRef]
- Wang, Q.; Li, J.; Yu, T.S.; Liu, Y.; Li, K.; Liu, S.; Liu, Y.; Feng, Q.; Zhang, L.; Li, G.S.; et al. Disrupted balance of CD4(+) T-cell subsets in bone marrow of patients with primary immune thrombocytopenia. Int. J. Biol. Sci. 2019, 15, 2798–2814. [Google Scholar] [CrossRef]
- Borst, S.; Sim, X.; Poncz, M.; French, D.L.; Gadue, P. Induced pluripotent stem cell-derived megakaryocytes and platelets for disease modeling and future clinical applications. Arter. Thromb. Vasc. Biol. 2017, 37, 2007–2013. [Google Scholar] [CrossRef]
- Shi, Q.; Montgomery, R.R. Platelets as delivery systems for disease treatments. Adv. Drug. Deliv. Rev. 2010, 62, 1196–1203. [Google Scholar] [CrossRef]
- Dutting, S.; Gaits-Iacovoni, F.; Stegner, D.; Popp, M.; Antkowiak, A.; van Eeuwijk, J.M.M.; Nurden, P.; Stritt, S.; Heib, T.; Aurbach, K.; et al. A Cdc42/RhoA regulatory circuit downstream of glycoprotein Ib guides transendothelial platelet biogenesis. Nat. Commun. 2017, 8, 15838. [Google Scholar] [CrossRef]
- Pan, J.; Lordier, L.; Meyran, D.; Rameau, P.; Lecluse, Y.; Kitchen-Goosen, S.; Badirou, I.; Mokrani, H.; Narumiya, S.; Alberts, A.S.; et al. The formin DIAPH1 (mDia1) regulates megakaryocyte proplatelet formation by remodeling the actin and microtubule cytoskeletons. Blood 2014, 124, 3967–3977. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zeng, J.; Liu, S.; Shen, X.; Jiang, N.; Wu, Y.S.; Li, H.; Wang, L.; Wu, J.M. DMAG, a novel countermeasure for the treatment of thrombocytopenia. Mol. Med. 2021, 27, 149. [Google Scholar] [CrossRef] [PubMed]
- Aslan, J.E.; McCarty, O.J. Rho GTPases in platelet function. J. Thromb. Haemost. 2013, 11, 35–46. [Google Scholar] [CrossRef]
- Kim, H.S.; Suh, J.S.; Jang, Y.K.; Ahn, S.H.; Raja, G.; Kim, J.C.; Jung, Y.; Jung, S.H.; Kim, T.J. Anti-cancer potential of persimmon (Diospyros kaki) leaves via the PDGFR-Rac-JNK pathway. Sci. Rep. 2020, 10, 18119. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Guo, Y.; Chang, Z.; Zhang, D.; Zhang, S.; Pei, H.; Pang, J.; Zhao, Z.J.; Chen, Y. Bidirectional Interaction Between Cancer Cells and Platelets Provides Potential Strategies for Cancer Therapies. Front. Oncol. 2021, 11, 764119. [Google Scholar] [CrossRef]
- Luff, S.A.; Papoutsakis, E.T. Megakaryocytic Maturation in Response to Shear Flow Is Mediated by the Activator Protein 1 (AP-1) Transcription Factor via Mitogen-activated Protein Kinase (MAPK) Mechanotransduction. J. Biol. Chem. 2016, 291, 7831–7843. [Google Scholar] [CrossRef]
- Wang, Z.; Shen, J.; Sun, W.; Zhang, T.; Zuo, D.; Wang, H.; Wang, G.; Xu, J.; Yin, F.; Mao, M.; et al. Antitumor activity of Raddeanin A is mediated by Jun amino-terminal kinase activation and signal transducer and activator of transcription 3 inhibition in human osteosarcoma. Cancer Sci. 2019, 110, 1746–1759. [Google Scholar] [CrossRef]
- Qing, Y.; Wang, X.; Wang, H.; Hu, P.; Li, H.; Yu, X.; Zhu, M.; Wang, Z.; Zhu, Y.; Xu, J.; et al. Pharmacologic targeting of the P-TEFb complex as a therapeutic strategy for chronic myeloid leukemia. Cell Commun. Signal. 2021, 19, 83. [Google Scholar] [CrossRef]
- Fogelman, D.; Cubillo, A.; Garcia-Alfonso, P.; Miron, M.L.L.; Nemunaitis, J.; Flora, D.; Borg, C.; Mineur, L.; Vieitez, J.M.; Cohn, A.; et al. Randomized, double-blind, phase two study of ruxolitinib plus regorafenib in patients with relapsed/refractory metastatic colorectal cancer. Cancer Med. 2018, 7, 5382–5393. [Google Scholar] [CrossRef]
- Ryan, M.M.; Patel, M.; Hogan, K.; Lipat, A.J.; Scandolara, R.; Das, R.; Bruker, C.; Galipeau, J.; Chinnadurai, R. Ruxolitinib Inhibits IFNgamma Licensing of Human Bone Marrow Derived Mesenchymal Stromal Cells. Transplant. Cell Ther. 2021, 27, 389.e381–389.e310. [Google Scholar] [CrossRef]
- Rossi, D.; Guerrini, A.; Bruni, R.; Brognara, E.; Borgatti, M.; Gambari, R.; Maietti, S.; Sacchetti, G. trans-Resveratrol in nutraceuticals: Issues in retail quality and effectiveness. Molecules 2012, 17, 12393–12405. [Google Scholar] [CrossRef] [PubMed]
- Xiong, P.; Huang, X.; Ye, N.; Lu, Q.; Zhang, G.; Peng, S.; Wang, H.; Liu, Y. Cytotoxicity of Metal-Based Nanoparticles: From Mechanisms and Methods of Evaluation to Pathological Manifestations. Adv. Sci. 2022, 9, e2106049. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Ma, L.; Chen, E.; Shaw, C.A.; Edelstein, L.C. Identification of the Regulatory Elements and Target Genes of Megakaryopoietic Transcription Factor MEF2C. Thromb. Haemost 2019, 119, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, K.; Khare, V.; Evstatiev, R.; Kulnigg-Dabsch, S.; Jambrich, M.; Strobl, H.; Gasche, C. Increased expression of HIF2alpha during iron deficiency-associated megakaryocytic differentiation. J. Thromb. Haemost 2015, 13, 1113–1127. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, J.; Bompard, G.; Cau, J.; Kunishima, S.; Rabeharivelo, G.; Mateos-Langerak, J.; Cazevieille, C.; Cavelier, P.; Boizet-Bonhoure, B.; Delsert, C.; et al. Microtubule polyglutamylation and acetylation drive microtubule dynamics critical for platelet formation. BMC Biol. 2018, 16, 116. [Google Scholar] [CrossRef]
- Venkateswaran, K.; Shrivastava, A.; Agrawala, P.K.; Prasad, A.; Kalra, N.; Pandey, P.R.; Manda, K.; Raj, H.G.; Parmar, V.S.; Dwarakanath, B.S. Mitigation of radiation-induced hematopoietic injury by the polyphenolic acetate 7, 8-diacetoxy-4-methylthiocoumarin in mice. Sci. Rep. 2016, 6, 37305. [Google Scholar] [CrossRef]
- Milano, F.; Merriam, F.; Nicoud, I.; Li, J.; Gooley, T.A.; Heimfeld, S.; Imren, S.; Delaney, C. Notch-Expanded Murine Hematopoietic Stem and Progenitor Cells Mitigate Death from Lethal Radiation and Convey Immune Tolerance in Mismatched Recipients. Stem. Cell Transl. Med. 2017, 6, 566–575. [Google Scholar] [CrossRef]
- Short, C.; Lim, H.K.; Tan, J.; O’Neill, H.C. Targeting the Spleen as an Alternative Site for Hematopoiesis. Bioessays 2019, 41, e1800234. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, B.; Wang, S.; Zhang, J.; Liu, Y.; Wang, J.; Fan, Z.; Lv, Y.; Zhang, X.; He, L.; et al. Recombinant human thrombopoietin promotes hematopoietic reconstruction after severe whole body irradiation. Sci. Rep. 2015, 5, 12993. [Google Scholar] [CrossRef]
- Suen, J.Y.; Gardiner, B.; Grimmond, S.; Fairlie, D.P. Profiling gene expression induced by protease-activated receptor 2 (PAR2) activation in human kidney cells. PLoS ONE 2010, 5, e13809. [Google Scholar] [CrossRef]
- Shi, J.; Tong, R.; Zhou, M.; Gao, Y.; Zhao, Y.; Chen, Y.; Liu, W.; Li, G.; Lu, D.; Meng, G.; et al. Circadian nuclear receptor Rev-erbalpha is expressed by platelets and potentiates platelet activation and thrombus formation. Eur. Heart. J. 2022, 43, 2317–2334. [Google Scholar] [CrossRef]
- Morel, A.; Rywaniak, J.; Bijak, M.; Miller, E.; Niwald, M.; Saluk, J. Flow cytometric analysis reveals the high levels of platelet activation parameters in circulation of multiple sclerosis patients. Mol. Cell Biochem. 2017, 430, 69–80. [Google Scholar] [CrossRef]
- Inamdar, V.V.; Kostyak, J.C.; Badolia, R.; Dangelmaier, C.A.; Manne, B.K.; Patel, A.; Kim, S.; Kunapuli, S.P. Impaired Glycoprotein VI-Mediated Signaling and Platelet Functional Responses in CD45 Knockout Mice. Thromb. Haemost 2019, 119, 1321–1331. [Google Scholar] [CrossRef]
- Karabicici, M.; Azbazdar, Y.; Iscan, E.; Ozhan, G. Misregulation of Wnt Signaling Pathways at the Plasma Membrane in Brain and Metabolic Diseases. Membranes 2021, 11, 844. [Google Scholar] [CrossRef]
- Bussel, J.; Arnold, D.M.; Grossbard, E.; Mayer, J.; Trelinski, J.; Homenda, W.; Hellmann, A.; Windyga, J.; Sivcheva, L.; Khalafallah, A.A.; et al. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: Results of two phase 3, randomized, placebo-controlled trials. Am. J. Hematol. 2018, 93, 921–930. [Google Scholar] [CrossRef]
- Wojciechowski, P.; Wilson, K.; Nazir, J.; Pustulka, I.; Tytula, A.; Smela, B.; Pochopien, M.; Vredenburg, M.; McCrae, K.R.; Jurczak, W. Efficacy and Safety of Avatrombopag in Patients with Chronic Immune Thrombocytopenia: A Systematic Literature Review and Network Meta-Analysis. Adv. Ther. 2021, 38, 3113–3128. [Google Scholar] [CrossRef]
- Robinson, S.; McGonigle, O.; Volin, S.; Sun, Y.C.; Moore, M.; Cassidy, C.; Smith, E. Comprehensive Look at Blood Transfusion Utilization in Total Joint Arthroplasty at a Single Academic Medical Center under a Single Surgeon. J. Blood. Transfus. 2013, 2013, 983250. [Google Scholar] [CrossRef]
- Liu, Y.; Zuo, X.; Chen, P.; Hu, X.; Sheng, Z.; Liu, A.; Liu, Q.; Leng, S.; Zhang, X.; Li, X.; et al. Deciphering transcriptome alterations in bone marrow hematopoiesis at single-cell resolution in immune thrombocytopenia. Signal Transduct. Target. Ther. 2022, 7, 347. [Google Scholar] [CrossRef]
- Kaur, J.; Rawat, Y.; Sood, V.; Periwal, N.; Rathore, D.K.; Kumar, S.; Kumar, N.; Bhattacharyya, S. Replication of Dengue Virus in K562-Megakaryocytes Induces Suppression in the Accumulation of Reactive Oxygen Species. Front. Microbiol. 2021, 12, 784070. [Google Scholar] [CrossRef]
- Ramasz, B.; Kruger, A.; Reinhardt, J.; Sinha, A.; Gerlach, M.; Gerbaulet, A.; Reinhardt, S.; Dahl, A.; Chavakis, T.; Wielockx, B.; et al. Hematopoietic stem cell response to acute thrombocytopenia requires signaling through distinct receptor tyrosine kinases. Blood 2019, 134, 1046–1058. [Google Scholar] [CrossRef]
- Wang, H.; He, J.; Xu, C.; Chen, X.; Yang, H.; Shi, S.; Liu, C.; Zeng, Y.; Wu, D.; Bai, Z.; et al. Decoding human megakaryocyte development. Cell Stem. Cell 2021, 28, 535–549.e538. [Google Scholar] [CrossRef] [PubMed]
- Clay, D.; Rubinstein, E.; Mishal, Z.; Anjo, A.; Prenant, M.; Jasmin, C.; Boucheix, C.; Le Bousse-Kerdilès, M.C. CD9 and megakaryocyte differentiation. Blood 2001, 97, 1982–1989. [Google Scholar] [CrossRef] [PubMed]
- Machlus, K.R.; Johnson, K.E.; Kulenthirarajan, R.; Forward, J.A.; Tippy, M.D.; Soussou, T.S.; El-Husayni, S.H.; Wu, S.K.; Wang, S.; Watnick, R.S.; et al. CCL5 derived from platelets increases megakaryocyte proplatelet formation. Blood 2016, 127, 921–926. [Google Scholar] [CrossRef]
- Beaulieu, L.M.; Lin, E.; Morin, K.M.; Tanriverdi, K.; Freedman, J.E. Regulatory effects of TLR2 on megakaryocytic cell function. Blood 2011, 117, 5963–5974. [Google Scholar] [CrossRef]
- Hernandez-Sanchez, J.M.; Bastida, J.M.; Alonso-Lopez, D.; Benito, R.; Gonzalez-Porras, J.R.; De Las Rivas, J.; Hernandez Rivas, J.M.; Rodriguez-Vicente, A.E. Transcriptomic analysis of patients with immune thrombocytopenia treated with eltrombopag. Platelets 2020, 31, 993–1000. [Google Scholar] [CrossRef]
- Chen, Q.; Xin, M.; Wang, L.; Li, L.; Shen, Y.; Geng, Y.; Jiang, H.; Wang, Y.; Zhang, L.; Xu, Y.; et al. Inhibition of LDHA to induce eEF2 release enhances thrombocytopoiesis. Blood 2022, 139, 2958–2971. [Google Scholar] [CrossRef]
- Su, H.; Jiang, M.; Senevirathne, C.; Aluri, S.; Zhang, T.; Guo, H.; Xavier-Ferrucio, J.; Jin, S.; Tran, N.-T.; Liu, S.-M.; et al. Methylation of dual-specificity phosphatase 4 controls cell differentiation. Cell Rep. 2021, 36, 109421. [Google Scholar] [CrossRef]
- Eckly, A.; Rinckel, J.Y.; Laeuffer, P.; Cazenave, J.P.; Lanza, F.; Gachet, C.; Leon, C. Proplatelet formation deficit and megakaryocyte death contribute to thrombocytopenia in Myh9 knockout mice. J. Thromb. Haemost 2010, 8, 2243–2251. [Google Scholar] [CrossRef]
- Jalagadugula, G.; Dhanasekaran, D.N.; Kim, S.; Kunapuli, S.P.; Rao, A.K. Early growth response transcription factor EGR-1 regulates Galphaq gene in megakaryocytic cells. Thromb. Haemost 2006, 4, 2678–2686. [Google Scholar] [CrossRef]
- Kruse, E.A.; Loughran, S.J.; Baldwin, T.M.; Josefsson, E.C.; Ellis, S.; Watson, D.K.; Nurden, P.; Metcalf, D.; Hilton, D.J.; Alexander, W.S.; et al. Dual requirement for the ETS transcription factors Fli-1 and Erg in hematopoietic stem cells and the megakaryocyte lineage. Proc. Natl. Acad. Sci. USA 2009, 106, 13814–13819. [Google Scholar] [CrossRef]
- Jiang, H.; Yu, Z.; Ding, N.; Yang, M.; Zhang, L.; Fan, X.; Zhou, Y.; Zou, Q.; Hou, J.; Zheng, J.; et al. The role of AGK in thrombocytopoiesis and possible therapeutic strategies. Blood 2020, 136, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Raghuwanshi, S.G.U.; Kandi, R.; Gutti, R.K. MicroRNA-9 promotes cell proliferation by regulating RUNX1 expression in human megakaryocyte development. Cell Prolif. 2018, 51, e12414. [Google Scholar] [CrossRef] [PubMed]
- Limb, J.K.; Yoon, S.; Lee, K.E.; Kim, B.H.; Lee, S.; Bae, Y.S.; Jhon, G.J.; Kim, J. Regulation of megakaryocytic differentiation of K562 cells by FosB, a member of the Fos family of AP-1 transcription factors. Cell Mol. Life Sci. 2009, 66, 1962–1973. [Google Scholar] [CrossRef]
- Yao, C.J.; Works, K.; Romagnoli, P.A.; Austin, G.E. Effects of overexpression of HBP1 upon growth and differentiation of leukemic myeloid cells. Leukemia 2005, 19, 1958–1968. [Google Scholar] [CrossRef][Green Version]
- Shavit, J.A.; Motohashi, H.; Onodera, K.; Akasaka, J.; Yamamoto, M.; Engel, J.D. Impaired megakaryopoiesis and behavioral defects in mafG-null mutant mice. Genes Dev. 1998, 12, 2164–2174. [Google Scholar] [CrossRef]
- Fan, H.; Lv, Z.; Gan, L.; Ning, C.; Li, Z.; Yang, M.; Zhang, B.; Song, B.; Li, G.; Tang, D.; et al. A Novel lncRNA Regulates the Toll-Like Receptor Signaling Pathway and Related Immune Function by Stabilizing FOS mRNA as a Competitive Endogenous RNA. Front. Immunol. 2019, 10, 838. [Google Scholar] [CrossRef]
- Palomer, X.; Capdevila-Busquets, E.; Botteri, G.; Davidson, M.M.; Rodriguez, C.; Martinez-Gonzalez, J.; Vidal, F.; Barroso, E.; Chan, T.O.; Feldman, A.M.; et al. miR-146a targets Fos expression in human cardiac cells. Dis. Model. Mech. 2015, 8, 1081–1091. [Google Scholar] [CrossRef]
- Eriksson, M.; Arminen, L.; Karjalainen-Lindsberg, M.L.; Leppa, S. AP-1 regulates alpha2beta1 integrin expression by ERK-dependent signals during megakaryocytic differentiation of K562 cells. Exp. Cell Res. 2005, 304, 175–186. [Google Scholar] [CrossRef]
- Duan, T.; Du, Y.; Xing, C.; Wang, H.Y.; Wang, R.F. Toll-Like Receptor Signaling and Its Role in Cell-Mediated Immunity. Front. Immunol. 2022, 13, 812774. [Google Scholar] [CrossRef]
- Gonzalez, C.R.; Gonzalez, B. Exploring the Stress Impact in the Paternal Germ Cells Epigenome: Can Catecholamines Induce Epigenetic Reprogramming? Front. Endocrinol. 2020, 11, 630948. [Google Scholar] [CrossRef]
- Koupenova, M.; Livada, A.C.; Morrell, C.N. Platelet and Megakaryocyte Roles in Innate and Adaptive Immunity. Circ. Res. 2022, 130, 288–308. [Google Scholar] [CrossRef] [PubMed]
- Kobatake, E.; Kabuki, T. S-Layer Protein of Lactobacillus helveticus SBT2171 Promotes Human beta-Defensin 2 Expression via TLR2-JNK Signaling. Front. Microbiol. 2019, 10, 2414. [Google Scholar] [CrossRef] [PubMed]
- Adorni, M.P.; Ronda, N.; Bernini, F.; Favari, E. Rac1 and cholesterol metabolism in macrophage. J. Cardiovasc. Pharmacol. 2013, 62, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Fotinos, A.; Klier, M.; Gowert, N.S.; Munzer, P.; Klatt, C.; Beck, S.; Borst, O.; Billuart, P.; Schaller, M.; Lang, F.; et al. Loss of oligophrenin1 leads to uncontrolled Rho activation and increased thrombus formation in mice. J. Thromb. Haemost 2015, 13, 619–630. [Google Scholar] [CrossRef]
- Coso, O.A.; Chiariello, M.; Yu, J.C.; Teramoto, H.; Crespo, P.; Xu, N.; Miki, T.; Gutkind, J.S. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell 1995, 81, 1137–1146. [Google Scholar] [CrossRef]
- Shi, H.; Lin, B.; Huang, Y.; Wu, J.; Zhang, H.; Lin, C.; Wang, Z.; Zhu, J.; Zhao, Y.; Fu, X.; et al. Basic fibroblast growth factor promotes melanocyte migration via activating PI3K/Akt-Rac1-FAK-JNK and ERK signaling pathways. IUBMB Life 2016, 68, 735–747. [Google Scholar] [CrossRef]
- Scuron, M.D.; Fay, B.L.; Connell, A.J.; Peel, M.T.; Smith, P.A. Ruxolitinib Cream Has Dual Efficacy on Pruritus and Inflammation in Experimental Dermatitis. Front. Immunol. 2020, 11, 620098. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, W.; Yin, P.; Gao, J.; Na, L.; Sun, Y.; Wang, Z.; Zhang, Z.; Zhao, C. Ruxolitinib Alleviates Renal Interstitial Fibrosis in UUO Mice. Int. J. Biol. Sci. 2020, 16, 194–203. [Google Scholar] [CrossRef]
- Yoon, H.R.; Chai, C.C.; Kim, C.H.; Kang, N.S. A Study on the Effect of the Substituent against PAK4 Inhibition Using In Silico Methods. Int. J. Mol. Sci. 2022, 23, 3337. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; Tang, X.; Wang, L.; Ni, C.; Wu, Y.; Zhou, L.; Zeng, Y.; Zhao, C.; Wu, A.; Wang, Q.; et al. Targeting TLR2/Rac1/cdc42/JNK Pathway to Reveal That Ruxolitinib Promotes Thrombocytopoiesis. Int. J. Mol. Sci. 2022, 23, 16137. https://doi.org/10.3390/ijms232416137
Yang S, Tang X, Wang L, Ni C, Wu Y, Zhou L, Zeng Y, Zhao C, Wu A, Wang Q, et al. Targeting TLR2/Rac1/cdc42/JNK Pathway to Reveal That Ruxolitinib Promotes Thrombocytopoiesis. International Journal of Molecular Sciences. 2022; 23(24):16137. https://doi.org/10.3390/ijms232416137
Chicago/Turabian StyleYang, Shuo, Xiaoqin Tang, Long Wang, Chengyang Ni, Yuesong Wu, Ling Zhou, Yueying Zeng, Chunling Zhao, Anguo Wu, Qiaozhi Wang, and et al. 2022. "Targeting TLR2/Rac1/cdc42/JNK Pathway to Reveal That Ruxolitinib Promotes Thrombocytopoiesis" International Journal of Molecular Sciences 23, no. 24: 16137. https://doi.org/10.3390/ijms232416137
APA StyleYang, S., Tang, X., Wang, L., Ni, C., Wu, Y., Zhou, L., Zeng, Y., Zhao, C., Wu, A., Wang, Q., Xu, X., Wang, Y., Chen, R., Zhang, X., Zou, L., Huang, X., & Wu, J. (2022). Targeting TLR2/Rac1/cdc42/JNK Pathway to Reveal That Ruxolitinib Promotes Thrombocytopoiesis. International Journal of Molecular Sciences, 23(24), 16137. https://doi.org/10.3390/ijms232416137