
Quo Vadis? Immunodynamics of Myeloid Cells after Myocardial Infarction


Abstract
1. Introduction
- (1)
- Production of leukocytes in the bone marrow and spleen;
- (2)
- Release of leukocytes from the bone marrow and spleen into the blood;
- (3)
- Recruitment of leukocytes from the blood to the heart;
- (4)
- Actions of leukocytes within the heart;
2. Bone Marrow Niches and Hematopoiesis
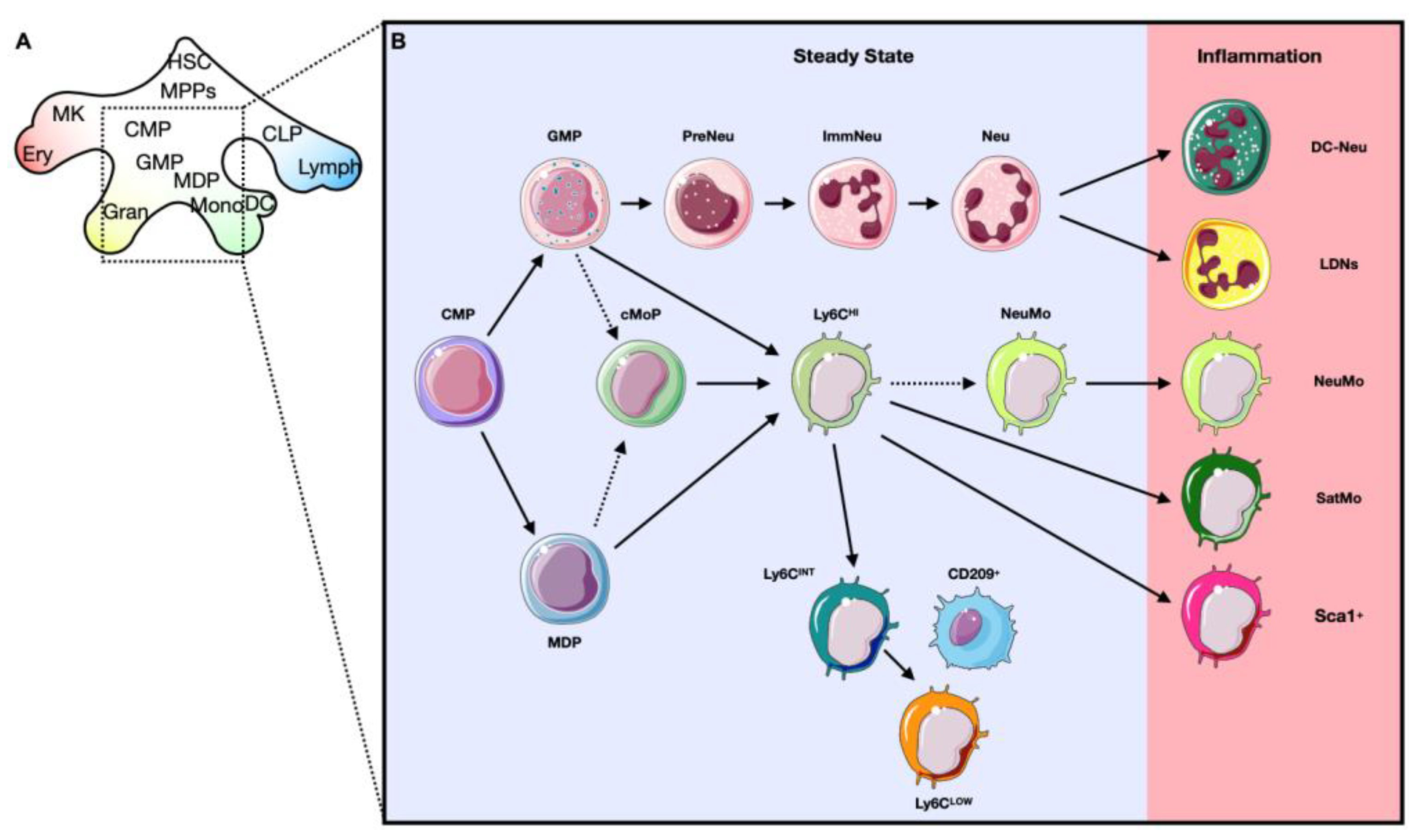
2.1. Hematopoiesis: The Generation of Blood Cells
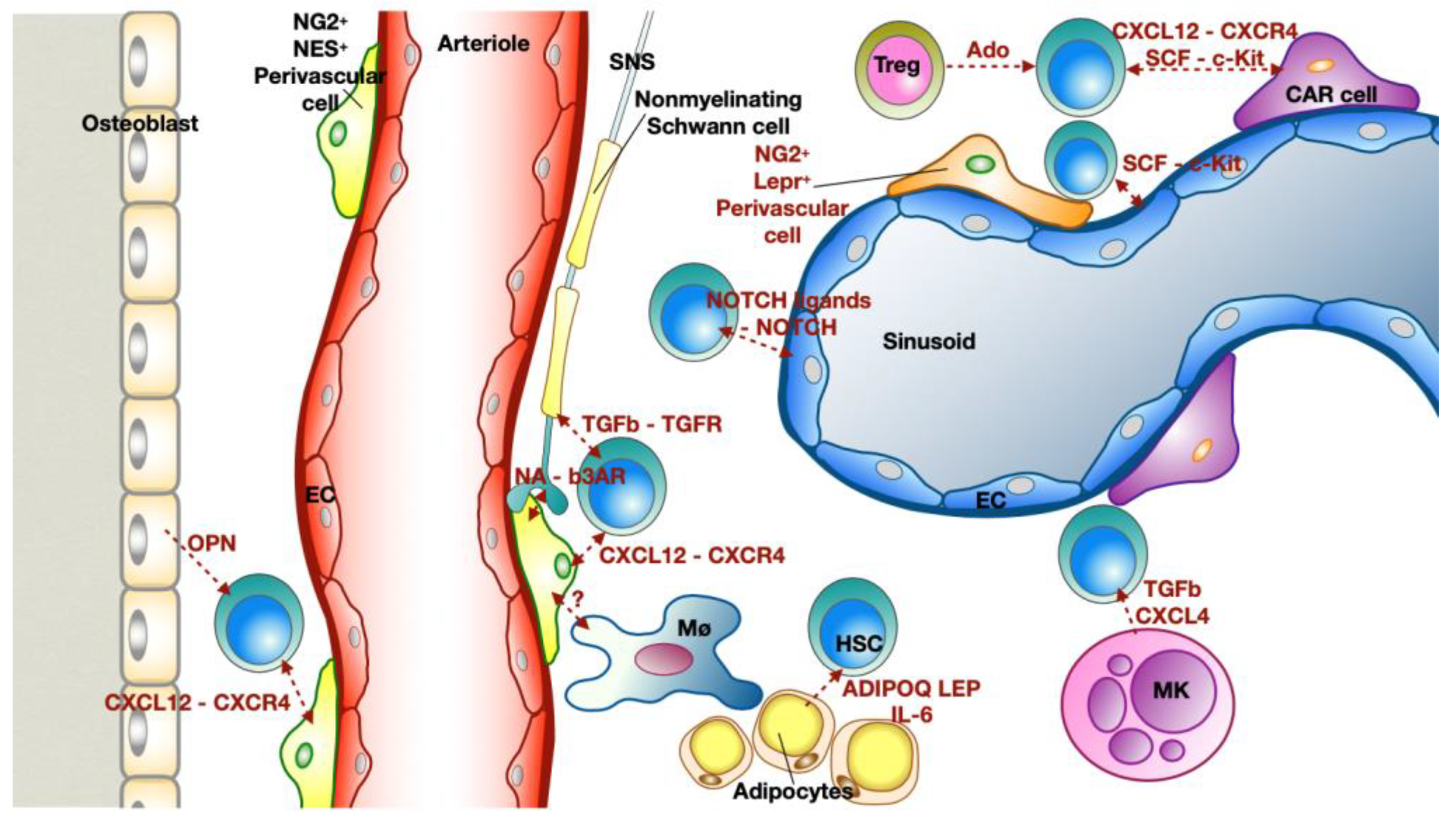
2.2. Bone Marrow Niche: Where Everything Starts
3. Myocardial Infarction Guided Emergency Hematopoiesis
3.1. Progenitor Amplification
3.2. Development of Alternative Phenotypes of Myeloid Cells (Qualitative Alterations)
3.3. Extramedullary Emergency Source of Myeloid Cells
4. Mobilization and Recruitment of Circulating Myeloid Cells
4.1. Chemokines
4.1.1. CC Chemokines
4.1.2. CX Chemokines
4.2. Inflammatory Cytokines: IL-1 Family, IL-6 and TNFa
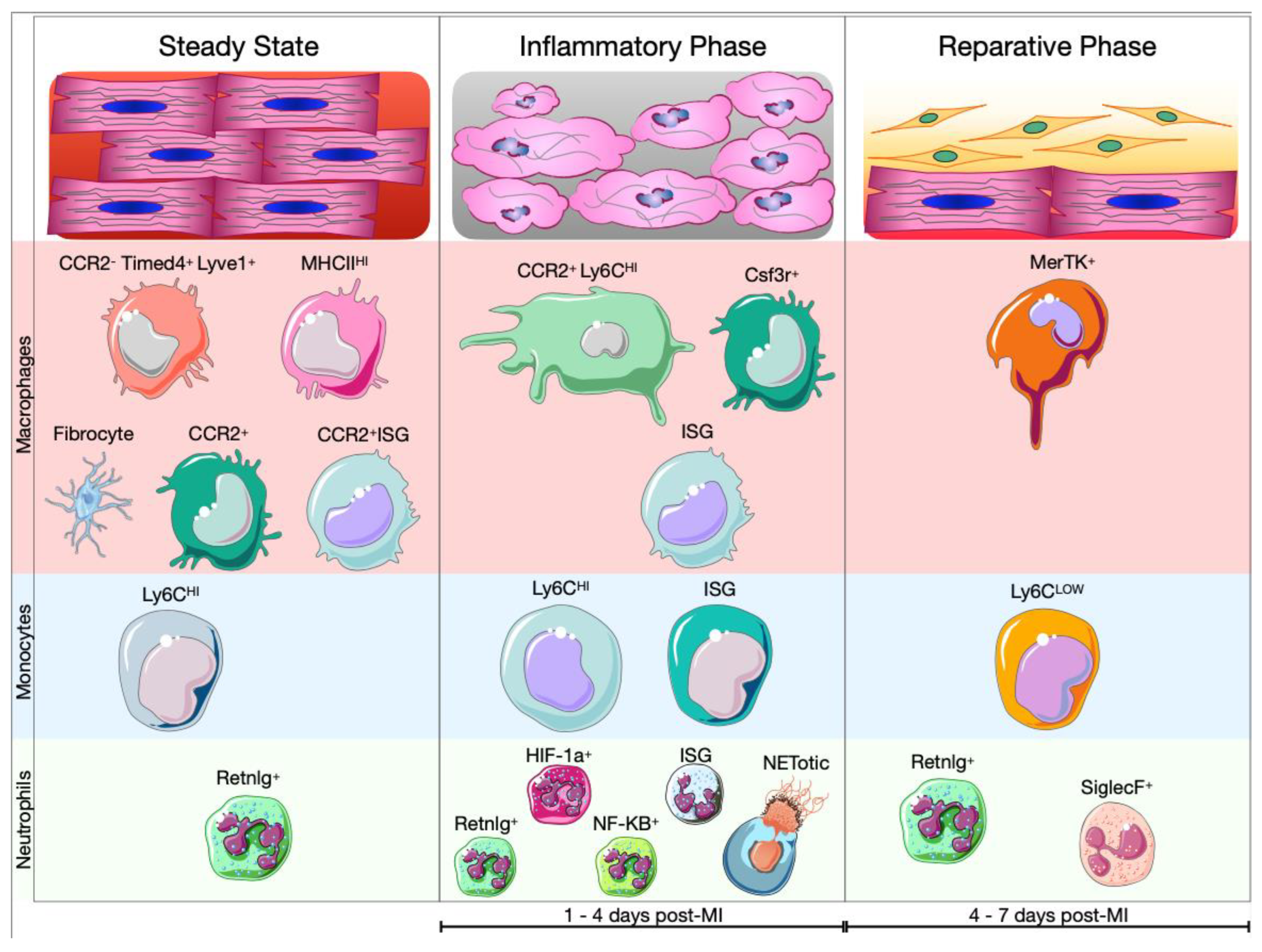
5. Myeloid Cells in Healthy and Infarcted Hearts
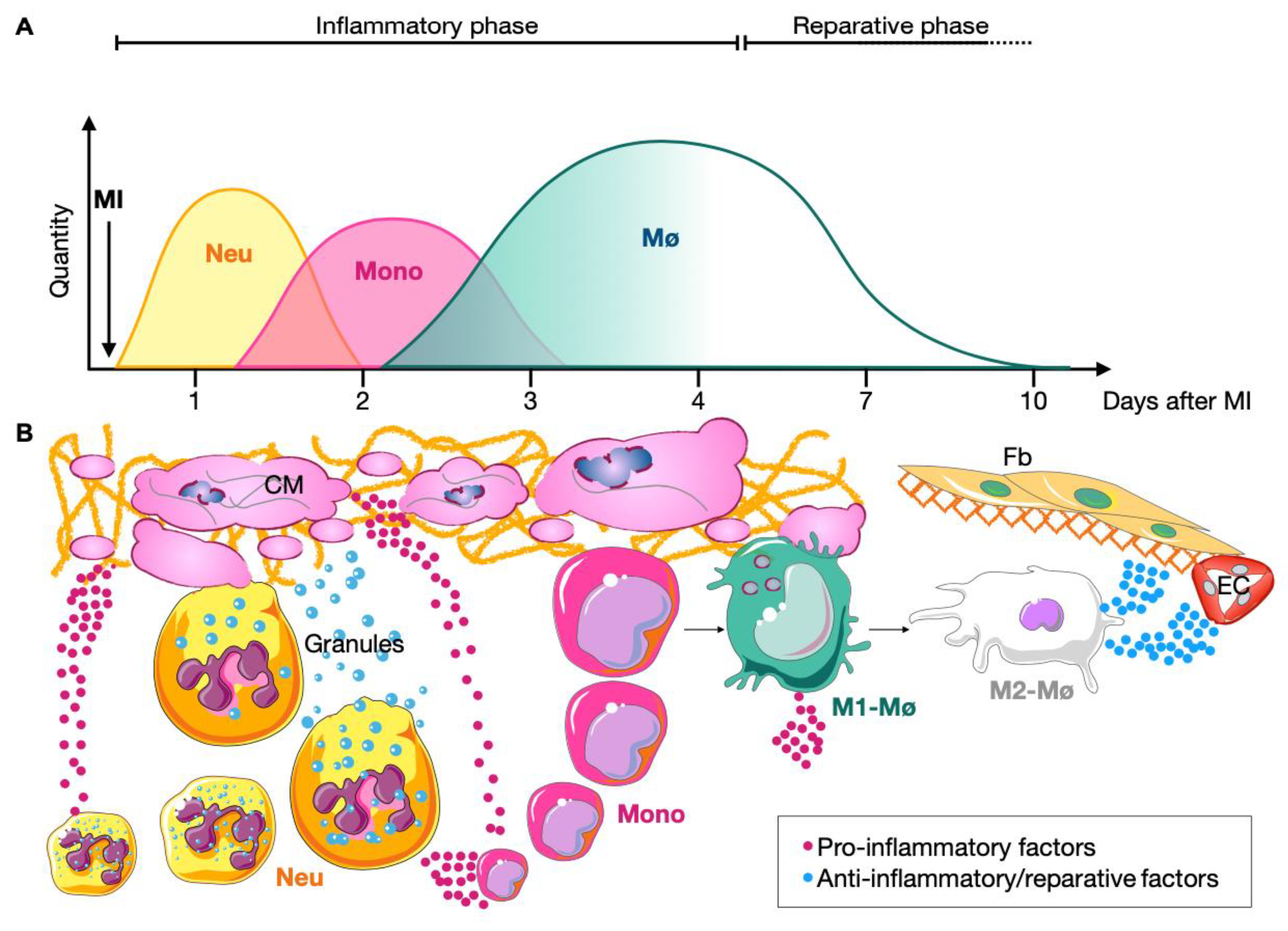
5.1. Cardiac Myeloid Cells during the Inflammatory Phase
- (1)
- Neutrophils expressing genes that are associated with NF-kB activation, including Nfkb1, Icam1, Il1a, Sod2, and Tnip1;
- (2)
- Neutrophils expressing genes linked with hypoxia-inducible factor 1α (HIF-1α) activation including Egln3, Hilpda, and Vegfa;
- (3)
- Neutrophils exhibiting an IFN response signature (Isg15+, Rsad2+, Ifit1+) [173,174]. It is not clear yet, whether the NETotic neutrophils belong to one of these clusters or have been excluded from the analysis since they are dying cells (Figure 5). A recent analysis of the transcriptome of neutrophils revealed that TFs like RELB, IRF5 and JUNB contribute to neutrophil survival and activation at the site of injury and promote phagocytosis, ROS production and NETosis. Finally, JUNB-deficient neutrophils showed reduced IL1β and ROS production leading to reduced infarct size [175].
Amplification of the Inflammation: DAMPs
5.2. Cardiac Myeloid Cells in the Resolution of Ischemic Injury
6. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO-CVD. Available online: https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 27 October 2022).
- Mauersberger, C.; Hinterdobler, J.; Schunkert, H.; Kessler, T.; Sager, H.B. Where the Action Is—Leukocyte Recruitment in Atherosclerosis. Front. Cardiovasc. Med. 2022, 8, 813984. [Google Scholar] [CrossRef] [PubMed]
- Palasubramaniam, J.; Wang, X.; Peter, K. Myocardial Infarction—From Atherosclerosis to Thrombosis: Uncovering New Diagnostic and Therapeutic Approaches. Arterioscler. Thromb. Vasc. Biol. 2019, 39, e176–e185. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K. Recent Advances in the Diagnosis and Treatment of Acute Myocardial Infarction. World J. Cardiol. 2015, 7, 243. [Google Scholar] [CrossRef]
- Hinterdobler, J.; Schott, S.; Jin, H.; Meesmann, A.; Steinsiek, A.-L.; Zimmermann, A.-S.; Wobst, J.; Müller, P.; Mauersberger, C.; Vilne, B.; et al. Acute Mental Stress Drives Vascular Inflammation and Promotes Plaque Destabilization in Mouse Atherosclerosis. Eur. Heart J. 2021, 42, 4077–4088. [Google Scholar] [CrossRef]
- Krittanawong, C.; Luo, Y.; Mahtta, D.; Narasimhan, B.; Wang, Z.; Jneid, H.; Tamis-Holland, J.E.; Mahboob, A.; Baber, U.; Mehran, R.; et al. Non-Traditional Risk Factors and the Risk of Myocardial Infarction in the Young in the US Population-Based Cohort. IJC Heart Vasc. 2020, 30, 100634. [Google Scholar] [CrossRef] [PubMed]
- Mauersberger, C.; Schunkert, H.; Sager, H.B. Inflammation-Related Risk Loci in Genome-Wide Association Studies of Coronary Artery Disease. Cells 2021, 10, 440. [Google Scholar] [CrossRef]
- Hinterdobler, J.; Schunkert, H.; Kessler, T.; Sager, H.B. Impact of Acute and Chronic Psychosocial Stress on Vascular Inflammation. Antioxid. Redox Signal. 2021, 35, 1531–1550. [Google Scholar] [CrossRef]
- Girard, D.; Vandiedonck, C. How Dysregulation of the Immune System Promotes Diabetes Mellitus and Cardiovascular Risk Complications. Front. Cardiovasc. Med. 2022, 9, 991716. [Google Scholar] [CrossRef]
- Frąk, W.; Wojtasińska, A.; Lisińska, W.; Młynarska, E.; Franczyk, B.; Rysz, J. Pathophysiology of Cardiovascular Diseases: New Insights into Molecular Mechanisms of Atherosclerosis, Arterial Hypertension, and Coronary Artery Disease. Biomedicines 2022, 10, 1938. [Google Scholar] [CrossRef]
- Nahrendorf, M. Myeloid Cell Contributions to Cardiovascular Health and Disease. Nat. Med. 2018, 24, 711–720. [Google Scholar] [CrossRef]
- Yerly, A.; van der Vorst, E.P.C.; Baumgartner, I.; Bernhard, S.M.; Schindewolf, M.; Döring, Y. Sex-specific and Hormone-related Differences in Vascular Remodelling in Atherosclerosis. Eur. J. Clin. Investig. 2022, e13885. [Google Scholar] [CrossRef]
- Ma, Y.; Long, Y.; Chen, Y. Roles of Inflammasome in Cigarette Smoke-Related Diseases and Physiopathological Disorders: Mechanisms and Therapeutic Opportunities. Front. Immunol. 2021, 12, 720049. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.N.; Wanjalla, C.N.; Mashayekhi, M.; Hasty, A.H. Immune Cell Activation in Obesity and Cardiovascular Disease. Curr. Hypertens. Rep. 2022, 24, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liu, J.; Zhao, K.; Gao, L.; Zhao, J. Cholesterol-Induced Toxicity: An Integrated View of the Role of Cholesterol in Multiple Diseases. Cell Metab. 2021, 33, 1911–1925. [Google Scholar] [CrossRef]
- Kelly, F.J.; Fussell, J.C. Linking Ambient Particulate Matter Pollution Effects with Oxidative Biology and Immune Responses: Oxidative Stress, Inflammation, and Particulate Matter Toxicity. Ann. N. Y. Acad. Sci. 2015, 1340, 84–94. [Google Scholar] [CrossRef]
- Frenis, K.; Kuntic, M.; Hahad, O.; Bayo Jimenez, M.T.; Oelze, M.; Daub, S.; Steven, S.; Münzel, T.; Daiber, A. Redox Switches in Noise-Induced Cardiovascular and Neuronal Dysregulation. Front. Mol. Biosci. 2021, 8, 784910. [Google Scholar] [CrossRef]
- Mozzini, C.; Pagani, M. Clonal Hematopoiesis and Cardiovascular Diseases: The Connection. Curr. Probl. Cardiol. 2022, 47, 100962. [Google Scholar] [CrossRef]
- Leal, L.G.; Lopes, M.A.; Batista, M.L. Physical Exercise-Induced Myokines and Muscle-Adipose Tissue Crosstalk: A Review of Current Knowledge and the Implications for Health and Metabolic Diseases. Front. Physiol. 2018, 9, 1307. [Google Scholar] [CrossRef]
- Besedovsky, L.; Lange, T.; Haack, M. The Sleep-Immune Crosstalk in Health and Disease. Physiol. Rev. 2019, 99, 1325–1380. [Google Scholar] [CrossRef]
- Li, B.; Xia, Y.; Hu, B. Infection and Atherosclerosis: TLR-Dependent Pathways. Cell. Mol. Life Sci. 2020, 77, 2751–2769. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Pathophysiology of Myocardial Infarction. Compr. Physiol. 2015, 5, 35. [Google Scholar]
- Swirski, F.K.; Nahrendorf, M. Cardioimmunology: The Immune System in Cardiac Homeostasis and Disease. Nat. Rev. Immunol. 2018, 18, 733–744. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, K.; Rankin, S.M. The Secretive Life of Neutrophils Revealed by Intravital Microscopy. Front. Cell Dev. Biol. 2020, 8, 603230. [Google Scholar] [CrossRef] [PubMed]
- Manz, M.G.; Boettcher, S. Emergency Granulopoiesis. Nat. Rev. Immunol. 2014, 14, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood Monocytes Consist of Two Principal Subsets with Distinct Migratory Properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef]
- Guilliams, M. Developmental and Functional Heterogeneity of Monocytes. Immunity 2018, 49, 595–613. [Google Scholar] [CrossRef]
- Robinson, A.; Han, C.Z.; Glass, C.K.; Pollard, J.W. Monocyte Regulation in Homeostasis and Malignancy. Trends Immunol. 2021, 42, 104–119. [Google Scholar] [CrossRef]
- Ma, Y. Role of Neutrophils in Cardiac Injury and Repair Following Myocardial Infarction. Cells 2021, 10, 1676. [Google Scholar] [CrossRef]
- Peet, C.; Ivetic, A.; Bromage, D.I.; Shah, A.M. Cardiac Monocytes and Macrophages after Myocardial Infarction. Cardiovasc. Res. 2020, 116, 1101–1112. [Google Scholar] [CrossRef]
- Peterson, E.A.; Sun, J.; Wang, J. Leukocyte-Mediated Cardiac Repair after Myocardial Infarction in Non-Regenerative vs. Regenerative Systems. J. Cardiovasc. Dev. Dis. 2022, 9, 63. [Google Scholar] [CrossRef]
- Sager, H.B.; Kessler, T.; Schunkert, H. Monocytes and Macrophages in Cardiac Injury and Repair. J. Thorac. Dis. 2017, 9, S30–S35. [Google Scholar] [CrossRef]
- Forte, E. Dynamic Interstitial Cell Response during Myocardial Infarction Predicts Resilience to Rupture in Genetically Diverse Mice. Cell Rep. 2022, 30, 3149–3163. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Tallquist, M.D. Cardiac Fibroblast Diversity. Annu. Rev. Physiol. 2020, 82, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized Fibroblast Differentiated States Underlie Scar Formation in the Infarcted Mouse Heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef]
- Pluijmert, N.J.; Atsma, D.E.; Quax, P.H.A. Post-Ischemic Myocardial Inflammatory Response: A Complex and Dynamic Process Susceptible to Immunomodulatory Therapies. Front. Cardiovasc. Med. 2021, 8, 647785. [Google Scholar] [CrossRef]
- De Villiers, C.; Riley, P.R. Mouse Models of Myocardial Infarction: Comparing Permanent Ligation and Ischaemia-Reperfusion. Dis. Models Mech. 2020, 13, dmm046565. [Google Scholar] [CrossRef]
- Fischer, M.; Weinberger, T.; Messerer, D.; Zacherl, M.J.; Schulz, C.; Massberg, S.; Bartenstein, P.; Lehner, S.; Boening, G.; Todica, A. Comparison of Transient and Permanent LAD Ligation in Mice Using 18F-FDG PET Imaging. Ann. Nucl. Med. 2022, 36, 533–543. [Google Scholar] [CrossRef]
- Schloss, M.J.; Horckmans, M.; Nitz, K.; Duchene, J.; Drechsler, M.; Bidzhekov, K.; Scheiermann, C.; Weber, C.; Soehnlein, O.; Steffens, S. The Time-of-day of Myocardial Infarction Onset Affects Healing through Oscillations in Cardiac Neutrophil Recruitment. EMBO Mol. Med. 2016, 8, 937–948. [Google Scholar] [CrossRef]
- du Pré, B.; Van Veen, T.; Crnko, S.; Vos, M.; Deddens, J.; Doevendans, P.; Van Laake, L. Variation within Variation: Comparison of 24-h Rhythm in Rodent Infarct Size between Ischemia Reperfusion and Permanent Ligation. Int. J. Mol. Sci. 2017, 18, 1670. [Google Scholar] [CrossRef]
- Pluijmert, N.J.; Bart, C.I.; Bax, W.H.; Quax, P.H.A.; Atsma, D.E. Effects on Cardiac Function, Remodeling and Inflammation Following Myocardial Ischemia–Reperfusion Injury or Unreperfused Myocardial Infarction in Hypercholesterolemic APOE*3-Leiden Mice. Sci. Rep. 2020, 10, 16601. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Anzai, A.; Katsumata, Y.; Matsuhashi, T.; Ito, K.; Endo, J.; Yamamoto, T.; Takeshima, A.; Shinmura, K.; Shen, W.; et al. Temporal Dynamics of Cardiac Immune Cell Accumulation Following Acute Myocardial Infarction. J. Mol. Cell. Cardiol. 2013, 62, 24–35. [Google Scholar] [CrossRef]
- Libby, P.; Nahrendorf, M.; Swirski, F.K. Leukocytes Link Local and Systemic Inflammation in Ischemic Cardiovascular Disease. J. Am. Coll. Cardiol. 2016, 67, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Poller, W.C.; Nahrendorf, M.; Swirski, F.K. Hematopoiesis and Cardiovascular Disease. Circ. Res. 2020, 126, 1061–1085. [Google Scholar] [CrossRef]
- Weinreb, C.; Rodriguez-Fraticelli, A.; Camargo, F.D.; Klein, A.M. Lineage Tracing on Transcriptional Landscapes Links State to Fate during Differentiation. Science 2020, 367, eaaw3381. [Google Scholar] [CrossRef]
- Jacobsen, S.E.W.; Nerlov, C. Haematopoiesis in the Era of Advanced Single-Cell Technologies. Nat. Cell Biol. 2019, 21, 2–8. [Google Scholar] [CrossRef]
- Laurenti, E.; Göttgens, B. From Haematopoietic Stem Cells to Complex Differentiation Landscapes. Nature 2018, 553, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.; Ssozi, D.; van Galen, P. Single-Cell RNA Sequencing to Disentangle the Blood System. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1012–1018. [Google Scholar] [CrossRef]
- Giladi, A.; Paul, F.; Herzog, Y.; Lubling, Y.; Weiner, A.; Yofe, I.; Jaitin, D.; Cabezas-Wallscheid, N.; Dress, R.; Ginhoux, F.; et al. Single-Cell Characterization of Haematopoietic Progenitors and Their Trajectories in Homeostasis and Perturbed Haematopoiesis. Nat. Cell Biol. 2018, 20, 836–846. [Google Scholar] [CrossRef]
- Nestorowa, S.; Hamey, F.K.; Pijuan Sala, B.; Diamanti, E.; Shepherd, M.; Laurenti, E.; Wilson, N.K.; Kent, D.G.; Göttgens, B. A Single-Cell Resolution Map of Mouse Hematopoietic Stem and Progenitor Cell Differentiation. Blood 2016, 128, e20–e31. [Google Scholar] [CrossRef]
- Kiel, M.J.; Yilmaz, Ö.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. SLAM Family Receptors Distinguish Hematopoietic Stem and Progenitor Cells and Reveal Endothelial Niches for Stem Cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Pietras, E.M.; Reynaud, D.; Kang, Y.-A.; Carlin, D.; Calero-Nieto, F.J.; Leavitt, A.D.; Stuart, J.M.; Göttgens, B.; Passegué, E. Functionally Distinct Subsets of Lineage-Biased Multipotent Progenitors Control Blood Production in Normal and Regenerative Conditions. Cell Stem Cell 2015, 17, 35–46. [Google Scholar] [CrossRef]
- Alberti-Servera, L.; Muenchow, L.; Tsapogas, P.; Capoferri, G.; Eschbach, K.; Beisel, C.; Ceredig, R.; Ivanek, R.; Rolink, A. Single-cell RNA Sequencing Reveals Developmental Heterogeneity among Early Lymphoid Progenitors. EMBO J. 2017, 36, 3619–3633. [Google Scholar] [CrossRef]
- Hettinger, J.; Richards, D.M.; Hansson, J.; Barra, M.M.; Joschko, A.-C.; Krijgsveld, J.; Feuerer, M. Origin of Monocytes and Macrophages in a Committed Progenitor. Nat. Immunol. 2013, 14, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Paul, F.; Arkin, Y.; Giladi, A.; Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Winter, D.; Lara-Astiaso, D.; Gury, M.; Weiner, A.; et al. Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell 2015, 163, 1663–1677. [Google Scholar] [CrossRef]
- Liu, Z.; Gu, Y.; Chakarov, S.; Bleriot, C.; Kwok, I.; Chen, X.; Shin, A.; Huang, W.; Dress, R.J.; Dutertre, C.-A.; et al. Fate Mapping via Ms4a3-Expression History Traces Monocyte-Derived Cells. Cell 2019, 178, 1509–1525.e19. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, J.; Scheitza, S.; Wernet, P.; Fischer, J.C.; Giebel, B. Asymmetric Cell Division within the Human Hematopoietic Stem and Progenitor Cell Compartment: Identification of Asymmetrically Segregating Proteins. Blood 2007, 109, 5494–5501. [Google Scholar] [CrossRef]
- Scadden, D.T. Nice Neighborhood: Emerging Concepts of the Stem Cell Niche. Cell 2014, 157, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, A.; Frenette, P.S. Hematopoietic Stem Cell Niche Maintenance during Homeostasis and Regeneration. Nat. Med. 2014, 20, 833–846. [Google Scholar] [CrossRef]
- Wei, Q.; Frenette, P.S. Niches for Hematopoietic Stem Cells and Their Progeny. Immunity 2018, 48, 632–648. [Google Scholar] [CrossRef]
- Crane, G.M.; Jeffery, E.; Morrison, S.J. Adult Haematopoietic Stem Cell Niches. Nat. Rev. Immunol. 2017, 17, 573–590. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, Y.-S.; Li, H.; Kang, Y.-L.; Chen, W.-C.; Cheng, W.-C.; Lai, D.-M. Loss of Cxcl12/Sdf-1 in Adult Mice Decreases the Quiescent State of Hematopoietic Stem/Progenitor Cells and Alters the Pattern of Hematopoietic Regeneration after Myelosuppression. Blood 2011, 117, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Comazzetto, S.; Murphy, M.M.; Berto, S.; Jeffery, E.; Zhao, Z.; Morrison, S.J. Restricted Hematopoietic Progenitors and Erythropoiesis Require SCF from Leptin Receptor+ Niche Cells in the Bone Marrow. Cell Stem Cell 2019, 24, 477–486.e6. [Google Scholar] [CrossRef]
- Baryawno, N.; Przybylski, D.; Kowalczyk, M.S.; Kfoury, Y.; Severe, N.; Gustafsson, K.; Kokkaliaris, K.D.; Mercier, F.; Tabaka, M.; Hofree, M.; et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell 2019, 177, 1915–1932.e16. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Gao, X.; Wei, Q.; Nakahara, F.; Zimmerman, S.E.; Mar, J.; Frenette, P.S. Stem Cell Factor Is Selectively Secreted by Arterial Endothelial Cells in Bone Marrow. Nat. Commun. 2018, 9, 2449. [Google Scholar] [CrossRef] [PubMed]
- Tikhonova, A.N.; Dolgalev, I.; Hu, H.; Sivaraj, K.K.; Hoxha, E.; Cuesta-Domínguez, Á.; Pinho, S.; Akhmetzyanova, I.; Gao, J.; Witkowski, M.; et al. The Bone Marrow Microenvironment at Single-Cell Resolution. Nature 2019, 569, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Poulos, M.G.; Guo, P.; Kofler, N.M.; Pinho, S.; Gutkin, M.C.; Tikhonova, A.; Aifantis, I.; Frenette, P.S.; Kitajewski, J.; Rafii, S.; et al. Endothelial Jagged-1 Is Necessary for Homeostatic and Regenerative Hematopoiesis. Cell Rep. 2013, 4, 1022–1034. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; MacArthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and Haematopoietic Stem Cells Form a Unique Bone Marrow Niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the Hematopoietic Stem Cell Pool by CXCL12-CXCR4 Chemokine Signaling in Bone Marrow Stromal Cell Niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef]
- Pinho, S.; Frenette, P.S. Haematopoietic Stem Cell Activity and Interactions with the Niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef] [PubMed]
- Baccin, C.; Al-Sabah, J.; Velten, L.; Helbling, P.M.; Grünschläger, F.; Hernández-Malmierca, P.; Nombela-Arrieta, C.; Steinmetz, L.M.; Trumpp, A.; Haas, S. Combined Single-Cell and Spatial Transcriptomics Reveal the Molecular, Cellular and Spatial Bone Marrow Niche Organization. Nat. Cell Biol. 2020, 22, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Kunisaki, Y.; Bruns, I.; Scheiermann, C.; Ahmed, J.; Pinho, S.; Zhang, D.; Mizoguchi, T.; Wei, Q.; Lucas, D.; Ito, K.; et al. Arteriolar Niches Maintain Haematopoietic Stem Cell Quiescence. Nature 2013, 502, 637–643. [Google Scholar] [CrossRef]
- Wang, H.; Leng, Y.; Gong, Y. Bone Marrow Fat and Hematopoiesis. Front. Endocrinol. 2018, 9, 694. [Google Scholar] [CrossRef]
- Zhou, B.O.; Yu, H.; Yue, R.; Zhao, Z.; Rios, J.J.; Naveiras, O.; Morrison, S.J. Bone Marrow Adipocytes Promote the Regeneration of Stem Cells and Hematopoiesis by Secreting SCF. Nat. Cell Biol. 2017, 19, 891–903. [Google Scholar] [CrossRef]
- Nilsson, S.K.; Johnston, H.M.; Whitty, G.A.; Williams, B.; Webb, R.J.; Denhardt, D.T.; Bertoncello, I.; Bendall, L.J.; Simmons, P.J.; Haylock, D.N. Osteopontin, a Key Component of the Hematopoietic Stem Cell Niche and Regulator of Primitive Hematopoietic Progenitor Cells. Blood 2005, 106, 1232–1239. [Google Scholar] [CrossRef]
- Maryanovich, M.; Takeishi, S.; Frenette, P.S. Neural Regulation of Bone and Bone Marrow. Cold Spring Harb. Perspect. Med. 2018, 8, a031344. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Ferrer, S.; Lucas, D.; Battista, M.; Frenette, P.S. Haematopoietic Stem Cell Release Is Regulated by Circadian Oscillations. Nature 2008, 452, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Ema, H.; Karlsson, G.; Yamaguchi, T.; Miyoshi, H.; Shioda, S.; Taketo, M.M.; Karlsson, S.; Iwama, A.; Nakauchi, H. Nonmyelinating Schwann Cells Maintain Hematopoietic Stem Cell Hibernation in the Bone Marrow Niche. Cell 2011, 147, 1146–1158. [Google Scholar] [CrossRef]
- Bruns, I.; Lucas, D.; Pinho, S.; Ahmed, J.; Lambert, M.P.; Kunisaki, Y.; Scheiermann, C.; Schiff, L.; Poncz, M.; Bergman, A.; et al. Megakaryocytes Regulate Hematopoietic Stem Cell Quiescence through CXCL4 Secretion. Nat. Med. 2014, 20, 1315–1320. [Google Scholar] [CrossRef]
- Zhao, M.; Perry, J.M.; Marshall, H.; Venkatraman, A.; Qian, P.; He, X.C.; Ahamed, J.; Li, L. Megakaryocytes Maintain Homeostatic Quiescence and Promote Post-Injury Regeneration of Hematopoietic Stem Cells. Nat. Med. 2014, 20, 1321–1326. [Google Scholar] [CrossRef]
- Chow, A.; Lucas, D.; Hidalgo, A.; Méndez-Ferrer, S.; Hashimoto, D.; Scheiermann, C.; Battista, M.; Leboeuf, M.; Prophete, C.; van Rooijen, N.; et al. Bone Marrow CD169+ Macrophages Promote the Retention of Hematopoietic Stem and Progenitor Cells in the Mesenchymal Stem Cell Niche. J. Exp. Med. 2011, 208, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Furuhashi, K.; Ishii, H.; Li, H.W.; Pinho, S.; Ding, L.; Robson, S.C.; Frenette, P.S.; Fujisaki, J. CD150high Bone Marrow Tregs Maintain Hematopoietic Stem Cell Quiescence and Immune Privilege via Adenosine. Cell Stem Cell 2018, 22, 445–453.e5. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Sager, H.B.; Stengel, K.R.; Naxerova, K.; Courties, G.; Saez, B.; Silberstein, L.; Heidt, T.; Sebas, M.; Sun, Y.; et al. Myocardial Infarction Activates CCR2+ Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2015, 16, 477–487.e5. [Google Scholar] [CrossRef] [PubMed]
- Sager, H.B.; Heidt, T.; Hulsmans, M.; Dutta, P.; Courties, G.; Sebas, M.; Wojtkiewicz, G.R.; Tricot, B.; Iwamoto, Y.; Sun, Y.; et al. Targeting Interleukin-1β Reduces Leukocyte Production After Acute Myocardial Infarction. Circulation 2015, 132, 1880–1890. [Google Scholar] [CrossRef] [PubMed]
- Sager, H.B.; Hulsmans, M.; Lavine, K.J.; Moreira, M.B.; Heidt, T.; Courties, G.; Sun, Y.; Iwamoto, Y.; Tricot, B.; Khan, O.F.; et al. Proliferation and Recruitment Contribute to Myocardial Macrophage Expansion in Chronic Heart Failure. Circ. Res. 2016, 119, 853–864. [Google Scholar] [CrossRef]
- Bronte, V.; Pittet, M.J. The Spleen in Local and Systemic Regulation of Immunity. Immunity 2013, 39, 806–818. [Google Scholar] [CrossRef]
- Abdali, A.; Marinković, G. Assessment of Medullary and Extramedullary Myelopoiesis in Cardiovascular Diseases. Pharmacol. Res. 2021, 169, 105663. [Google Scholar] [CrossRef]
- Grieshaber-Bouyer, R.; Nigrovic, P.A. Neutrophil Heterogeneity as Therapeutic Opportunity in Immune-Mediated Disease. Front. Immunol. 2019, 10, 346. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.A.; Yáñez, A.; Barman, P.K.; Goodridge, H.S. The Ontogeny of Monocyte Subsets. Front. Immunol. 2019, 10, 1642. [Google Scholar] [CrossRef]
- Assmus, B.; Iwasaki, M.; Schachinger, V.; Roexe, T.; Koyanagi, M.; Iekushi, K.; Xu, Q.; Tonn, T.; Seifried, E.; Liebner, S.; et al. Acute Myocardial Infarction Activates Progenitor Cells and Increases Wnt Signalling in the Bone Marrow. Eur. Heart J. 2012, 33, 1911–1919. [Google Scholar] [CrossRef]
- Tobin, S.W.; Alibhai, F.J.; Weisel, R.D.; Li, R.-K. Considering Cause and Effect of Immune Cell Aging on Cardiac Repair after Myocardial Infarction. Cells 2020, 9, 1894. [Google Scholar] [CrossRef]
- Sanganalmath, S.K.; Abdel-Latif, A.; Bolli, R.; Xuan, Y.-T.; Dawn, B. Hematopoietic Cytokines for Cardiac Repair: Mobilization of Bone Marrow Cells and Beyond. Basic Res. Cardiol. 2011, 106, 709–733. [Google Scholar] [CrossRef]
- Dutta, P.; Courties, G.; Wei, Y.; Leuschner, F.; Gorbatov, R.; Robbins, C.S.; Iwamoto, Y.; Thompson, B.; Carlson, A.L.; Heidt, T.; et al. Myocardial Infarction Accelerates Atherosclerosis. Nature 2012, 487, 325–329. [Google Scholar] [CrossRef]
- Delgaudine, M.; Gothot, A.; Beguin, Y. Spontaneous and Granulocyte–Colony-Stimulating Factor-Enhanced Marrow Response and Progenitor Cell Mobilization in Mice after Myocardial Infarction. Cytotherapy 2010, 12, 909–918. [Google Scholar] [CrossRef] [PubMed]
- D’Amario, D.; Leone, A.M.; Borovac, J.A.; Cannata, F.; Siracusano, A.; Niccoli, G.; Crea, F. Granulocyte Colony-Stimulating Factor for the Treatment of Cardiovascular Diseases: An Update with a Critical Appraisal. Pharmacol. Res. 2018, 127, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Dawn, B.; Guo, Y.; Rezazadeh, A.; Huang, Y.; Stein, A.B.; Hunt, G.; Tiwari, S.; Varma, J.; Gu, Y.; Prabhu, S.D.; et al. Postinfarct Cytokine Therapy Regenerates Cardiac Tissue and Improves Left Ventricular Function. Circ. Res. 2006, 98, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Anzai, A.; Choi, J.L.; He, S.; Fenn, A.M.; Nairz, M.; Rattik, S.; McAlpine, C.S.; Mindur, J.E.; Chan, C.T.; Iwamoto, Y.; et al. The Infarcted Myocardium Solicits GM-CSF for the Detrimental Oversupply of Inflammatory Leukocytes. J. Exp. Med. 2017, 214, 3293–3310. [Google Scholar] [CrossRef] [PubMed]
- Mossadegh-Keller, N.; Sarrazin, S.; Kandalla, P.K.; Espinosa, L.; Stanley, E.R.; Nutt, S.L.; Moore, J.; Sieweke, M.H. M-CSF Instructs Myeloid Lineage Fate in Single Haematopoietic Stem Cells. Nature 2013, 497, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, D. Hematopoietic Cytokines. Blood 2008, 111, 485–491. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, Q.; Johnson, C.; Olsson, A.; Slaughter, A.; May, M.L.; Weinhaus, B.; Bowers, E.; Engel, J.D.; Jiang, J.X.; et al. Spatial Mapping of Myelopoiesis Reveals the Bone Marrow Niche for Monocyte Dendritic Cell Progenitors. Blood 2019, 134, 528. [Google Scholar] [CrossRef]
- Dai, X.-M.; Ryan, G.R.; Hapel, A.J.; Dominguez, M.G.; Russell, R.G.; Kapp, S.; Sylvestre, V.; Stanley, E.R. Targeted Disruption of the Mouse Colony-Stimulating Factor 1 Receptor Gene Results in Osteopetrosis, Mononuclear Phagocyte Deficiency, Increased Primitive Progenitor Cell Frequencies, and Reproductive Defects. Blood 2002, 99, 111–120. [Google Scholar] [CrossRef]
- Dewald, O.; Ren, G.; Duerr, G.D.; Zoerlein, M.; Klemm, C.; Gersch, C.; Tincey, S.; Michael, L.H.; Entman, M.L.; Frangogiannis, N.G. Of Mice and Dogs. Am. J. Pathol. 2004, 164, 665–677. [Google Scholar] [CrossRef]
- Morimoto, H.; Takahashi, M.; Shiba, Y.; Izawa, A.; Ise, H.; Hongo, M.; Hatake, K.; Motoyoshi, K.; Ikeda, U. Bone Marrow-Derived CXCR4+ Cells Mobilized by Macrophage Colony-Stimulating Factor Participate in the Reduction of Infarct Area and Improvement of Cardiac Remodeling after Myocardial Infarction in Mice. Am. J. Pathol. 2007, 171, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Hibbs, M.L.; Quilici, C.; Kountouri, N.; Seymour, J.F.; Armes, J.E.; Burgess, A.W.; Dunn, A.R. Mice Lacking Three Myeloid Colony-Stimulating Factors (G-CSF, GM-CSF, and M-CSF) Still Produce Macrophages and Granulocytes and Mount an Inflammatory Response in a Sterile Model of Peritonitis. J. Immunol. 2007, 178, 6435–6443. [Google Scholar] [CrossRef]
- Iqbal, A.J.; Fisher, E.A.; Greaves, D.R. Inflammation—A Critical Appreciation of the Role of Myeloid Cells. Myeloid Cells Health Dis. A Synth. 2017, 17, 325–342. [Google Scholar]
- Trzebanski, S.; Jung, S. Plasticity of Monocyte Development and Monocyte Fates. Immunol. Lett. 2020, 227, 66–78. [Google Scholar] [CrossRef]
- Sagiv, J.Y.; Michaeli, J.; Assi, S.; Mishalian, I.; Kisos, H.; Levy, L.; Damti, P.; Lumbroso, D.; Polyansky, L.; Sionov, R.V.; et al. Phenotypic Diversity and Plasticity in Circulating Neutrophil Subpopulations in Cancer. Cell Rep. 2015, 10, 562–573. [Google Scholar] [CrossRef]
- Fites, J.S.; Gui, M.; Kernien, J.F.; Negoro, P.; Dagher, Z.; Sykes, D.B.; Nett, J.E.; Mansour, M.K.; Klein, B.S. An Unappreciated Role for Neutrophil-DC Hybrids in Immunity to Invasive Fungal Infections. PLoS Pathog. 2018, 14, e1007073. [Google Scholar] [CrossRef]
- Yáñez, A.; Coetzee, S.G.; Olsson, A.; Muench, D.E.; Berman, B.P.; Hazelett, D.J.; Salomonis, N.; Grimes, H.L.; Goodridge, H.S. Granulocyte-Monocyte Progenitors and Monocyte-Dendritic Cell Progenitors Independently Produce Functionally Distinct Monocytes. Immunity 2017, 47, 890–902.e4. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Nakagawa, K.; Sugihara, F.; Kuwahara, R.; Ashihara, M.; Yamane, F.; Minowa, Y.; Fukushima, K.; Ebina, I.; Yoshioka, Y.; et al. Identification of an Atypical Monocyte and Committed Progenitor Involved in Fibrosis. Nature 2017, 541, 96–101. [Google Scholar] [CrossRef]
- Askenase, M.H.; Han, S.-J.; Byrd, A.L.; Morais da Fonseca, D.; Bouladoux, N.; Wilhelm, C.; Konkel, J.E.; Hand, T.W.; Lacerda-Queiroz, N.; Su, X.; et al. Bone-Marrow-Resident NK Cells Prime Monocytes for Regulatory Function during Infection. Immunity 2015, 42, 1130–1142. [Google Scholar] [CrossRef]
- Ikeda, N.; Asano, K.; Kikuchi, K.; Uchida, Y.; Ikegami, H.; Takagi, R.; Yotsumoto, S.; Shibuya, T.; Makino-Okamura, C.; Fukuyama, H.; et al. Emergence of Immunoregulatory Ym1 + Ly6C hi Monocytes during Recovery Phase of Tissue Injury. Sci. Immunol. 2018, 3, eaat0207. [Google Scholar] [CrossRef]
- Calcagno, D.M.; Ng, R.P.; Toomu, A.; Zhang, C.; Huang, K.; Aguirre, A.D.; Weissleder, R.; Daniels, L.B.; Fu, Z.; King, K.R. The Myeloid Type I Interferon Response to Myocardial Infarction Begins in Bone Marrow and Is Regulated by Nrf2-Activated Macrophages. Sci. Immunol. 2020, 5, eaaz1974. [Google Scholar] [CrossRef]
- Fraccarollo, D.; Neuser, J.; Möller, J.; Riehle, C.; Galuppo, P.; Bauersachs, J. Expansion of CD10neg Neutrophils and CD14+HLA-DRneg/Low Monocytes Driving Proinflammatory Responses in Patients with Acute Myocardial Infarction. eLife 2021, 10, e66808. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Nahrendorf, M.; Etzrodt, M.; Wildgruber, M.; Cortez-Retamozo, V.; Panizzi, P.; Figueiredo, J.-L.; Kohler, R.H.; Chudnovskiy, A.; Waterman, P.; et al. Identification of Splenic Reservoir Monocytes and Their Deployment to Inflammatory Sites. Science 2009, 325, 612–616. [Google Scholar] [CrossRef]
- Leuschner, F.; Panizzi, P.; Chico-Calero, I.; Lee, W.W.; Ueno, T.; Cortez-Retamozo, V.; Waterman, P.; Gorbatov, R.; Marinelli, B.; Iwamoto, Y.; et al. Angiotensin-Converting Enzyme Inhibition Prevents the Release of Monocytes From Their Splenic Reservoir in Mice With Myocardial Infarction. Circ. Res. 2010, 107, 1364–1373. [Google Scholar] [CrossRef]
- Senders, M.L.; Meerwaldt, A.E.; van Leent, M.M.T.; Sanchez-Gaytan, B.L.; van de Voort, J.C.; Toner, Y.C.; Maier, A.; Klein, E.D.; Sullivan, N.A.T.; Sofias, A.M.; et al. Probing Myeloid Cell Dynamics in Ischaemic Heart Disease by Nanotracer Hot-Spot Imaging. Nat. Nanotechnol. 2020, 15, 398–405. [Google Scholar] [CrossRef]
- Toor, I.S.; Rückerl, D.; Mair, I.; Thomson, A.; Rossi, A.G.; Newby, D.E.; Allen, J.E.; Gray, G.A. Enhanced Monocyte Recruitment and Delayed Alternative Macrophage Polarization Accompanies Impaired Repair Following Myocardial Infarction in C57BL/6 Compared to BALB/c Mice. Clin. Exp. Immunol. 2019, 198, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Hoyer, F.F.; Grigoryeva, L.S.; Sager, H.B.; Leuschner, F.; Courties, G.; Borodovsky, A.; Novobrantseva, T.; Ruda, V.M.; Fitzgerald, K.; et al. Macrophages Retain Hematopoietic Stem Cells in the Spleen via VCAM-1. J. Exp. Med. 2015, 212, 497–512. [Google Scholar] [CrossRef]
- Grisanti, L.A.; Gumpert, A.M.; Traynham, C.J.; Gorsky, J.E.; Repas, A.A.; Gao, E.; Carter, R.L.; Yu, D.; Calvert, J.W.; García, A.P.; et al. Leukocyte-Expressed β2-Adrenergic Receptors Are Essential for Survival After Acute Myocardial Injury. Circulation 2016, 134, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Grisanti, L.A.; de Lucia, C.; Thomas, T.P.; Stark, A.; Strony, J.T.; Myers, V.D.; Beretta, R.; Yu, D.; Sardu, C.; Marfella, R.; et al. Prior Beta Blocker Treatment Decreases Leukocyte Responsiveness to Injury. JCI Insight 2019, 4, e99485. [Google Scholar] [CrossRef] [PubMed]
- Grisanti, L.A.; Traynham, C.J.; Repas, A.A.; Gao, E.; Koch, W.J.; Tilley, D.G. B2-Adrenergic Receptor-Dependent Chemokine Receptor 2 Expression Regulates Leukocyte Recruitment to the Heart Following Acute Injury. Proc. Natl. Acad. Sci. USA 2016, 113, 15126–15131. [Google Scholar] [CrossRef]
- Tanner, M.A.; Maitz, C.A.; Grisanti, L.A. Immune Cell β 2 -Adrenergic Receptors Contribute to the Development of Heart Failure. Am. J. Physiol.-Heart Circ. Physiol. 2021, 321, H633–H649. [Google Scholar] [CrossRef]
- Leuschner, F.; Rauch, P.J.; Ueno, T.; Gorbatov, R.; Marinelli, B.; Lee, W.W.; Dutta, P.; Wei, Y.; Robbins, C.; Iwamoto, Y.; et al. Rapid Monocyte Kinetics in Acute Myocardial Infarction Are Sustained by Extramedullary Monocytopoiesis. J. Exp. Med. 2012, 209, 123–137. [Google Scholar] [CrossRef]
- Deniset, J.F.; Surewaard, B.G.; Lee, W.; Kubes, P. Splenic Ly6Ghigh Mature and Ly6Gint Immature Neutrophils Contribute to Eradication of S. Pneumoniae. J. Exp. Med. 2017, 214, 1333–1350. [Google Scholar] [CrossRef]
- Evrard, M.; Kwok, I.W.H.; Chong, S.Z.; Teng, K.W.W.; Becht, E.; Chen, J.; Sieow, J.L.; Penny, H.L.; Ching, G.C.; Devi, S.; et al. Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity 2018, 48, 364–379.e8. [Google Scholar] [CrossRef]
- O’Connell, K.E.; Mikkola, A.M.; Stepanek, A.M.; Vernet, A.; Hall, C.D.; Sun, C.C.; Yildirim, E.; Staropoli, J.F.; Lee, J.T.; Brown, D.E. Practical Murine Hematopathology: A Comparative Review and Implications for Research. Comp. Med. 2015, 65, 18. [Google Scholar]
- Noels, H.; Weber, C.; Koenen, R.R. Chemokines as Therapeutic Targets in Cardiovascular Disease: The Road Behind, The Road Ahead. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Bartekova, M.; Radosinska, J.; Jelemensky, M.; Dhalla, N.S. Role of Cytokines and Inflammation in Heart Function during Health and Disease. Heart Fail. Rev. 2018, 23, 733–758. [Google Scholar] [CrossRef]
- Serbina, N.V.; Pamer, E.G. Monocyte Emigration from Bone Marrow during Bacterial Infection Requires Signals Mediated by Chemokine Receptor CCR2. Nat. Immunol. 2006, 7, 311–317. [Google Scholar] [CrossRef]
- Leuschner, F.; Dutta, P.; Gorbatov, R.; Novobrantseva, T.I.; Donahoe, J.S.; Courties, G.; Lee, K.M.; Kim, J.I.; Markmann, J.F.; Marinelli, B.; et al. Therapeutic SiRNA Silencing in Inflammatory Monocytes in Mice. Nat. Biotechnol. 2011, 29, 1005–1010. [Google Scholar] [CrossRef]
- Jiao, J.; He, S.; Wang, Y.; Lu, Y.; Gu, M.; Li, D.; Tang, T.; Nie, S.; Zhang, M.; Lv, B.; et al. Regulatory B Cells Improve Ventricular Remodeling after Myocardial Infarction by Modulating Monocyte Migration. Basic Res. Cardiol. 2021, 116, 46. [Google Scholar] [CrossRef] [PubMed]
- Gschwandtner, M.; Derler, R.; Midwood, K.S. More Than Just Attractive: How CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front. Immunol. 2019, 10, 2759. [Google Scholar] [CrossRef] [PubMed]
- Dewald, O.; Zymek, P.; Winkelmann, K.; Koerting, A.; Ren, G.; Abou-Khamis, T.; Michael, L.H.; Rollins, B.J.; Entman, M.L.; Frangogiannis, N.G. CCL2/Monocyte Chemoattractant Protein-1 Regulates Inflammatory Responses Critical to Healing Myocardial Infarcts. Circ. Res. 2005, 96, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Zouggari, Y.; Ait-Oufella, H.; Bonnin, P.; Simon, T.; Sage, A.P.; Guérin, C.; Vilar, J.; Caligiuri, G.; Tsiantoulas, D.; Laurans, L.; et al. B Lymphocytes Trigger Monocyte Mobilization and Impair Heart Function after Acute Myocardial Infarction. Nat. Med. 2013, 19, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Vajen, T.; Koenen, R.R.; Werner, I.; Staudt, M.; Projahn, D.; Curaj, A.; Sönmez, T.T.; Simsekyilmaz, S.; Schumacher, D.; Möllmann, J.; et al. Blocking CCL5-CXCL4 Heteromerization Preserves Heart Function after Myocardial Infarction by Attenuating Leukocyte Recruitment and NETosis. Sci. Rep. 2018, 8, 10647. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, F.; Braunersreuther, V.; Lenglet, S.; Delattre, B.M.A.; Pelli, G.; Buatois, V.; Guilhot, F.; Galan, K.; Vuilleumier, N.; Ferlin, W.; et al. CC Chemokine CCL5 Plays a Central Role Impacting Infarct Size and Post-Infarction Heart Failure in Mice. Eur. Heart J. 2012, 33, 1964–1974. [Google Scholar] [CrossRef] [PubMed]
- Eash, K.J.; Means, J.M.; White, D.W.; Link, D.C. CXCR4 Is a Key Regulator of Neutrophil Release from the Bone Marrow under Basal and Stress Granulopoiesis Conditions. Blood 2009, 113, 4711–4719. [Google Scholar] [CrossRef]
- Casanova-Acebes, M.; Pitaval, C.; Weiss, L.A.; Nombela-Arrieta, C.; Chèvre, R.; A-González, N.; Kunisaki, Y.; Zhang, D.; van Rooijen, N.; Silberstein, L.E.; et al. Rhythmic Modulation of the Hematopoietic Niche through Neutrophil Clearance. Cell 2013, 153, 1025–1035. [Google Scholar] [CrossRef]
- Abbott, J.D.; Huang, Y.; Liu, D.; Hickey, R.; Krause, D.S.; Giordano, F.J. Stromal Cell–Derived Factor-1α Plays a Critical Role in Stem Cell Recruitment to the Heart After Myocardial Infarction but Is Not Sufficient to Induce Homing in the Absence of Injury. Circulation 2004, 110, 3300–3305. [Google Scholar] [CrossRef]
- Veltman, D.; Wu, M.; Pokreisz, P.; Claus, P.; Gillijns, H.; Caluwé, E.; Vanhaverbeke, M.; Gsell, W.; Himmelreich, U.; Sinnaeve, P.R.; et al. Clec4e-Receptor Signaling in Myocardial Repair After Ischemia-Reperfusion Injury. JACC Basic Transl. Sci. 2021, 6, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Hettwer, J.; Hinterdobler, J.; Miritsch, B.; Deutsch, M.-A.; Li, X.; Mauersberger, C.; Moggio, A.; Braster, Q.; Gram, H.; Robertson, A.A.B.; et al. Interleukin-1b Suppression Dampens Inflammatory Leucocyte Production and Uptake in Atherosclerosis. Cardiovasc. Res. 2022, 118, 2778–2791. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lian, K.; Zhang, L.; Wang, R.; Yi, F.; Gao, C.; Xin, C.; Zhu, D.; Li, Y.; Yan, W.; et al. TXNIP Mediates NLRP3 Inflammasome Activation in Cardiac Microvascular Endothelial Cells as a Novel Mechanism in Myocardial Ischemia/Reperfusion Injury. Basic Res. Cardiol. 2014, 109, 415. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome Activation of Cardiac Fibroblasts Is Essential for Myocardial Ischemia/Reperfusion Injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef]
- Sandanger, Ø.; Ranheim, T.; Vinge, L.E.; Bliksøen, M.; Alfsnes, K.; Finsen, A.V.; Dahl, C.P.; Askevold, E.T.; Florholmen, G.; Christensen, G.; et al. The NLRP3 Inflammasome Is Up-Regulated in Cardiac Fibroblasts and Mediates Myocardial Ischaemia–Reperfusion Injury. Cardiovasc. Res. 2013, 99, 164–174. [Google Scholar] [CrossRef]
- Bujak, M.; Dobaczewski, M.; Chatila, K.; Mendoza, L.H.; Li, N.; Reddy, A.; Frangogiannis, N.G. Interleukin-1 Receptor Type I Signaling Critically Regulates Infarct Healing and Cardiac Remodeling. Am. J. Pathol. 2008, 173, 57–67. [Google Scholar] [CrossRef]
- Saxena, A.; Chen, W.; Su, Y.; Rai, V.; Uche, O.U.; Li, N.; Frangogiannis, N.G. IL-1 Induces Proinflammatory Leukocyte Infiltration and Regulates Fibroblast Phenotype in the Infarcted Myocardium. J. Immunol. 2013, 191, 4838–4848. [Google Scholar] [CrossRef]
- Schunk, S.J.; Triem, S.; Schmit, D.; Zewinger, S.; Sarakpi, T.; Becker, E.; Hütter, G.; Wrublewsky, S.; Küting, F.; Hohl, M.; et al. Interleukin-1α Is a Central Regulator of Leukocyte-Endothelial Adhesion in Myocardial Infarction and in Chronic Kidney Disease. Circulation 2021, 144, 893–908. [Google Scholar] [CrossRef]
- Aukrust, P.; Kleveland, O.; Gullestad, L. Targeting IL-6 Trans-Signaling. JACC Basic Transl. Sci. 2021, 6, 444–446. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The Pro- and Anti-Inflammatory Properties of the Cytokine Interleukin-6. Biochim. Biophys. Acta-Mol. Cell Res. 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Su, J.-H.; Luo, M.-Y.; Liang, N.-; Gong, S.-X.; Chen, W.; Huang, W.-Q.; Tian, Y.; Wang, A.-P. Interleukin-6: A Novel Target for Cardio-Cerebrovascular Diseases. Front. Pharmacol. 2021, 12, 745061. [Google Scholar] [CrossRef] [PubMed]
- George, M.J.; Jasmin, N.H.; Cummings, V.T.; Richard-Loendt, A.; Launchbury, F.; Woollard, K.; Turner-Stokes, T.; Garcia Diaz, A.I.; Lythgoe, M.; Stuckey, D.J.; et al. Selective Interleukin-6 Trans-Signaling Blockade Is More Effective Than Panantagonism in Reperfused Myocardial Infarction. JACC Basic Transl. Sci. 2021, 6, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Kobara, M.; Noda, K.; Kitamura, M.; Okamoto, A.; Shiraishi, T.; Toba, H.; Matsubara, H.; Nakata, T. Antibody against Interleukin-6 Receptor Attenuates Left Ventricular Remodelling after Myocardial Infarction in Mice. Cardiovasc. Res. 2010, 87, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Yuan, Y.-C.; Li, J.-Y.; Gionfriddo, M.R.; Huang, R.-C. Tumor Necrosis Factor-α and Its Role as a Mediator in Myocardial Infarction: A Brief Review. Chronic Dis. Transl. Med. 2015, 1, 18–26. [Google Scholar] [CrossRef]
- Schulz, R.; Heusch, G. Tumor Necrosis Factor-α and Its Receptors 1 and 2: Yin and Yang in Myocardial Infarction? Circulation 2009, 119, 1355–1357. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Lindsey, M.L.; Michael, L.H.; Youker, K.A.; Bressler, R.B.; Mendoza, L.H.; Spengler, R.N.; Smith, C.W.; Entman, M.L. Resident Cardiac Mast Cells Degranulate and Release Preformed TNF-α, Initiating the Cytokine Cascade in Experimental Canine Myocardial Ischemia/Reperfusion. Circulation 1998, 98, 699–710. [Google Scholar] [CrossRef]
- Sun, M.; Dawood, F.; Wen, W.-H.; Chen, M.; Dixon, I.; Kirshenbaum, L.A.; Liu, P.P. Excessive Tumor Necrosis Factor Activation After Infarction Contributes to Susceptibility of Myocardial Rupture and Left Ventricular Dysfunction. Circulation 2004, 110, 3221–3228. [Google Scholar] [CrossRef]
- Kacimi, R.; Karliner, J.S.; Koudssi, F.; Long, C.S. Expression and Regulation of Adhesion Molecules in Cardiac Cells by Cytokines: Response to Acute Hypoxia. Circ. Res. 1998, 82, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Targeting Inflammatory Pathways in Cardiovascular Disease: The Inflammasome, Interleukin-1, Interleukin-6 and Beyond. Cells 2021, 10, 951. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-H.; Do, Y.; Cheong, C.; Koh, H.; Boscardin, S.B.; Oh, Y.-S.; Bozzacco, L.; Trumpfheller, C.; Park, C.G.; Steinman, R.M. Identification of Antigen-Presenting Dendritic Cells in Mouse Aorta and Cardiac Valves. J. Exp. Med. 2009, 206, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Skelly, D.A.; Squiers, G.T.; McLellan, M.A.; Bolisetty, M.T.; Robson, P.; Rosenthal, N.A.; Pinto, A.R. Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep. 2018, 22, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-Renewing Resident Cardiac Macrophages Limit Adverse Remodeling Following Myocardial Infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential Contribution of Monocytes to Heart Macrophages in Steady-State and After Myocardial Infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and Adult-Derived Resident Cardiac Macrophages Are Maintained through Distinct Mechanisms at Steady State and during Inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef]
- Nahrendorf, M. Myeloid Cells in Cardiovascular Organs. J. Intern. Med. 2019, 285, 491–502. [Google Scholar] [CrossRef]
- Hilgendorf, I.; Gerhardt, L.M.S.; Tan, T.C.; Winter, C.; Holderried, T.A.W.; Chousterman, B.G.; Iwamoto, Y.; Liao, R.; Zirlik, A.; Scherer-Crosbie, M.; et al. Ly-6Chigh Monocytes Depend on Nr4a1 to Balance Both Inflammatory and Reparative Phases in the Infarcted Myocardium. Circ. Res. 2014, 114, 1611–1622. [Google Scholar] [CrossRef]
- Puhl, S.-L.; Steffens, S. Neutrophils in Post-Myocardial Infarction Inflammation: Damage vs. Resolution? Front. Cardiovasc. Med. 2019, 6, 25. [Google Scholar] [CrossRef]
- Franck, G.; Mawson, T.L.; Folco, E.J.; Molinaro, R.; Ruvkun, V.; Engelbertsen, D.; Liu, X.; Tesmenitsky, Y.; Shvartz, E.; Sukhova, G.K.; et al. Roles of PAD4 and NETosis in Experimental Atherosclerosis and Arterial Injury: Implications for Superficial Erosion. Circ. Res. 2018, 123, 33–42. [Google Scholar] [CrossRef]
- Savchenko, A.S.; Borissoff, J.I.; Martinod, K.; De Meyer, S.F.; Gallant, M.; Erpenbeck, L.; Brill, A.; Wang, Y.; Wagner, D.D. VWF-Mediated Leukocyte Recruitment with Chromatin Decondensation by PAD4 Increases Myocardial Ischemia/Reperfusion Injury in Mice. Blood 2014, 123, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Vafadarnejad, E.; Rizzo, G.; Krampert, L.; Arampatzi, P.; Arias-Loza, A.-P.; Nazzal, Y.; Rizakou, A.; Knochenhauer, T.; Bandi, S.R.; Nugroho, V.A.; et al. Dynamics of Cardiac Neutrophil Diversity in Murine Myocardial Infarction. Circ. Res. 2020, 127. [Google Scholar] [CrossRef] [PubMed]
- Calcagno, D.M.; Zhang, C.; Toomu, A.; Huang, K.; Ninh, V.K.; Miyamoto, S.; Aguirre, A.D.; Fu, Z.; Heller Brown, J.; King, K.R. SiglecF(HI) Marks Late-Stage Neutrophils of the Infarcted Heart: A Single-Cell Transcriptomic Analysis of Neutrophil Diversification. J. Am. Heart Assoc. 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Khoyratty, T.E.; Ai, Z.; Ballesteros, I.; Eames, H.L.; Mathie, S.; Martín-Salamanca, S.; Wang, L.; Hemmings, A.; Willemsen, N.; von Werz, V.; et al. Distinct Transcription Factor Networks Control Neutrophil-Driven Inflammation. Nat. Immunol. 2021, 22, 1093–1106. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.-M.; Weinheimer, C.; et al. Tissue Resident CCR2− and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ. Res. 2019, 124, 263–278. [Google Scholar] [CrossRef]
- Farbehi, N.; Patrick, R.; Dorison, A.; Xaymardan, M.; Janbandhu, V.; Wystub-Lis, K.; Ho, J.W.; Nordon, R.E.; Harvey, R.P. Single-Cell Expression Profiling Reveals Dynamic Flux of Cardiac Stromal, Vascular and Immune Cells in Health and Injury. eLife 2019, 8, e43882. [Google Scholar] [CrossRef] [PubMed]
- King, K.R.; Aguirre, A.D.; Ye, Y.-X.; Sun, Y.; Roh, J.D.; Ng, R.P.; Kohler, R.H.; Arlauckas, S.P.; Iwamoto, Y.; Savol, A.; et al. IRF3 and Type I Interferons Fuel a Fatal Response to Myocardial Infarction. Nat. Med. 2017, 23, 1481–1487. [Google Scholar] [CrossRef]
- Zhuang, L.; Lu, L.; Zhang, R.; Chen, K.; Yan, X. Comprehensive Integration of Single-Cell Transcriptional Profiling Reveals the Heterogeneities of Non-Cardiomyocytes in Healthy and Ischemic Hearts. Front. Cardiovasc. Med. 2020, 7, 615161. [Google Scholar] [CrossRef]
- Silvis, M.J.M.; Kaffka genaamd Dengler, S.E.; Odille, C.A.; Mishra, M.; van der Kaaij, N.P.; Doevendans, P.A.; Sluijter, J.P.G.; de Kleijn, D.P.V.; de Jager, S.C.A.; Bosch, L.; et al. Damage-Associated Molecular Patterns in Myocardial Infarction and Heart Transplantation: The Road to Translational Success. Front. Immunol. 2020, 11, 599511. [Google Scholar] [CrossRef]
- Cai, Z.; Xie, Q.; Hu, T.; Yao, Q.; Zhao, J.; Wu, Q.; Tang, Q. S100A8/A9 in Myocardial Infarction: A Promising Biomarker and Therapeutic Target. Front. Cell Dev. Biol. 2020, 8, 603902. [Google Scholar] [CrossRef]
- Epelman, S.; Liu, P.P.; Mann, D.L. Role of Innate and Adaptive Immune Mechanisms in Cardiac Injury and Repair. Nat. Rev. Immunol. 2015, 15, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Wahid, A.; Chen, W.; Wang, X.; Tang, X. High-Mobility Group Box 1 Serves as an Inflammation Driver of Cardiovascular Disease. Biomed. Pharmacother. 2021, 139, 111555. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Anzai, T.; Naito, K.; Miyasho, T.; Okamoto, M.; Yokota, H.; Yamada, S.; Maekawa, Y.; Takahashi, T.; Yoshikawa, T.; et al. Role of High-Mobility Group Box 1 Protein in Post-Infarction Healing Process and Left Ventricular Remodelling. Cardiovasc. Res. 2008, 81, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Charles, E.J.; Yan, Z.; Wu, D.; French, B.A.; Kron, I.L.; Yang, Z. The Myocardial Infarct-Exacerbating Effect of Cell-Free DNA Is Mediated by the High-Mobility Group Box 1–Receptor for Advanced Glycation End Products–Toll-like Receptor 9 Pathway. J. Thorac. Cardiovasc. Surg. 2019, 157, 2256–2269.e3. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Yellon, D.M.; Davidson, S.M. The Role of Extracellular DNA and Histones in Ischaemia-Reperfusion Injury of the Myocardium. Cardiovasc. Drugs 2020, 34, 123–131. [Google Scholar] [CrossRef]
- Qin, C.-Y.; Zhang, H.-W.; Gu, J.; Xu, F.; Liang, H.-M.; Fan, K.-J.; Shen, J.-Y.; Xiao, Z.-H.; Zhang, E.-Y.; Hu, J. Mitochondrial DNA-Induced Inflammatory Damage Contributes to Myocardial Ischemia Reperfusion Injury in Rats: Cardioprotective Role of Epigallocatechin. Mol. Med. Rep. 2017, 16, 7569–7576. [Google Scholar] [CrossRef]
- Sreejit, G.; Abdel-Latif, A.; Athmanathan, B.; Annabathula, R.; Dhyani, A.; Noothi, S.K.; Quaife-Ryan, G.A.; Al-Sharea, A.; Pernes, G.; Dragoljevic, D.; et al. Neutrophil-Derived S100A8/A9 Amplify Granulopoiesis After Myocardial Infarction. Circulation 2020, 141, 1080–1094. [Google Scholar] [CrossRef]
- Marinković, G.; Grauen Larsen, H.; Yndigegn, T.; Szabo, I.A.; Mares, R.G.; de Camp, L.; Weiland, M.; Tomas, L.; Goncalves, I.; Nilsson, J.; et al. Inhibition of Pro-Inflammatory Myeloid Cell Responses by Short-Term S100A9 Blockade Improves Cardiac Function after Myocardial Infarction. Eur. Heart J. 2019, 40, 2713–2723. [Google Scholar] [CrossRef]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils Orchestrate Post-Myocardial Infarction Healing by Polarizing Macrophages towards a Reparative Phenotype. Eur. Heart J. 2016, 38, 187–197. [Google Scholar] [CrossRef]
- Chen, B.; Huang, S.; Su, Y.; Wu, Y.-J.; Hanna, A.; Brickshawana, A.; Graff, J.; Frangogiannis, N.G. Macrophage Smad3 Protects the Infarcted Heart, Stimulating Phagocytosis and Regulating Inflammation. Circ. Res. 2019, 125, 55–70. [Google Scholar] [CrossRef]
- Daseke, M.J.; Tenkorang-Impraim, M.A.A.; Ma, Y.; Chalise, U.; Konfrst, S.R.; Garrett, M.R.; DeLeon-Pennell, K.Y.; Lindsey, M.L. Exogenous IL-4 Shuts off pro-Inflammation in Neutrophils While Stimulating Anti-Inflammation in Macrophages to Induce Neutrophil Phagocytosis Following Myocardial Infarction. J. Mol. Cell. Cardiol. 2020, 145, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Huse, C.; Anstensrud, A.K.; Michelsen, A.E.; Ueland, T.; Broch, K.; Woxholt, S.; Yang, K.; Sharma, K.; Tøllefsen, I.M.; Bendz, B.; et al. Interleukin-6 Inhibition in ST-Elevation Myocardial Infarction: Immune Cell Profile in the Randomised ASSAIL-MI Trial. eBioMedicine 2022, 80, 104013. [Google Scholar] [CrossRef] [PubMed]
- Imazio, M.; Nidorf, M. Colchicine and the Heart. Eur. Heart J. 2021, 42, 2745–2760. [Google Scholar] [CrossRef]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Bouabdallaoui, N.; Tardif, J.-C.; Waters, D.D.; Pinto, F.J.; Maggioni, A.P.; Diaz, R.; Berry, C.; Koenig, W.; Lopez-Sendon, J.; Gamra, H.; et al. Time-to-Treatment Initiation of Colchicine and Cardiovascular Outcomes after Myocardial Infarction in the Colchicine Cardiovascular Outcomes Trial (COLCOT). Eur. Heart J. 2020, 41, 4092–4099. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef]
- Meyer-Lindemann, U.; Mauersberger, C.; Schmidt, A.-C.; Moggio, A.; Hinterdobler, J.; Li, X.; Khangholi, D.; Hettwer, J.; Gräßer, C.; Dutsch, A.; et al. Colchicine Impacts Leukocyte Trafficking in Atherosclerosis and Reduces Vascular Inflammation. Front. Immunol. 2022, 13, 898690. [Google Scholar] [CrossRef]
- D’Amario, D.; Cappetta, D.; Cappannoli, L.; Princi, G.; Migliaro, S.; Diana, G.; Chouchane, K.; Borovac, J.A.; Restivo, A.; Arcudi, A.; et al. Colchicine in Ischemic Heart Disease: The Good, the Bad and the Ugly. Clin. Res. Cardiol. 2021, 110, 1531–1542. [Google Scholar] [CrossRef]
- Ridker, P.M. From RESCUE to ZEUS: Will Interleukin-6 Inhibition with Ziltivekimab Prove Effective for Cardiovascular Event Reduction? Cardiovasc. Res. 2021, 117, e138–e140. [Google Scholar] [CrossRef]
- Ridker, P.M.; Devalaraja, M.; Baeres, F.M.M.; Engelmann, M.D.M.; Hovingh, G.K.; Ivkovic, M.; Lo, L.; Kling, D.; Pergola, P.; Raj, D.; et al. IL-6 Inhibition with Ziltivekimab in Patients at High Atherosclerotic Risk (RESCUE): A Double-Blind, Randomised, Placebo-Controlled, Phase 2 Trial. Lancet 2021, 397, 2060–2069. [Google Scholar] [CrossRef]
- Goldberger, J.J.; Subačius, H.; Marroquin, O.C.; Beau, S.L.; Simonson, J.; Desai, P.; Betzen, M.; DeLuna, D.; Whitehill, J.; Hatch, J.; et al. One-Year Landmark Analysis of the Effect of Beta-Blocker Dose on Survival After Acute Myocardial Infarction. J. Am. Heart Assoc. 2021, 10, e019017. [Google Scholar] [CrossRef] [PubMed]
- García-Prieto, J.; Villena-Gutiérrez, R.; Gómez, M.; Bernardo, E.; Pun-García, A.; García-Lunar, I.; Crainiciuc, G.; Fernández-Jiménez, R.; Sreeramkumar, V.; Bourio-Martínez, R.; et al. Neutrophil Stunning by Metoprolol Reduces Infarct Size. Nat. Commun. 2017, 8, 14780. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Moragón, A.; Gómez, M.; Villena-Gutiérrez, R.; Lalama, D.V.; García-Prieto, J.; Martínez, F.; Sánchez-Cabo, F.; Fuster, V.; Oliver, E.; Ibáñez, B. Metoprolol Exerts a Non-Class Effect against Ischaemia–Reperfusion Injury by Abrogating Exacerbated Inflammation. Eur. Heart J. 2020, 41, 4425–4440. [Google Scholar] [CrossRef]
- Leone, A.M.; D’Amario, D.; Cannata, F.; Graziani, F.; Borovac, J.A.; Leone, G.; De Stefano, V.; Basile, E.; Siracusano, A.; Galiuto, L.; et al. The Effects of Granulocyte Colony-Stimulating Factor in Patients with a Large Anterior Wall Acute Myocardial Infarction to Prevent Left Ventricular Remodeling: A 10-Year Follow-Up of the RIGENERA Study. J. Clin. Med. 2020, 9, 1214. [Google Scholar] [CrossRef] [PubMed]
- Leone, A.M.; Galiuto, L.; Garramone, B.; Rutella, S.; Giannico, M.B.; Brugaletta, S.; Perfetti, M.; Liuzzo, G.; Porto, I.; Burzotta, F.; et al. Usefulness of Granulocyte Colony-Stimulating Factor in Patients With a Large Anterior Wall Acute Myocardial Infarction to Prevent Left Ventricular Remodeling (The Rigenera Study). Am. J. Cardiol. 2007, 100, 397–403. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
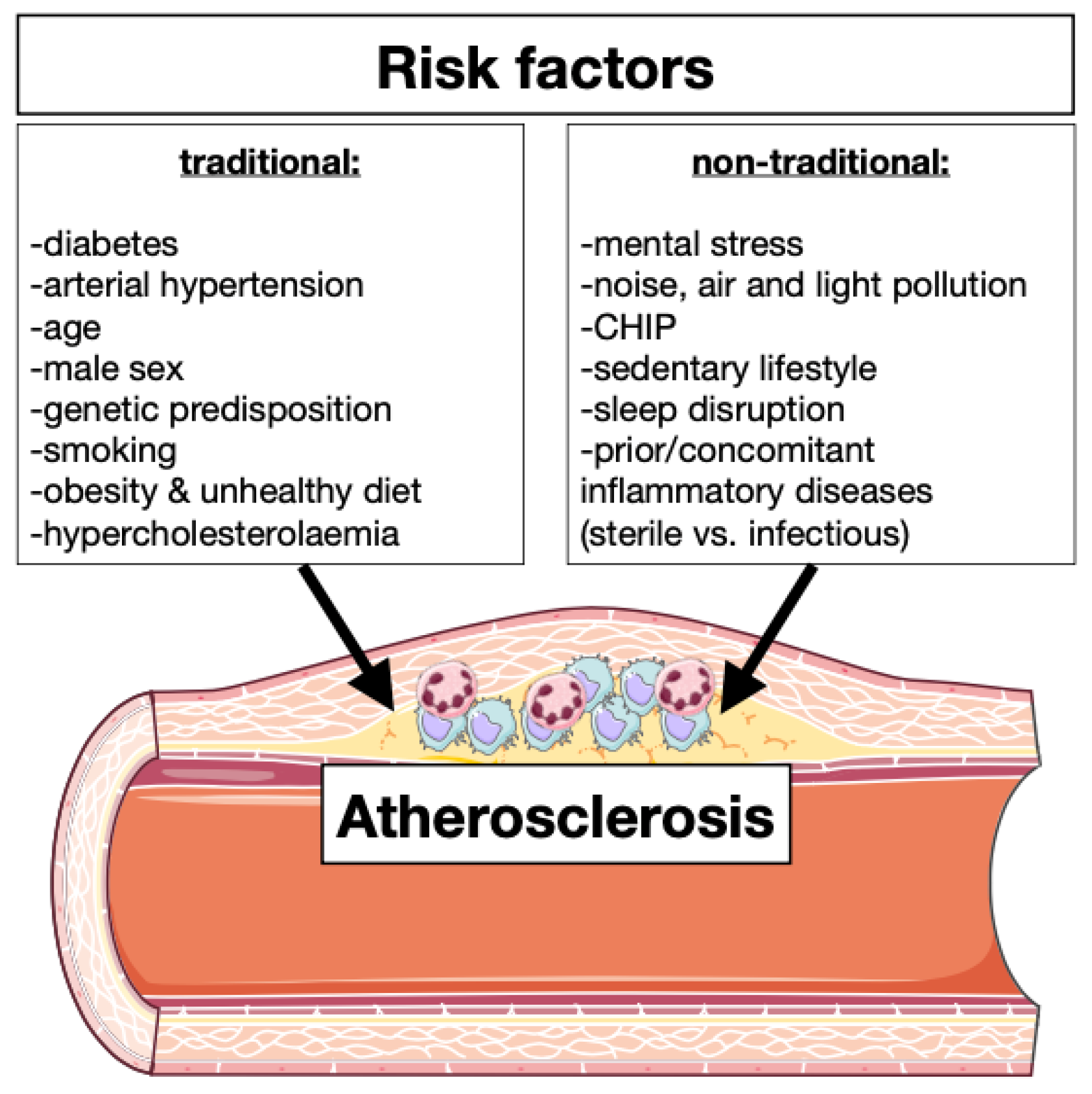
| Traditional Risk Factors | Effects on Immune System |
|---|---|
| Diabetes | Hyperglycemia leads to enhanced myelopoiesis [9] |
| Arterial hypertension | Increased quantity of circulating leukocytes and cytokine concentration [10] |
| Age | Clonal hematopoiesis, enhanced inflammation and endothelial disfunction [11] |
| Male sex | Testosterone induced inflammation and sexual immune dimorphism [12] |
| Genetic predisposition | Immunity modulation and leukocytes production [7] |
| Smoking | Leukocytes production and inflammasome activation [13] |
| Obesity and unhealthy diet | Adipose tissue macrophages induce leukocyte production [14] |
| Hypercholesterolemia | Production of foam cells, activates inflammasomes, enhanced immune signaling and oxidative stress [15] |
| Non-Traditional Risk Factors | |
| Mental stress | Leukocytes expansion and infiltration [5,8] |
| Noise, air, and light pollution | Oxidative stress, enhanced inflammation and endothelial disfunction [16,17] |
| CHIP | Increased of circulating leukocytes [11,18] |
| Sedentary life | Lower level of myokine and possible regulation of immunity [11,19] |
| Sleep disruption | Chronic systemic low-grade inflammation [20] |
| Prior/concomitant inflammatory disease | Modulation of leukocyte production, phenotype and activation [21] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moggio, A.; Schunkert, H.; Kessler, T.; Sager, H.B. Quo Vadis? Immunodynamics of Myeloid Cells after Myocardial Infarction. Int. J. Mol. Sci. 2022, 23, 15814. https://doi.org/10.3390/ijms232415814
Moggio A, Schunkert H, Kessler T, Sager HB. Quo Vadis? Immunodynamics of Myeloid Cells after Myocardial Infarction. International Journal of Molecular Sciences. 2022; 23(24):15814. https://doi.org/10.3390/ijms232415814
Chicago/Turabian StyleMoggio, Aldo, Heribert Schunkert, Thorsten Kessler, and Hendrik B. Sager. 2022. "Quo Vadis? Immunodynamics of Myeloid Cells after Myocardial Infarction" International Journal of Molecular Sciences 23, no. 24: 15814. https://doi.org/10.3390/ijms232415814
APA StyleMoggio, A., Schunkert, H., Kessler, T., & Sager, H. B. (2022). Quo Vadis? Immunodynamics of Myeloid Cells after Myocardial Infarction. International Journal of Molecular Sciences, 23(24), 15814. https://doi.org/10.3390/ijms232415814