
When Two Maladies Meet: Disease Burden and Pathophysiology of Stroke in Cancer



Abstract
:1. Introduction
2. The Burden of Stroke and Cancer
2.1. Burden of Stroke
2.2. Burden of Cancer
3. Risk Factors for Stroke in Cancer
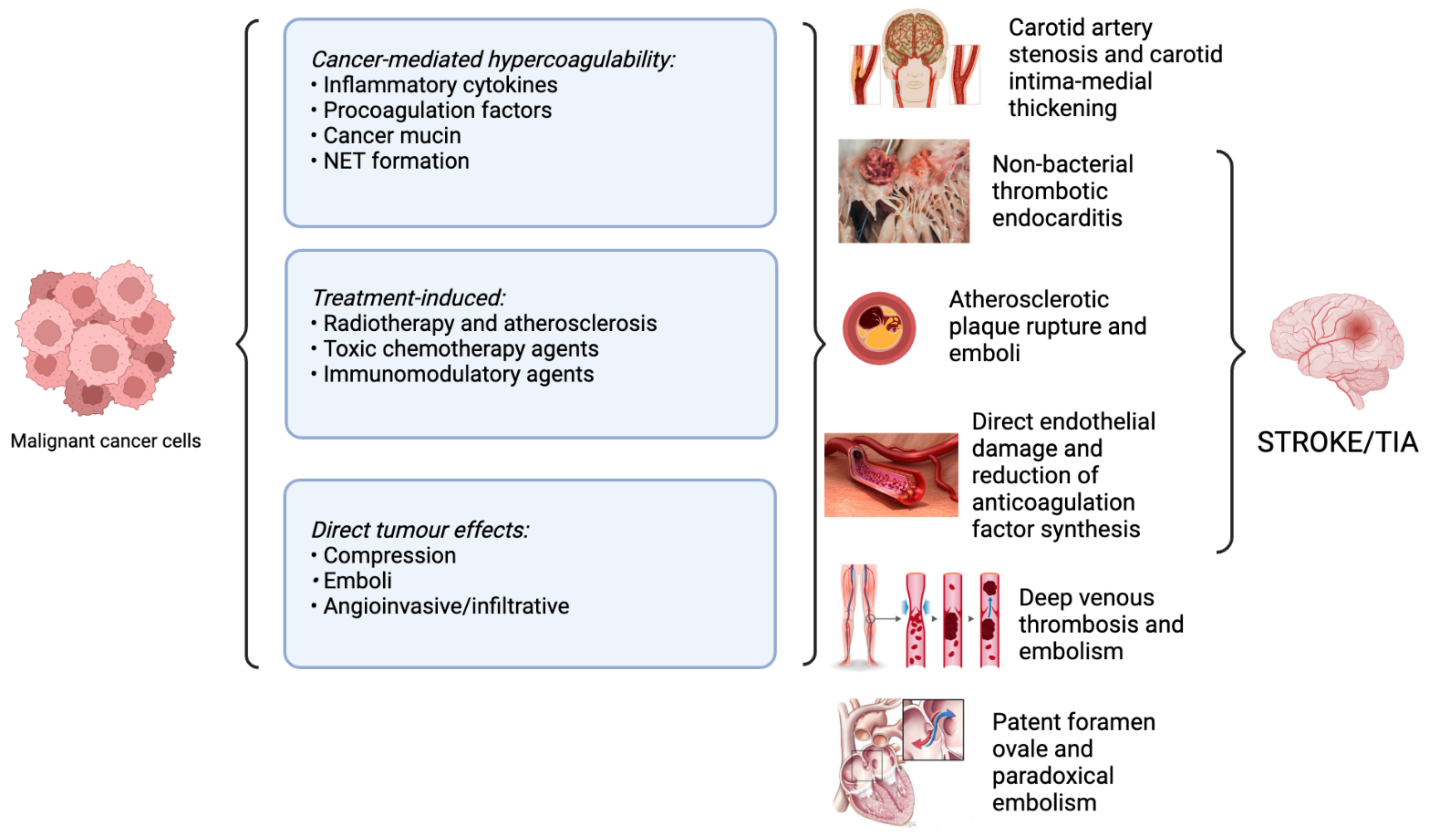
4. Pathophysiological Mechanisms Underlying Stroke in Cancer
4.1. Direct Impact of the Tumour
4.2. Compression
4.3. Emboli
4.4. Angioinvasive/Infiltrative
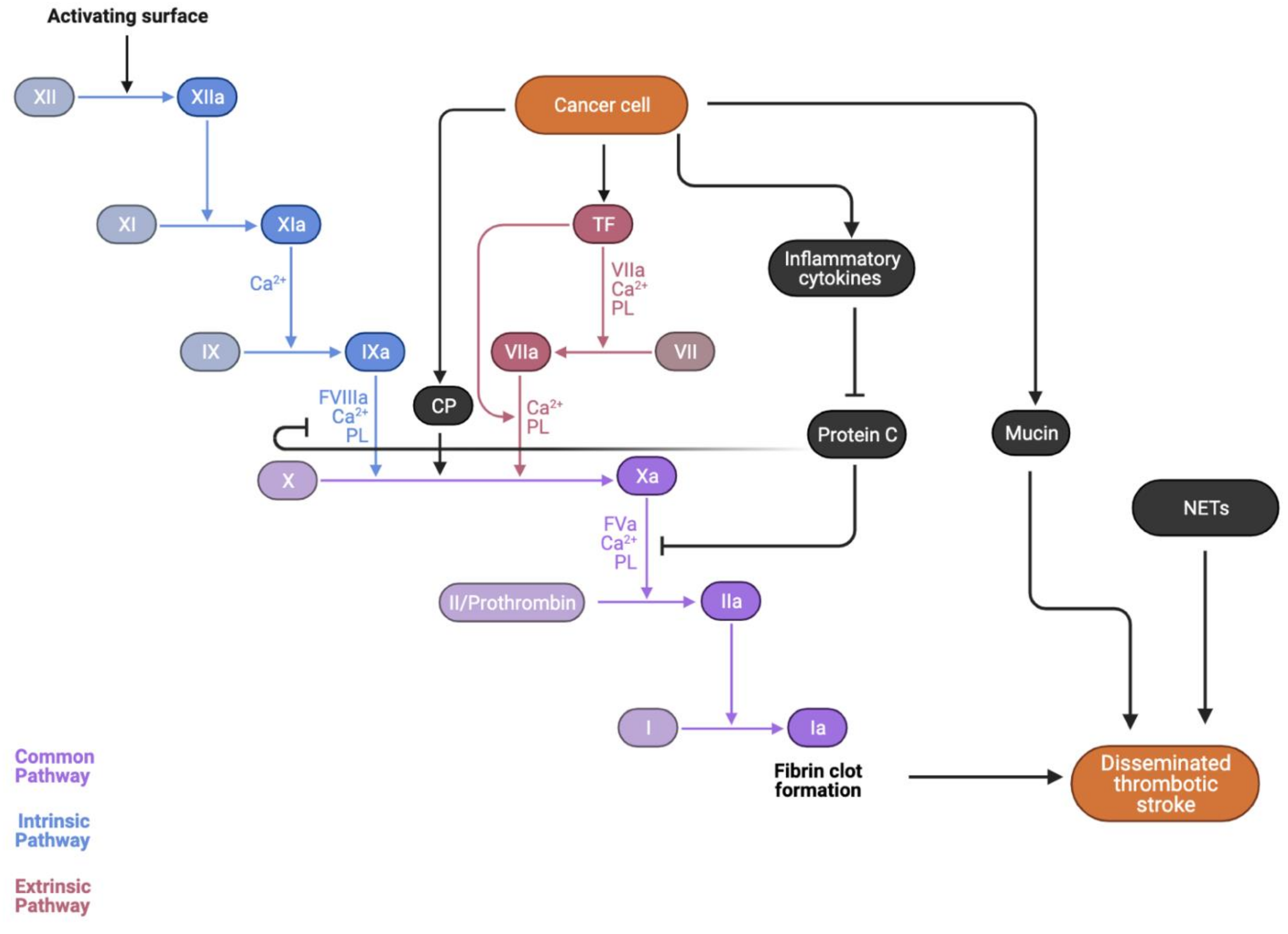
4.5. Cancer-Mediated Hypercoagulability
4.6. Inflammatory Cytokines
4.7. Procoagulation Factors
4.8. Angiogenesis Inhibitors
4.9. Cancer Mucin
4.10. Non-bacterial Thrombotic Endocarditis (NBTE)
4.11. Neutrophil Extracellular Trap (NET) Formation
4.12. Paradoxical Embolism and Patent Foramen Ovale (PFO)
5. Cancer Treatments and Stroke Risk
5.1. Radiotherapy and Stroke
5.2. Chemotherapy and Stroke
5.2.1. L-Asparaginase (L-Asp)
5.2.2. Cisplatin
5.2.3. Fluorouracil (5-FU)
5.2.4. Immunomodulatory Drugs
6. Timing and Type of Stroke
7. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar]
- Feigin, V.L.; Stark, B.A.; Johnson, C.O.; Roth, G.A.; Bisignano, C.; Abady, G.G.; Abbasifard, M.; Abbasi-Kangevari, M.; Abd-Allah, F.; Abedi, V.; et al. Global, regional, and national burden of stroke and its risk factors, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021, 20, 795–820. [Google Scholar]
- Deloitte Australia. The Economic Impact of Stroke in Australia: Estimating the Magnitude and Impacts of Stroke in Australia; Deloitte Australia: Sydney, Australia, 2020. [Google Scholar]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar]
- Union for International Cancer Control. The Economics of Cancer Prevention & Control; Union for International Cancer Control: Geneva, Switzerland, 2014. [Google Scholar]
- Gujral, D.M.; Chahal, N.; Senior, R.; Harrington, K.J.; Nutting, C.M. Radiation-induced carotid artery atherosclerosis. Radiother. Oncol. 2014, 110, 31–38. [Google Scholar]
- Koene, R.J.; Prizment, A.E.; Blaes, A.; Konety, S.H. Shared Risk Factors in Cardiovascular Disease and Cancer. Circulation 2016, 133, 1104–1114. [Google Scholar]
- Lanas, F.; Seron, P. Facing the stroke burden worldwide. Lancet Glob. Health 2021, 9, e235–e236. [Google Scholar]
- Kamp, D.W.; Shacter, E.; Weitzman, S.A. Chronic inflammation and cancer: The role of the mitochondria. Oncology 2011, 25, 400–410, 413. [Google Scholar]
- Australian Institute of Health and Welfare. Stroke; Australian Institute of Health and Welfare: Canberra, Australia, 2020. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar]
- Venketasubramanian, N. Stroke Epidemiology in Oceania: A Review. Neuroepidemiology 2021, 55, 1–10. [Google Scholar]
- Tripodi, A. D-dimer testing in laboratory practice. Clin. Chem. 2011, 57, 1256–1262. [Google Scholar]
- Luengo-Fernandez, R.; Violato, M.; Candio, P.; Leal, J. Economic burden of stroke across Europe: A population-based cost analysis. Eur. Stroke J. 2020, 5, 17–25. [Google Scholar]
- Girotra, T.; Lekoubou, A.; Bishu, K.G.; Ovbiagele, B. A contemporary and comprehensive analysis of the costs of stroke in the United States. J. Neurol. Sci. 2020, 410, 116643. [Google Scholar]
- Australian Institute of Health and Welfare. Cancer in Australia 2021; Australian Institute of Health and Welfare: Canberra, Australia, 2021. [Google Scholar]
- Global Burden of Disease Cancer Collaboration. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2019, 5, 1749–1768. [Google Scholar]
- Shah, S.C.; Kayamba, V.; Peek, R.M., Jr.; Heimburger, D. Cancer Control in Low- and Middle-Income Countries: Is It Time to Consider Screening? J. Glob. Oncol. 2019, 5, 1–8. [Google Scholar]
- Goldsbury, D.E.; Yap, S.; Weber, M.F.; Veerman, L.; Rankin, N.; Banks, E.; Canfell, K.; O’Connell, D.L. Health services costs for cancer care in Australia: Estimates from the 45 and Up Study. PLoS ONE 2018, 13, e0201552. [Google Scholar]
- Hofmarcher, T.; Lindgren, P.; Wilking, N.; Jonsson, B. The cost of cancer in Europe 2018. Eur. J. Cancer 2020, 129, 41–49. [Google Scholar]
- Guy, G.P., Jr.; Ekwueme, D.U.; Yabroff, K.R.; Dowling, E.C.; Li, C.; Rodriguez, J.L.; de Moor, J.S.; Virgo, K.S. Economic burden of cancer survivorship among adults in the United States. J. Clin. Oncol. 2013, 31, 3749–3757. [Google Scholar]
- Bradley, C.J.; Yabroff, K.R.; Dahman, B.; Feuer, E.J.; Mariotto, A.; Brown, M.L. Productivity costs of cancer mortality in the United States: 2000–2020. J. Natl. Cancer Inst. 2008, 100, 1763–1770. [Google Scholar]
- Zaorsky, N.G.; Zhang, Y.; Tchelebi, L.T.; Mackley, H.B.; Chinchilli, V.M.; Zacharia, B.E. Stroke among cancer patients. Nat. Commun. 2019, 10, 5172. [Google Scholar]
- Krishnamurthi, R.V.; Ikeda, T.; Feigin, V.L. Global, Regional and Country-Specific Burden of Ischaemic Stroke, Intracerebral Haemorrhage and Subarachnoid Haemorrhage: A Systematic Analysis of the Global Burden of Disease Study 2017. Neuroepidemiology 2020, 54, 171–179. [Google Scholar]
- Pandian, J.D.; William, A.G.; Kate, M.P.; Norrving, B.; Mensah, G.A.; Davis, S.; Roth, G.A.; Thrift, A.G.; Kengne, A.P.; Kissela, B.M.; et al. Strategies to Improve Stroke Care Services in Low- and Middle-Income Countries: A Systematic Review. Neuroepidemiology 2017, 49, 45–61. [Google Scholar]
- Gardiner, F.W.; Rallah-Baker, K.; Dos Santos, A.; Sharma, P.; Churilov, L.; Donnan, G.A.; Davis, S.M.; Quinlan, F.; Worley, P. Indigenous Australians have a greater prevalence of heart, stroke, and vascular disease, are younger at death, with higher hospitalisation and more aeromedical retrievals from remote regions. eClinicalMedicine 2021, 42, 101181. [Google Scholar]
- Bhaskar, S.; Thomas, P.; Cheng, Q.; Clement, N.; McDougall, A.; Hodgkinson, S.; Cordato, D. Trends in acute stroke presentations to an emergency department: Implications for specific communities in accessing acute stroke care services. Postgrad. Med. J. 2019, 95, 258–264. [Google Scholar]
- Venkat, A.; Cappelen-Smith, C.; Askar, S.; Thomas, P.R.; Bhaskar, S.; Tam, A.; McDougall, A.J.; Hodgkinson, S.J.; Cordato, D.J. Factors Associated with Stroke Misdiagnosis in the Emergency Department: A Retrospective Case-Control Study. Neuroepidemiology 2018, 51, 123–127. [Google Scholar]
- Santana Baskar, P.; Cordato, D.; Wardman, D.; Bhaskar, S. In-hospital acute stroke workflow in acute stroke—Systems-based approaches. Acta Neurol. Scand. 2021, 143, 111–120. [Google Scholar]
- Chowdhury, S.Z.; Baskar, P.S.; Bhaskar, S. Effect of prehospital workflow optimization on treatment delays and clinical outcomes in acute ischemic stroke: A systematic review and meta-analysis. Acad. Emerg. Med. 2021, 28, 781–801. [Google Scholar]
- Australian Institute of Health and Welfare. Health System Expenditure on Cancer and Other Neoplasms in Australia, 2015–2016; Australian Institute of Health and Welfare: Canberra, Australia, 2021. [Google Scholar]
- Diaz, A.; Whop, L.J.; Valery, P.C.; Moore, S.P.; Cunningham, J.; Garvey, G.; Condon, J.R. Cancer outcomes for Aboriginal and Torres Strait Islander Australians in rural and remote areas. Aust. J. Rural. Health 2015, 23, 4–18. [Google Scholar]
- World Health Organization. Global Action Plan for the Prevention and Control of NCDs 2013–2020; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- Wells, C.R.; Galvani, A.P. Impact of the COVID-19 pandemic on cancer incidence and mortality. Lancet Public Health 2022, 7, e490–e491. [Google Scholar]
- Gourd, E. COVID-19 pandemic causes cervical cancer screening crisis. Lancet Oncol. 2021, 22, 1060. [Google Scholar]
- Bhaskar, S.; Rastogi, A.; Menon, K.V.; Kunheri, B.; Balakrishnan, S.; Howick, J. Call for Action to Address Equity and Justice Divide During COVID-19. Front. Psychiatry 2020, 11, 559905. [Google Scholar]
- Libby, P. Inflammation and cardiovascular disease mechanisms. Am. J. Clin. Nutr. 2006, 83, 456s–460s. [Google Scholar]
- Dearborn, J.L.; Urrutia, V.C.; Zeiler, S.R. Stroke and Cancer- A Complicated Relationship. J. Neurol. Transl. Neurosci. 2014, 2, 1039. [Google Scholar]
- Rogers, L.R. Cerebrovascular complications in patients with cancer. In Seminars in Neurology; Thieme Medical Publishers: Leipzig, Germany, 2010; Volume 30, pp. 311–319. [Google Scholar]
- Bick, R.L. Cancer-associated thrombosis. N. Engl. J. Med. 2003, 349, 109–111. [Google Scholar]
- Schwarzbach, C.J.; Schaefer, A.; Ebert, A.; Held, V.; Bolognese, M.; Kablau, M.; Hennerici, M.G.; Fatar, M. Stroke and cancer: The importance of cancer-associated hypercoagulation as a possible stroke etiology. Stroke 2012, 43, 3029–3034. [Google Scholar]
- Savarapu, P.; Abdelazeem, B.; Isa, S.; Baral, N.; Hassan, M. Cancer-Related Non-Bacterial Thrombotic Endocarditis Presenting as Acute Ischemic Stroke. Cureus 2021, 13, e14953. [Google Scholar]
- Dardiotis, E.; Aloizou, A.M.; Markoula, S.; Siokas, V.; Tsarouhas, K.; Tzanakakis, G.; Libra, M.; Kyritsis, A.P.; Brotis, A.G.; Aschner, M.; et al. Cancer-associated stroke: Pathophysiology, detection and management (Review). Int. J. Oncol. 2019, 54, 779–796. [Google Scholar]
- Kitano, T.; Sasaki, T.; Gon, Y.; Todo, K.; Okazaki, S.; Kitamura, T.; Kitamura, Y.; Sakaguchi, M.; Sobue, T.; Matsumura, Y.; et al. The Effect of Chemotherapy on Stroke Risk in Cancer Patients. Thromb. Haemost. 2020, 120, 714–723. [Google Scholar]
- Jiang, D.; Lee, A.I. Thrombotic Risk from Chemotherapy and Other Cancer Therapies. In Thrombosis and Hemostasis in Cancer; Soff, G., Ed.; Springer: Berlin/Heidelberg, Germany, 2019; pp. 87–101. [Google Scholar]
- Katz, J.M.; Segal, A.Z. Incidence and etiology of cerebrovascular disease in patients with malignancy. Curr. Atheroscler. Rep. 2005, 7, 280–288. [Google Scholar]
- Grisold, W.; Oberndorfer, S.; Struhal, W. Stroke and cancer: A review. Acta Neurol. Scand. 2009, 119, 1–16. [Google Scholar]
- Kase, C.S. Intracerebral hemorrhage: Non-hypertensive causes. Stroke 1986, 17, 590–595. [Google Scholar]
- Navi, B.B.; Kasner, S.E.; Elkind, M.S.V.; Cushman, M.; Bang, O.Y.; DeAngelis, L.M. Cancer and Embolic Stroke of Undetermined Source. Stroke 2021, 52, 1121–1130. [Google Scholar]
- Woock, M.; Martinez-Majander, N.; Seiffge, D.J.; Selvik, H.A.; Nordanstig, A.; Redfors, P.; Lindgren, E.; Sanchez van Kammen, M.; Rentzos, A.; Coutinho, J.M.; et al. Cancer and stroke: Commonly encountered by clinicians, but little evidence to guide clinical approach. Ther. Adv. Neurol. Disord. 2022, 15, 17562864221106362. [Google Scholar]
- Holmøy, T.; Nakstad, P.H.; Fredø, H.L.; Kumar, T. Intravascular Large B-Cell Lymphoma Presenting as Cerebellar and Cerebral Infarction. Arch. Neurol. 2007, 64, 754–755. [Google Scholar]
- Ravindran, A.V.; Killingsworth, M.C.; Bhaskar, S. Cerebral collaterals in acute ischaemia: Implications for acute ischaemic stroke patients receiving reperfusion therapy. Eur. J. Neurosci. 2021, 53, 1238–1261. [Google Scholar]
- Huang, J.C.; Bhaskar, S.M.M. Clot Morphology in Acute Ischemic Stroke Decision Making. Int. J. Mol. Sci. 2022, 23, 12373. [Google Scholar] [CrossRef]
- Schwarzbach, C.J.; Fatar, M.; Eisele, P.; Ebert, A.D.; Hennerici, M.G.; Szabo, K. DWI Lesion Patterns in Cancer-Related Stroke—Specifying the Phenotype. Cerebrovasc. Dis. Extra 2015, 5, 139–145. [Google Scholar]
- Caine, G.J.; Stonelake, P.S.; Lip, G.Y.; Kehoe, S.T. The hypercoagulable state of malignancy: Pathogenesis and current debate. Neoplasia 2002, 4, 465–473. [Google Scholar]
- Dahlbäck, B.; Villoutreix, B.O. Regulation of Blood Coagulation by the Protein C Anticoagulant Pathway. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1311–1320. [Google Scholar]
- Dittman, W.A.; Majerus, P.W. Structure and function of thrombomodulin: A natural anticoagulant. Blood 1990, 75, 329–336. [Google Scholar]
- Nemerson, Y. The tissue factor pathway of blood coagulation. In Seminars in Hematology; Elsevier: Amsterdam, The Netherlands, 1992; Volume 29, pp. 170–176. [Google Scholar]
- Gordon, S.G.; Cross, B.A. An Enzyme-linked Immunosorbent Assay for Cancer Procoagulant and Its Potential as a New Tumor Marker. Cancer Res. 1990; 50, 6229–6234. [Google Scholar]
- Conti, E.; Romiti, A.; Musumeci, M.B.; Passerini, J.; Zezza, L.; Mastromarino, V.; D’Antonio, C.; Marchetti, P.; Paneni, F.; Autore, C.; et al. Arterial thrombotic events and acute coronary syndromes with cancer drugs: Are growth factors the missed link? What both cardiologist and oncologist should know about novel angiogenesis inhibitors. Int. J. Cardiol. 2013, 167, 2421–2429. [Google Scholar]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar]
- Choueiri, T.K.; Schutz, F.A.; Je, Y.; Rosenberg, J.E.; Bellmunt, J. Risk of arterial thromboembolic events with sunitinib and sorafenib: A systematic review and meta-analysis of clinical trials. J. Clin. Oncol. 2010, 28, 2280–2285. [Google Scholar]
- Schmidinger, M.; Zielinski, C.C.; Vogl, U.M.; Bojic, A.; Bojic, M.; Schukro, C.; Ruhsam, M.; Hejna, M.; Schmidinger, H. Cardiac toxicity of sunitinib and sorafenib in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2008, 26, 5204–5212. [Google Scholar]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Staehler, M.; Negrier, S.; Chevreau, C.; Desai, A.A.; Rolland, F.; et al. Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J. Clin. Oncol. 2009, 27, 3312–3318. [Google Scholar]
- Bombeli, T.; Karsan, A.; Tait, J.F.; Harlan, J.M. Apoptotic vascular endothelial cells become procoagulant. Blood 1997, 89, 2429–2442. [Google Scholar]
- Conti, E.; Musumeci, M.B.; De Giusti, M.; Dito, E.; Mastromarino, V.; Autore, C.; Volpe, M. IGF-1 and atherothrombosis: Relevance to pathophysiology and therapy. Clin. Sci. 2011, 120, 377–402. [Google Scholar]
- De Cicco, M. The prothrombotic state in cancer: Pathogenic mechanisms. Crit. Rev. Oncol./Hematol. 2004, 50, 187–196. [Google Scholar]
- Merten, M.; Thiagarajan, P. P-selectin in arterial thrombosis. Z. Kardiol. 2004, 93, 855–863. [Google Scholar]
- Mitma, A.A.; Varghese, J.G.; Witt, D.; Zarich, S.W. Stroke and a valvular lesion in a patient with stage IV non-small cell lung cancer. BMJ Case Rep. 2016, 2016, bcr2016215317. [Google Scholar]
- Morimoto, S.; Tanaka, J.; Saito, Y.; Tsuyama, N.; Nishimura, T.; Komiya, T.; Kyo, S.; Arai, T.; Kanemaru, A.; Kanemaru, K.; et al. Non-bacterial thrombotic endocarditis in a Trousseau syndrome patient with stomach cancer: A case report. Geriatr. Gerontol. Int. 2016, 16, 1171–1172. [Google Scholar]
- Wigger, O.; Windecker, S.; Bloechlinger, S. Nonbacterial thrombotic endocarditis presenting as intracerebral hemorrhage. Wien. Klin. Wochenschr. 2016, 128, 922–924. [Google Scholar]
- Giray, S.; Sarica, F.B.; Arlier, Z.; Bal, N. Recurrent ischemic stroke as an initial manifestation of an concealed pancreatic adenocarcinoma: Trousseau’s syndrome. Chin. Med. J. 2011, 124, 637–640. [Google Scholar]
- Green, K.B.; Silverstein, R.L. Hypercoagulability in cancer. Hematol. Oncol. Clin. North Am. 1996, 10, 499–530. [Google Scholar]
- Kooiker, J.C.; MacLean, J.M.; Sumi, S.M. Cerebral embolism, marantic endocarditis, and cancer. Arch. Neurol. 1976, 33, 260–264. [Google Scholar]
- Singhal, A.B.; Topcuoglu, M.A.; Buonanno, F.S. Acute ischemic stroke patterns in infective and nonbacterial thrombotic endocarditis: A diffusion-weighted magnetic resonance imaging study. Stroke 2002, 33, 1267–1273. [Google Scholar]
- Uchino, K.; Shimizu, T.; Mizukami, H.; Isahaya, K.; Ogura, H.; Shinohara, K.; Hasegawa, Y. Nonbacterial Thrombotic Endocarditis Complicated by Cerebral Infarction in a Patient with Adenomyosis with High Serum CA125 Level; A Case Report. J. Stroke Cerebrovasc. Dis. 2018, 27, e42–e45. [Google Scholar]
- Thålin, C.; Demers, M.; Blomgren, B.; Wong, S.L.; von Arbin, M.; von Heijne, A.; Laska, A.C.; Wallén, H.; Wagner, D.D.; Aspberg, S. NETosis promotes cancer-associated arterial microthrombosis presenting as ischemic stroke with troponin elevation. Thromb. Res. 2016, 139, 56–64. [Google Scholar]
- Bang, O.Y.; Chung, J.W.; Cho, Y.H.; Oh, M.J.; Seo, W.K.; Kim, G.M.; Ahn, M.J. Circulating DNAs, a Marker of Neutrophil Extracellular Traposis and Cancer-Related Stroke: The OASIS-Cancer Study. Stroke 2019, 50, 2944–2947. [Google Scholar]
- Blom, J.W.; Doggen, C.J.; Osanto, S.; Rosendaal, F.R. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. Jama 2005, 293, 715–722. [Google Scholar]
- Iguchi, Y.; Kimura, K.; Kobayashi, K.; Ueno, Y.; Inoue, T. Ischaemic stroke with malignancy may often be caused by paradoxical embolism. J. Neurol. Neurosurg. Psychiatry 2006, 77, 1336–1339. [Google Scholar]
- Thompson, C.M.; Rodgers, L.R. Analysis of the autopsy records of 157 cases of carcinoma of the pancreas with particular reference to the incidence of thromboembolism. Am. J. Med. Sci. 1952, 223, 469–478. [Google Scholar]
- Huang, R.; Zhou, Y.; Hu, S.; Ren, G.; Cui, F.; Zhou, P.-K. Radiotherapy Exposure in Cancer Patients and Subsequent Risk of Stroke: A Systematic Review and Meta-Analysis. Front. Neurol. 2019, 10, 233. [Google Scholar]
- Van Aken, E.S.M.; van der Laan, H.P.; Bijl, H.P.; Van den Bosch, L.; van den Hoek, J.G.M.; Dieters, M.; Steenbakkers, R.; Langendijk, J.A. Risk of ischaemic cerebrovascular events in head and neck cancer patients is associated with carotid artery radiation dose. Radiother. Oncol. 2021, 157, 182–187. [Google Scholar]
- Nilsson, G.; Holmberg, L.; Garmo, H.; Terent, A.; Blomqvist, C. Radiation to supraclavicular and internal mammary lymph nodes in breast cancer increases the risk of stroke. Br. J. Cancer 2009, 100, 811–816. [Google Scholar]
- Makita, C.; Okada, S.; Kajiura, Y.; Tanaka, O.; Asahi, Y.; Yamada, N.; Yanagida, M.; Kumagai, M.; Murase, S.; Ito, M.; et al. Vascular events from carotid artery atherosclerosis after radiation therapy for laryngeal and hypopharyngeal cancer: The incidence and risk factors. Nagoya J. Med. Sci. 2020, 82, 747–761. [Google Scholar]
- Kim, B.J.; Kang, H.G.; Lee, S.W.; Jung, J.; Lee, M.H.; Kang, D.W.; Kim, J.S.; Kwon, S.U. Changes in the Common Carotid Artery after Radiotherapy: Wall Thickness, Calcification, and Atherosclerosis. J. Clin. Neurol. 2018, 14, 35–42. [Google Scholar]
- Mueller, S.; Sear, K.; Hills, N.K.; Chettout, N.; Afghani, S.; Gastelum, E.; Haas-Kogan, D.; Fullerton, H.J. Risk of first and recurrent stroke in childhood cancer survivors treated with cranial and cervical radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 643–648. [Google Scholar]
- Mueller, S.; Fullerton, H.J.; Stratton, K.; Leisenring, W.; Weathers, R.E.; Stovall, M.; Armstrong, G.T.; Goldsby, R.E.; Packer, R.J.; Sklar, C.A.; et al. Radiation, atherosclerotic risk factors, and stroke risk in survivors of pediatric cancer: A report from the Childhood Cancer Survivor Study. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 649–655. [Google Scholar]
- Bashar, K.; Healy, D.; Clarke-Moloney, M.; Burke, P.; Kavanagh, E.; Walsh, S.R. Effects of neck radiation therapy on extra-cranial carotid arteries atherosclerosis disease prevalence: Systematic review and a meta-analysis. PLoS ONE 2014, 9, e110389. [Google Scholar]
- Muzaffar, K.; Collins, S.L.; Labropoulos, N.; Baker, W.H. A prospective study of the effects of irradiation on the carotid artery. Laryngoscope 2000, 110, 1811–1814. [Google Scholar]
- Chang, Y.J.; Chang, T.C.; Lee, T.H.; Ryu, S.J. Predictors of carotid artery stenosis after radiotherapy for head and neck cancers. J. Vasc. Surg. 2009, 50, 280–285. [Google Scholar]
- Dorresteijn, L.D.A.; Kappelle, A.C.; Boogerd, W.; Klokman, W.J.; Balm, A.J.M.; Keus, R.B.; van Leeuwen, F.E.; Bartelink, H. Increased risk of ischemic stroke after radiotherapy on the neck in patients younger than 60 years. J. Clin. Oncol. 2002, 20, 282–288. [Google Scholar]
- Darby, S.C.; McGale, P.; Taylor, C.W.; Peto, R. Long-term mortality from heart disease and lung cancer after radiotherapy for early breast cancer: Prospective cohort study of about 300,000 women in US SEER cancer registries. Lancet Oncol. 2005, 6, 557–565. [Google Scholar]
- Taylor, C.W.; Nisbet, A.; McGale, P.; Darby, S.C. Cardiac exposures in breast cancer radiotherapy: 1950s–1990s. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, 1484–1495. [Google Scholar]
- Robbins, M.E.; Zhao, W. Chronic oxidative stress and radiation-induced late normal tissue injury: A review. Int. J. Radiat. Biol. 2004, 80, 251–259. [Google Scholar]
- Cheng, S.W.; Ting, A.C.; Wu, L.L. Ultrasonic analysis of plaque characteristics and intimal-medial thickness in radiation-induced atherosclerotic carotid arteries. Eur. J. Vasc. Endovasc. Surg. 2002, 24, 499–504. [Google Scholar]
- Nilsson, G.; Holmberg, L.; Garmo, H.; Terent, A.; Blomqvist, C. Increased incidence of stroke in women with breast cancer. Eur. J. Cancer 2005, 41, 423–429. [Google Scholar]
- Woodward, W.A.; Durand, J.B.; Tucker, S.L.; Strom, E.A.; Perkins, G.H.; Oh, J.; Arriaga, L.; Domain, D.; Buchholz, T.A. Prospective analysis of carotid artery flow in breast cancer patients treated with supraclavicular irradiation 8 or more years previously: No increase in ipsilateral carotid stenosis after radiation noted. Cancer 2008, 112, 268–273. [Google Scholar]
- Jairam, V.; Roberts, K.B.; Yu, J.B. Historical trends in the use of radiation therapy for pediatric cancers: 1973–2008. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, e151–e155. [Google Scholar]
- Van Dijk, I.W.; van der Pal, H.J.; van Os, R.M.; Roos, Y.B.; Sieswerda, E.; van Dalen, E.C.; Ronckers, C.M.; Oldenburger, F.; van Leeuwen, F.E.; Caron, H.N.; et al. Risk of Symptomatic Stroke After Radiation Therapy for Childhood Cancer: A Long-Term Follow-Up Cohort Analysis. Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, 597–605. [Google Scholar]
- Albergotti, W.G.; Aylward, A.; Chera, B.S. Risk of Stroke After Definitive Radiotherapy—Cause for Concern or Modest Risk? JAMA Otolaryngol. Head Neck Surg. 2022, 148, 747–748. [Google Scholar]
- Bavle, A.; Srinivasan, A.; Choudhry, F.; Anderson, M.; Confer, M.; Simpson, H.; Gavula, T.; Thompson, J.S.; Clifton, S.; Gross, N.L.; et al. Systematic review of the incidence and risk factors for cerebral vasculopathy and stroke after cranial proton and photon radiation for childhood brain tumors. Neurooncol. Pract. 2021, 8, 31–39. [Google Scholar]
- Arthurs, E.; Hanna, T.P.; Zaza, K.; Peng, Y.; Hall, S.F. Stroke After Radiation Therapy for Head and Neck Cancer: What Is the Risk? Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, 589–596. [Google Scholar]
- Baker, J.E.; Moulder, J.E.; Hopewell, J.W. Radiation as a Risk Factor for Cardiovascular Disease. Antioxid. Redox Signal. 2010, 15, 1945–1956. [Google Scholar]
- Turner, M.; Murchie, P.; Derby, S.; Ong, A.Y.; Walji, L.; McLernon, D.; Macleod, M.-J.; Adam, R. Is stroke incidence increased in survivors of adult cancers? A systematic review and meta-analysis. J. Cancer Surviv. 2022, 16, 1414–1448. [Google Scholar]
- Armstrong, G.T.; Stovall, M.; Robison, L.L. Long-term effects of radiation exposure among adult survivors of childhood cancer: Results from the childhood cancer survivor study. Radiat. Res. 2010, 174, 840–850. [Google Scholar]
- Jang, H.-S.; Choi, J.; Shin, J.; Chung, J.-W.; Bang, O.Y.; Kim, G.-M.; Seo, W.-K.; Lee, J. The Long-Term Effect of Cancer on Incident Stroke: A Nationwide Population-Based Cohort Study in Korea. Front. Neurol. 2019, 10, 52. [Google Scholar]
- Caruso, V.; Iacoviello, L.; Di Castelnuovo, A.; Storti, S.; Mariani, G.; de Gaetano, G.; Donati, M.B. Thrombotic complications in childhood acute lymphoblastic leukemia: A meta-analysis of 17 prospective studies comprising 1752 pediatric patients. Blood 2006, 108, 2216–2222. [Google Scholar]
- Grace, R.F.; Dahlberg, S.E.; Neuberg, D.; Sallan, S.E.; Connors, J.M.; Neufeld, E.J.; Deangelo, D.J.; Silverman, L.B. The frequency and management of asparaginase-related thrombosis in paediatric and adult patients with acute lymphoblastic leukaemia treated on Dana-Farber Cancer Institute consortium protocols. Br. J. Haematol. 2011, 152, 452–459. [Google Scholar]
- Li, S.H.; Chen, W.H.; Tang, Y.; Rau, K.M.; Chen, Y.Y.; Huang, T.L.; Liu, J.S.; Huang, C.H. Incidence of ischemic stroke post-chemotherapy: A retrospective review of 10,963 patients. Clin. Neurol. Neurosurg. 2006, 108, 150–156. [Google Scholar]
- Zahir, M.N.; Shaikh, Q.; Shabbir-Moosajee, M.; Jabbar, A.A. Incidence of Venous Thromboembolism in cancer patients treated with Cisplatin based chemotherapy—A cohort study. BMC Cancer 2017, 17, 57. [Google Scholar]
- Seng, S.; Liu, Z.; Chiu, S.K.; Proverbs-Singh, T.; Sonpavde, G.; Choueiri, T.K.; Tsao, C.K.; Yu, M.; Hahn, N.M.; Oh, W.K.; et al. Risk of venous thromboembolism in patients with cancer treated with Cisplatin: A systematic review and meta-analysis. J. Clin. Oncol. 2012, 30, 4416–4426. [Google Scholar]
- Hoy, J.; Neeman, T.; Stuart-Harris, R.; Davis, A. Risk of venous thromboembolism in patients receiving adjuvant chemotherapy with 5-fluorouracil, epirubicin and cyclophosphamide for early breast cancer. Asia-Pac. J. Clin. Oncol. 2009, 5, 129–136. [Google Scholar]
- Watanabe, K.; Arakawa, Y.; Oguma, E.; Uehara, T.; Yanagi, M.; Oyama, C.; Ikeda, Y.; Sasaki, K.; Isobe, K.; Mori, M.; et al. Characteristics of methotrexate-induced stroke-like neurotoxicity. Int. J. Hematol. 2018, 108, 630–636. [Google Scholar]
- Auer, T.A.; Renovanz, M.; Marini, F.; Brockmann, M.A.; Tanyildizi, Y. Ischemic stroke and intracranial hemorrhage in patients with recurrent glioblastoma multiforme, treated with bevacizumab. J. Neurooncol. 2017, 133, 571–579. [Google Scholar]
- Ranpura, V.; Hapani, S.; Chuang, J.; Wu, S. Risk of cardiac ischemia and arterial thromboembolic events with the angiogenesis inhibitor bevacizumab in cancer patients: A meta-analysis of randomized controlled trials. Acta Oncol. 2010, 49, 287–297. [Google Scholar]
- Barni, S.; Labianca, R.; Agnelli, G.; Bonizzoni, E.; Verso, M.; Mandalà, M.; Brighenti, M.; Petrelli, F.; Bianchini, C.; Perrone, T.; et al. Chemotherapy-associated thromboembolic risk in cancer outpatients and effect of nadroparin thromboprophylaxis: Results of a retrospective analysis of the PROTECHT study. J. Transl. Med. 2011, 9, 179. [Google Scholar]
- Sun, M.-Y.; Bhaskar, S.M.M. Venous Thromboembolism in Cancer Patients Undergoing Chemotherapy: A Systematic Review and Meta-Analysis. Diagnostics 2022, 12, 2954. [Google Scholar] [CrossRef]
- Kanjanapan, Y.; Gilbourd, D.; Pranavan, G. Acute ischaemic stroke following cisplatin-based chemotherapy for testicular cancer. BMJ Case Rep. 2020, 13, e235005. [Google Scholar]
- Osman, K.; Comenzo, R.; Rajkumar, S.V. Deep venous thrombosis and thalidomide therapy for multiple myeloma. N. Engl. J. Med. 2001, 344, 1951–1952. [Google Scholar]
- Schutt, P.; Ebeling, P.; Buttkereit, U.; Brandhorst, D.; Opalka, B.; Hoiczyk, M.; Flasshove, M.; Hense, J.; Bojko, P.; Metz, K.; et al. Thalidomide in combination with vincristine, epirubicin and dexamethasone (VED) for previously untreated patients with multiple myeloma. Eur J. Haematol. 2005, 74, 40–46. [Google Scholar]
- Zervas, K.; Dimopoulos, M.A.; Hatzicharissi, E.; Anagnostopoulos, A.; Papaioannou, M.; Mitsouli, C.; Panagiotidis, P.; Korantzis, J.; Tzilianos, M.; Maniatis, A. Primary treatment of multiple myeloma with thalidomide, vincristine, liposomal doxorubicin and dexamethasone (T-VAD doxil): A phase II multicenter study. Ann. Oncol. 2004, 15, 134–138. [Google Scholar]
- Baz, R.; Li, L.; Kottke-Marchant, K.; Srkalovic, G.; McGowan, B.; Yiannaki, E.; Karam, M.A.; Faiman, B.; Jawde, R.A.; Andresen, S.; et al. The role of aspirin in the prevention of thrombotic complications of thalidomide and anthracycline-based chemotherapy for multiple myeloma. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2005; Volume 80, pp. 1568–1574. [Google Scholar]
- Stone, J.B.; DeAngelis, L.M. Cancer-treatment-induced neurotoxicity—focus on newer treatments. Nat. Rev. Clin. Oncol. 2016, 13, 92–105. [Google Scholar]
- Abdol Razak, N.B.; Jones, G.; Bhandari, M.; Berndt, M.C.; Metharom, P. Cancer-Associated Thrombosis: An Overview of Mechanisms, Risk Factors, and Treatment. Cancers 2018, 10, 380. [Google Scholar]
- Van den Berg, H. Asparaginase revisited. Leuk. Lymphoma 2011, 52, 168–178. [Google Scholar]
- Asselin, B.L.; Ryan, D.; Frantz, C.N.; Bernal, S.D.; Leavitt, P.; Sallan, S.E.; Cohen, H.J. In vitro and in vivo killing of acute lymphoblastic leukemia cells by L-asparaginase. Cancer Res. 1989, 49, 4363–4368. [Google Scholar]
- Truelove, E.; Fielding, A.K.; Hunt, B.J. The coagulopathy and thrombotic risk associated with L-asparaginase treatment in adults with acute lymphoblastic leukaemia. Leukemia 2013, 27, 553–559. [Google Scholar]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar]
- Grem, J.L. 5-Fluorouracil: Forty-plus and still ticking. A review of its preclinical and clinical development. Investig. New Drugs 2000, 18, 299–313. [Google Scholar]
- Zhang, N.; Yin, Y.; Xu, S.J.; Chen, W.S. 5-Fluorouracil: Mechanisms of resistance and reversal strategies. Molecules 2008, 13, 1551–1569. [Google Scholar]
- Ceilley, R.I. Mechanisms of action of topical 5-fluorouracil: Review and implications for the treatment of dermatological disorders. J. Dermatolog. Treat. 2012, 23, 83–89. [Google Scholar]
- Grover, S.P.; Hisada, Y.M.; Kasthuri, R.S.; Reeves, B.N.; Mackman, N. Cancer Therapy–Associated Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1291–1305. [Google Scholar]
- Zeldis, J.B.; Knight, R.; Hussein, M.; Chopra, R.; Muller, G. A review of the history, properties, and use of the immunomodulatory compound lenalidomide. Ann. N. Y. Acad. Sci. 2011, 1222, 76–82. [Google Scholar]
- Luptakova, K.; Rosenblatt, J.; Glotzbecker, B.; Mills, H.; Stroopinsky, D.; Kufe, T.; Vasir, B.; Arnason, J.; Tzachanis, D.; Zwicker, J.I.; et al. Lenalidomide enhances anti-myeloma cellular immunity. Cancer Immunol. Immunother. 2013, 62, 39–49. [Google Scholar]
- Rajkumar, S.V.; Hayman, S.; Gertz, M.A.; Dispenzieri, A.; Lacy, M.Q.; Greipp, P.R.; Geyer, S.; Iturria, N.; Fonseca, R.; Lust, J.A.; et al. Combination therapy with thalidomide plus dexamethasone for newly diagnosed myeloma. J. Clin. Oncol. 2002, 20, 4319–4323. [Google Scholar]
- Zangari, M.; Anaissie, E.; Barlogie, B.; Badros, A.; Desikan, R.; Gopal, A.V.; Morris, C.; Toor, A.; Siegel, E.; Fink, L.; et al. Increased risk of deep-vein thrombosis in patients with multiple myeloma receiving thalidomide and chemotherapy. Blood 2001, 98, 1614–1615. [Google Scholar]
- Palumbo, A.; Rajkumar, S.V.; Dimopoulos, M.A.; Richardson, P.G.; San Miguel, J.; Barlogie, B.; Harousseau, J.; Zonder, J.A.; Cavo, M.; Zangari, M.; et al. Prevention of thalidomide- and lenalidomide-associated thrombosis in myeloma. Leukemia 2008, 22, 414–423. [Google Scholar]
- Zangari, M.; Saghafifar, F.; Mehta, P.; Barlogie, B.; Fink, L.; Tricot, G. The blood coagulation mechanism in multiple myeloma. In Seminars in Thrombosis and Hemostasis; Thieme Medical Publishers, Inc.: New York, NY, USA, 2003; Volume 29, pp. 275–282. [Google Scholar]
- Richardson, P.; Schlossman, R.; Munshi, N.; Avigan, D.; Jagannath, S.; Alsina, M.; Doss, D.; McKenney, M.; Hande, K.; Farrell, M.; et al. A Phase 1 Trial of Lenalidomide (REVLIMID®) with Bortezomib (VELCADE®) in Relapsed and Refractory Multiple Myeloma. Blood 2005, 106, 365. [Google Scholar]
- Stefan, O.; Vera, N.; Otto, B.; Heinz, L.; Wolfgang, G. Stroke in cancer patients: A risk factor analysis. J. Neurooncol. 2009, 94, 221–226. [Google Scholar]
- Navi, B.B.; Iadecola, C. Ischemic stroke in cancer patients: A review of an underappreciated pathology. Ann. Neurol. 2018, 83, 873–883. [Google Scholar]
- Zöller, B.; Ji, J.; Sundquist, J.; Sundquist, K. Risk of haemorrhagic and ischaemic stroke in patients with cancer: A nationwide follow-up study from Sweden. Eur. J. Cancer 2012, 48, 1875–1883. [Google Scholar]
- Navi, B.B.; Singer, S.; Merkler, A.E.; Cheng, N.T.; Stone, J.B.; Kamel, H.; Iadecola, C.; Elkind, M.S.; DeAngelis, L.M. Recurrent thromboembolic events after ischemic stroke in patients with cancer. Neurology 2014, 83, 26–33. [Google Scholar]
- Navi, B.B.; Kamel, H.; Sidney, S.; Klingman, J.G.; Nguyen-Huynh, M.N.; Johnston, S.C. Validation of the Stroke Prognostic Instrument-II in a large, modern, community-based cohort of ischemic stroke survivors. Stroke 2011, 42, 3392–3396. [Google Scholar]
- Lun, R.; Roy, D.C.; Ramsay, T.; Siegal, D.; Shorr, R.; Fergusson, D.; Dowlatshahi, D. Incidence of stroke in the first year after diagnosis of cancer-A protocol for systematic review and meta-analysis. PLoS ONE 2021, 16, e0256825. [Google Scholar]
- Selvik, H.A.; Thomassen, L.; Bjerkreim, A.T.; Næss, H. Cancer-Associated Stroke: The Bergen NORSTROKE Study. Cerebrovasc. Dis. Extra 2015, 5, 107–113. [Google Scholar]
- Salazar-Camelo, R.A.; Moreno-Vargas, E.A.; Cardona, A.F.; Bayona-Ortiz, H.F. Ischemic stroke: A paradoxical manifestation of cancer. Crit. Rev. Oncol./Hematol. 2021, 157, 103181. [Google Scholar]
- Donkor, A.; Luckett, T.; Aranda, S.; Phillips, J. Barriers and facilitators to implementation of cancer treatment and palliative care strategies in low- and middle-income countries: Systematic review. Int. J. Public Health 2018, 63, 1047–1057. [Google Scholar]
- Blacker, D.; Armstrong, E. Indigenous stroke care: Differences, challenges and a need for change. Intern. Med. J. 2019, 49, 945–947. [Google Scholar]
- Balabanski, A.H.; Dos Santos, A.; Woods, J.A.; Thrift, A.G.; Kleinig, T.J.; Suchy-Dicey, A.; Siri, S.R.; Boden-Albala, B.; Krishnamurthi, R.; Feigin, V.L.; et al. The Incidence of Stroke in Indigenous Populations of Countries With a Very High Human Development Index: A Systematic Review Protocol. Front. Neurol. 2021, 12, 532. [Google Scholar]
- Katzenellenbogen, J.M.; Atkins, E.R.; Thompson, S.C.; Hersh, D.; Coffin, J.; Flicker, L.; Hayward, C.; Ciccone, N.; Woods, D.; McAllister, M.; et al. Missing voices: Profile and extent of acquired communication disorders in Aboriginal and non-Aboriginal adult stroke survivors in Western Australia using linked administrative records. Int. J. Stroke 2016, 11, 103–116. [Google Scholar]
- Kocarnik, J.M.; Compton, K.; Dean, F.E.; Fu, W.; Gaw, B.L.; Harvey, J.D.; Henrikson, H.J.; Lu, D.; Pennini, A.; Xu, R.; et al. Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life Years for 29 Cancer Groups From 2010 to 2019: A Systematic Analysis for the Global Burden of Disease Study 2019. JAMA Oncol. 2022, 8, 420–444. [Google Scholar]
- Mohr, J.P.; Thompson, J.L.; Lazar, R.M.; Levin, B.; Sacco, R.L.; Furie, K.L.; Kistler, J.P.; Albers, G.W.; Pettigrew, L.C.; Adams, H.P., Jr.; et al. A comparison of warfarin and aspirin for the prevention of recurrent ischemic stroke. N. Engl. J. Med. 2001, 345, 1444–1451. [Google Scholar]


{kind=link}
{kind=link}
| Burden of Disease | Prevalence | DALY | Financial Burden | |
|---|---|---|---|---|
| Stroke | In Australia [9]: 2.7% of the total burden of disease (2015) 38,000 incident cases (2017) 27,428 incident cases (2020) Globally [2]: 12.2 million incident cases (2019) 2nd leading cause of death, 11.6% of total deaths (2019) LMICs [8]: 70% of strokes and 87% of stroke-related deaths occur in LMICs (2017) | In Australia: 387,000 stroke survivors (2018) [10] 445,087 stroke survivors (2020) [3] Globally [2]: 101 million stroke survivors | In Australia: 4.6 DALYs per 1000 population (2015) [11] = 118,220 DALYs across the Australian population (2015) * 398.22 DALYs/100,000 population (2021) [12] = 102,342 DALYs across the Australian population (2021) * Globally [2]: 143 million DALYs, 5.7% of total DALYs (2019) LMICs: 87% of DALYs occur in LMICs [8] | In Australia: AUD 633 million was spent on health system expenditure (2015–16) [13] AUD 6.2 billion in direct financial costs (2020) [3] AUD 26 billion in lost well-being and premature mortality (2020) [3] Globally: Worldwide data N/R In the EU: EUR 27 billion in health system expenditure, EUR 1.3 billion in cost for informal care, EUR 12 billion in lost productivity (2017) [14] In the US: USD 156.8 billion in total cost, $103.5 billion in indirect costs, USD 38.1 billion from productivity loss, and USD 30.4 billion from premature death (2020) [15] |
| Cancer | In Australia [16]: 151,000 new diagnoses (2021) Estimated that 43% will be diagnosed with cancer by the age of 85 Globally: 23.6 million new cancer cases (17.2 excluding nonmelanoma skin cancer) (2019) [17] | In Australia [16]: 456,978 people living (who had been diagnosed with cancer) at the end of 2016 (diagnosed in 2012–2016) 1,176,285 people living (who had been diagnosed with cancer) at the end of 2016 (diagnosed from 1982 to 2016) Globally [17]: 85.8 million cases (2019) LMICs: 65% of all cancer deaths occurred in LMICs (2012), projected to be 75% in 2030 [18] Projection of 75% of the world’s cancers to occur in LMICs by 2040 [17] | In Australia [16]: 18% of total burden (2018) 881,094 DALYs (2018) Globally [17]: 250 million DALYs (2019) | In Australia: 9% of health system expenditure (2021) AUD 6.3 billion in yearly cost to the Australian healthcare system (for Australians diagnosed in 2009–13) (2013) [19] Globally: Worldwide data N/R In the EU: EUR 199 billion in total cost of cancer, EUR 103 billion on health expenditure on cancer care (including EUR 32 billion on cancer drugs), EUR 26 billion in informal care, EUR 70 billion in total productivity loss (EUR 50 billion from premature mortality, EUR 20 billion lost due to morbidity) (2018) [20] In the US: estimates of USD 161.2 billion on healthcare spending, USD 30.3 billion spent on productivity loss due to morbidity, USD 150.7 billion lost to premature mortality (2017) [21,22] |
| Stroke in Cancer | Country-specific data N/R A US population-based study of 7,529,481 patients found the risk of fatal stroke to be 21.64 per 100,000-person-years. Standardized mortality ratio is 2.17 (95% CI, 2.15–2.19) (2019) [23] | In Australia: Total number of cases of stroke in cancer (2019) = 0.06 × 456,978 = ~27,419 * Globally: The rate of non-fatal strokes in cancer patients is 5%, rate of fatal strokes in cancer patients is 1%. The combined rate of fatal plus non-fatal stroke is 6% for cancer cohort [23]. Total number of cases of stroke in cancer (2019) = 0.06 × 85.8 million = ~5.148 million * | In Australia: Total number of DALYs due to stroke in cancer (2011) = 0.06 × 833,250 = ~49,995 * Globally: Global DALYs due to stroke in cancer (2019) = 0.06 × 250 million = ~15 million * | In Australia: 0.06 × USD 6.3 billion of direct costs = ~USD 378 million of Australian health system expenditure on stroke in cancer (2013) * |
| Study | Cancer Phenotype | No. Controls vs. No. Patients | Incidence | Site of Therapy | Radiation Dose (Gy) | Interval from Radiotherapy (Months) | Overall Findings |
|---|---|---|---|---|---|---|---|
| Radiation-induced carotid artery atherosclerosis Gujral et al., 2014 [6] | Breast cancer Head and neck cancer Hodgkin’s lymphoma | Varied | Stroke/TIA: RR of stroke is 1.12 in patients with breast cancer. RR of stroke is 5.6 in head and neck cancer patients. Carotid artery stenosis (CAS): Increased prevalence of 16–55% in irradiated patients. Carotid intima-medial thickness (CIMT): Thickness increased in irradiated carotid arteries by 18–40%. | Varied | Varied | Varied | Measured a significant increase in stroke incidence in irradiated patients, and consistent differences in CAS and CIMT in irradiated and unirradiated carotid arteries. |
| Risk of ischaemic cerebrovascular events in head and neck cancer patients is associated with carotid artery radiation dose Van Aken et al., 2021 [83] | Head and neck cancer | n = 750 No control group. | A total of 27 patients (3.6%) experienced an ischaemic cerebrovascular event (ICVE). The 5-year cumulative risk of stroke is 4.6%. The 8-year cumulative risk of stroke is 7.4%. | Solid tumours located in the larynx (45%) and the oropharynx (36%). | Mean dose to carotid arteries = 39.8 ± 0.5 Gy. | The mean time to event is 22.8 months. Mean age at ICVE = 63 years. | The increased risk is associated with an increased dose to carotid arteries. Maximum dose significantly associated with ICVE risk. The absolute volume of the carotid arteries that received at least a radiation dose of 10 Gy is most important prognostic factor. |
| Radiation to supraclavicular and internal mammary lymph nodes in breast cancer increases the risk of stroke Nilsson et al., 2009 [84] | Breast cancer | Nested case-control study of a cohort of 4689 women with invasive breast cancer. n = 282 women diagnosed with breast cancer, hospitalized for stroke. Control group = 282 women with breast cancer. | 282 cases of stroke = 6% of cohort. 10% of cases were hemorrhagic stroke, 2% subarachnoidal, and 8% intracerebral. 62% of cases had ischaemic stroke, 48% were infarction, 14% had transient cerebral ischemia. 80% of strokes originated in carotid arteries, 20% from the vertebrobasilar system. | Combination of the thoracic wall, internal mammary chain, supraclavicular, and axillary lymph nodes. | Different RT regimens across 1970–2003, varying from 20 Gy to 54 Gy. | Not specified | Radiotherapy to the internal mammary chain and supraclavicular lymph nodes was associated with a higher risk of stroke compared to no radiotherapy, OR = 1.3 (95% CI, 1.1–2.8), however not statistically significant. Statistically significant trend for increased risk of stroke with higher daily fraction dose. |
| Vascular events from carotid artery atherosclerosis after radiation therapy for laryngeal and hypopharyngeal cancer: the incidence and risk factors Makita et al., 2020 [85] | Laryngeal and hypopharyngeal cancer | n = 111 patients (95 laryngeal, 16 hypopharyngeal) No control group. | 5.4% (6 of 111 patients) The 5-year occurrence rate is 5.5% (95% CI, 0–10.5%). The 8-year occurrence rate is 10.7% (95% CI, 1.4–19.1%). | For laryngeal cancer and dependent on spread of disease-vocal cords, 1 cm margin surrounding the tumour For hypopharyngeal cancer–gross tumor plus 1 cm margin, prophylactic lymph node areas. | Median of 66 Gy. Range of 60–74 Gy. | Median occurred at 51.7 months. Range of 0.3–78.3 months after radiotherapy initiation. | Dyslipidaemia, diabetes mellitus and carotid calcification are important factors for event occurrence. As 3 of 6 cases occurred out of the field of irradiation, no carotid artery parameters were significantly correlated with a vascular event. |
| Changes in the Common Carotid Artery after Radiotherapy: Wall Thickness, Calcification, and Atherosclerosis Kim et al., 2017 [86] | Laryngeal cancer patients | n = 125 No control group. | Calcification in 37 patients (29.6%) and atherosclerosis in 71 patients (56.8%). | Primary tumor site, neck region, entire common carotid artery area. | Cumulative radiation dose ranged from 27 to 82.6 Gy. | Mean of 62.7 ± 32.1 months after radiotherapy for radiation-induced atherosclerotic changes. | Atherosclerosis in the middle portion of the common carotid artery occurred in 24.6% of patients, at the proximal CCA at the intrathoracic level in 20.6% of patients, and at the distal CCA in 4.8% of patients. Demographic, risk factors and radiation doses were not found to be associated with the change in CCA wall thickness. |
| Risk of First and Recurrent Stroke in Childhood Cancer Survivors Treated with Cranial and Cervical Radiation Therapy Mueller et al., 2013 [87] | Childhood cancer | n = 325 No control group. | A total of 19 first strokes occurred over the period of the study from 1980–2009. 13 ischaemic, 4 haemorrhagic, 2 unknown. The cumulative incidence of the first stroke is 2% at 5 years after irradiation, 4% at 10 years after irradiation. A total of 6 recurrent strokes. The median time to recurrence is 15 months after the first stroke; 38% at 5 years, 59% at 10 years | Cranial and cervical radiotherapy | Range from 29.5 Gy–72 Gy. | The median time to develop the first stroke after RT was 12 years (144 months), IQR is 5–18 years. Recurrent stroke occurred with a median time of 15 months. | Each additional year of age at the time of radiation increased stroke risk by 12%. With each 100-cGy increase in radiation dose, stroke hazard increase by 5%. |
| Radiation, Atherosclerotic Risk Factors, and Stroke Risk in Survivors of Pediatric Cancer: A Report from the Childhood Cancer Survivor Study Mueller et al., 2013 [88] | Childhood cancer | Retrospective cohort study n = 14,358 five-year survivors of childhood cancer Control group = 4023 random selection sibling controls. | A total of 292 survivors reported late-occurring stroke, 23.3 years mean follow-up. Stroke rate of 77 per 100,000 person-years, 9.3 in siblings RR is 7.8 (95% CI, 4.7–13.0) compared to control siblings. | Varied | Varied Range from 0–50 + Gy | Time after radiotherapy unable to be found The mean time from diagnosis to late-occurring stroke was 18.6 years Range time was 5.2–38.1 years Cumulative incidence in survivors treated with 50+ Gy CRT is 1.1% at 10 years post-diagnosis and 12% at 30 years post-diagnosis. The median age of stroke was at 29 years of age. | Dose-dependent hazard ratio, 5.9 for 30–49 Gy CRT, 11.0 for 50+ Gy CRT. Among young adult survivors with 50+ Gy CRT, 12fold cumulative incidence of stroke 10–30 years post-diagnosis. |
| Radiotherapy Exposure in Cancer Patients and Subsequent Risk of Stroke: A Systematic Review and Meta-Analysis Huang et al., 2019 [82] | Cancer survivors | From 12 eligible studies, n = 57,881 Control groups existed of cancer patients receiving non-radiotherapy treatments. | RR 2.09 (95% CI, 1.45–3.16) in radiotherapy vs. no radiotherapy cancer patients. | Varied | 13–80 Gy | N/A | Overall doubling of stroke risk in cancer patients. |
| Effects of Neck Radiation Therapy on Extra-Cranial Carotid Arteries Atherosclerosis Disease Prevalence: Systematic Review and a Meta-Analysis Bashar et al., 2014 [89] | Malignant head and neck tumors - squamous cell carcinoma, nasopharyngeal carcinoma, etc. | Combination of 8 studies, 1070 patients total n = 596 Control group = 474 non-irradiated cancer patients. | Risk ratio is 4.38 for overall stenosis, 7.51 for severe stenosis. Abnormal scan: 6 studies, 908 patients total; 237/534 of patients in the RT group had some degree of stenosis, vs. 33/374 in the control group High-grade stenosis: 5 studies, 717 patients. >70% stenosis in 51/406 of the RT group vs. 3/311 of controls. Low-grade stenosis: 5 studies, 770 patients; 89/454 in RT group, vs. 21/316 in the control group. | Head and neck | Variable as protocols differ across institutions and diseases. | N/A | Extracranial carotid artery stenosis is higher in patients receiving radiotherapy for neck malignancies. Progressive thickening of intima-media early in the first 12 months following radiotherapy initiation. Acceleration of the process of thickening is estimated to be 21 times higher than control groups undergoing no radiotherapy [90]. |
| Predictors of carotid artery stenosis after radiotherapy for head and neck cancers Chang et al., 2009 [91] | Head and neck cancer | n = 192 Control group = 98 patients not undergoing radiotherapy. | Carotid plaque score significantly higher in the irradiated group, p < 0.001. | Whole pharynx, skull base, whole neck lymphatic system. | All upper neck areas at least 6000 cGy of radiation 18–20 Gy daily fraction, 5 fractions a week. The total median value of 7060 cGy to the initial area of gross disease. | Time intervals only available for the RT group The average time after RT is 2 years to develop carotid artery stenosis. | Bilateral plaque score for carotid artery stenosis is significantly correlated with age, hyperlipidemia and radiotherapy. |
| Increased risk of ischemic stroke after radiotherapy on the neck in patients younger than 60 years Dorresteijn et al., 2002 [92] | Head and neck cancer | n = 367 patients No control group. | In 14 cases of stroke, RR is 5.6 (95% CI, 3.1–9.4), and 15-year cumulative stroke risk is 12%. | Varied | 50–66 Gy | Mean of 10.9 years. | Analysis of risk factors revealed hypertension and DM to increase RR after RT. After more than 10 years of follow-up, RR elevated to 10.1 (95% CI, 4.4–20). |
| Study | Cancer Phenotype | Therapeutic Drug | Cohort Size | Stroke Incidence | Impact/Other Findings |
|---|---|---|---|---|---|
| Caruso et al., 2006 [108] | Children with acute lymphoblastic leukemia (ALL) | L-asparaginase/anthracyclines/prednisone | n = 1752 children from 17 prospective studies No control group. | Rate of thrombosis 5.2% (95% CI 4.2–6.4) | Most events occurred during the induction phase of therapy. Lower doses for longer periods are associated with the highest thrombotic incidence. |
| Grace et al., 2010 [109] | Pediatric and Adult patients with ALL | L-asparaginase | n = 548 patients No control group. | 8% experienced VTE 5% of pediatric and 34% of adult patients | Timing: for 8 patients, VTE occurred during induction, post-induction in 35 patients Median time to VTE was 3.5 months from initiation of therapy (range 0.5 to 10.1 months) Age is a significant predictor of risk, >30 years deemed very high risk with a VTE rate of 42% |
| Li et al., 2006 [110] | Various | Various | n = 10,963 patients with malignancies followed up at 1-month post chemotherapy No control group. | 15 patients experienced 16 ischaemic strokes within first month after the latest chemotherapy Incidence of a post-chemotherapy stroke at 0.137%. | Drug type: Cisplatin was administered prior to 8 ischaemic strokes, oxaliplatin to one. 75% occurred within 10 days of the latest chemotherapy, 10 happened after first cycle of chemotherapy. |
| Zahir et al., 2017 [111] | Various | Cisplatin | n = 200 patients receiving cisplatin Control group = 200 patients on non-cisplatin-based regimens | 31 VTE events in the cisplatin group, the cumulative dose was 471 mg/m2 Group without events had a mean cumulative dose of 322 mg/m2 The crude RR of VTE in the cisplatin group is 2.8 (95% CI, 1.4–4.2) compared to the non-cisplatin group Adjusted for gender, ECOG, and presence of central venous catheter, the adjusted RR is 3.32. | Baseline characteristics of diabetes mellitus, hypertension and coronary artery disease were similar across both cisplatin and non-cisplatin groups. High incidence of VTE in patients receiving cisplatin-based chemotherapy. |
| Seng et al., 2012 [112] | Various | Cisplatin | n = 8216 patients from 38 RCTs No control group. | A 1.92% incidence of VTEs in cisplatin-receiving patients vs. 0.79% incidence in non-cisplatin-based regimens. RR of 1.67 (95% CI, 1.25–2.23) of VTE for the cisplatin-receiving group | Dose-dependent effect noted. Cisplatin is associated with a significant increase in VTE risk in patients with advanced solid tumours. |
| Hoy et al., 2009 [113] | Early breast cancer | 5-fluorouracil, epirubicin, cyclophosphamide (FEC) | n = s 176 patients receiving FEC regimen Control group = 149 patients receiving other chemotherapy regimens. | 27% incidence in FEC receiving group (47/176), with 5% of patients on other regiments experiencing VTE (7/149). | Adjuvant FEC chemotherapy is associated with increased VTE incidence for patients with early breast cancer. |
| Watanabe et al., 2018 [114] | Leukaemia or lymphoma | Methotrexate (MTX) | n = 9 patients No control group. | Of 44.4% (4/9) of patients with leukaemia/lymphoma and episodes of stroke-like presentation were diagnosed with MTX-induced stroke-like neurotoxicity, 22.2% (2/9) had disturbed consciousness, speech disorders, hemiparalysis. | MTX-induced neurotoxicity may manifest in a stroke-like presentation, difficult to distinguish from stroke. Neurological events occurred 10–13 days after the 4th or later MTX treatment. |
| Auer et al., 2017 [115] | Recurrent glioblastoma multiforme (GBM)-1st/2nd/3rd relapse | Bevacizumab (BVZ) | n = 40 treated with BVZ (178 scans) Control group = 42 patients matched for age and gender receiving basic treatment (186 scans). | An 8% (7/82) incidence of vascular events from MRI scans, with 4 events recorded in BVZ group, 3 ischaemic stroke, and 1 intracranial haemorrhage; 3 events in the control group 1 ischaemic stroke, and 2 intracranial haemorrhages. | BVZ treatment not associated with increased risk for vascular events in recurrent GBM patients. |
| Ranpura et al., 2009 [116] | Variety of solid tumours | Bevacizumab | n = 12,617 patients from 20 RCTs No control group. | The incidence of all-grade arterial thromboembolic events was 3.3% (95% CI, 2.0–5.6). The incidence of high-grade ATE is 2.0% (95% CI, 1.7–2.5). With BVZ, RR of 1.44 compared to controls of having an arterial thromboembolic event. | The risk did not increase with BVZ doses, with 2.5 mg and 5 mg/kg/week having an RR of 1.52 (95% CI, 1.10–2.09) and 1.50 (95% CI, 0.84–2.69), respectively. Increased risks for patients with renal cell cancer (RR 3.71) and colorectal cancer (RR 1.89). High-grade cardiac ischemia is higher than controls at RR 2.14, but the risk of ischaemic stroke is not significantly different from controls with an RR 1.37. The median time to the first event is 2.6 months in BVZ treated group, vs. 2.1 months in the control group. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, M.-Y.; Bhaskar, S.M.M. When Two Maladies Meet: Disease Burden and Pathophysiology of Stroke in Cancer. Int. J. Mol. Sci. 2022, 23, 15769. https://doi.org/10.3390/ijms232415769
Sun M-Y, Bhaskar SMM. When Two Maladies Meet: Disease Burden and Pathophysiology of Stroke in Cancer. International Journal of Molecular Sciences. 2022; 23(24):15769. https://doi.org/10.3390/ijms232415769
Chicago/Turabian StyleSun, Ming-Yee, and Sonu M. M. Bhaskar. 2022. "When Two Maladies Meet: Disease Burden and Pathophysiology of Stroke in Cancer" International Journal of Molecular Sciences 23, no. 24: 15769. https://doi.org/10.3390/ijms232415769
APA StyleSun, M.-Y., & Bhaskar, S. M. M. (2022). When Two Maladies Meet: Disease Burden and Pathophysiology of Stroke in Cancer. International Journal of Molecular Sciences, 23(24), 15769. https://doi.org/10.3390/ijms232415769