

Anticancer Activity, Reduction Mechanism and G-Quadruplex DNA Binding of a Redox-Activated Platinum(IV)–Salphen Complex

 ,
,  , and
, and 
Abstract
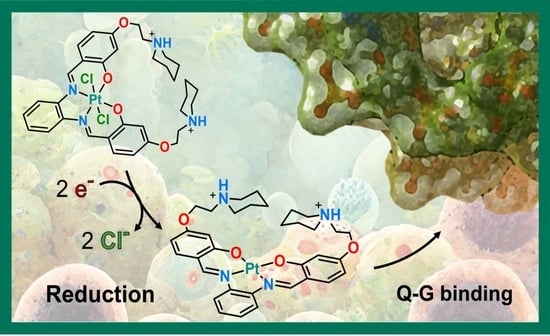
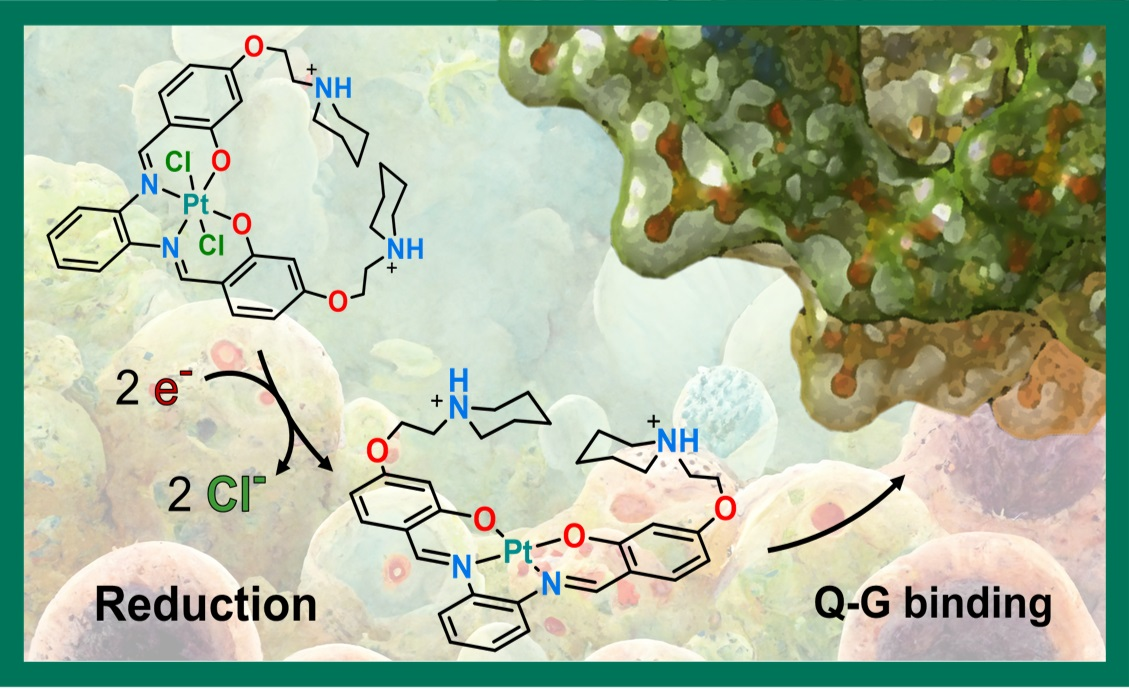{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
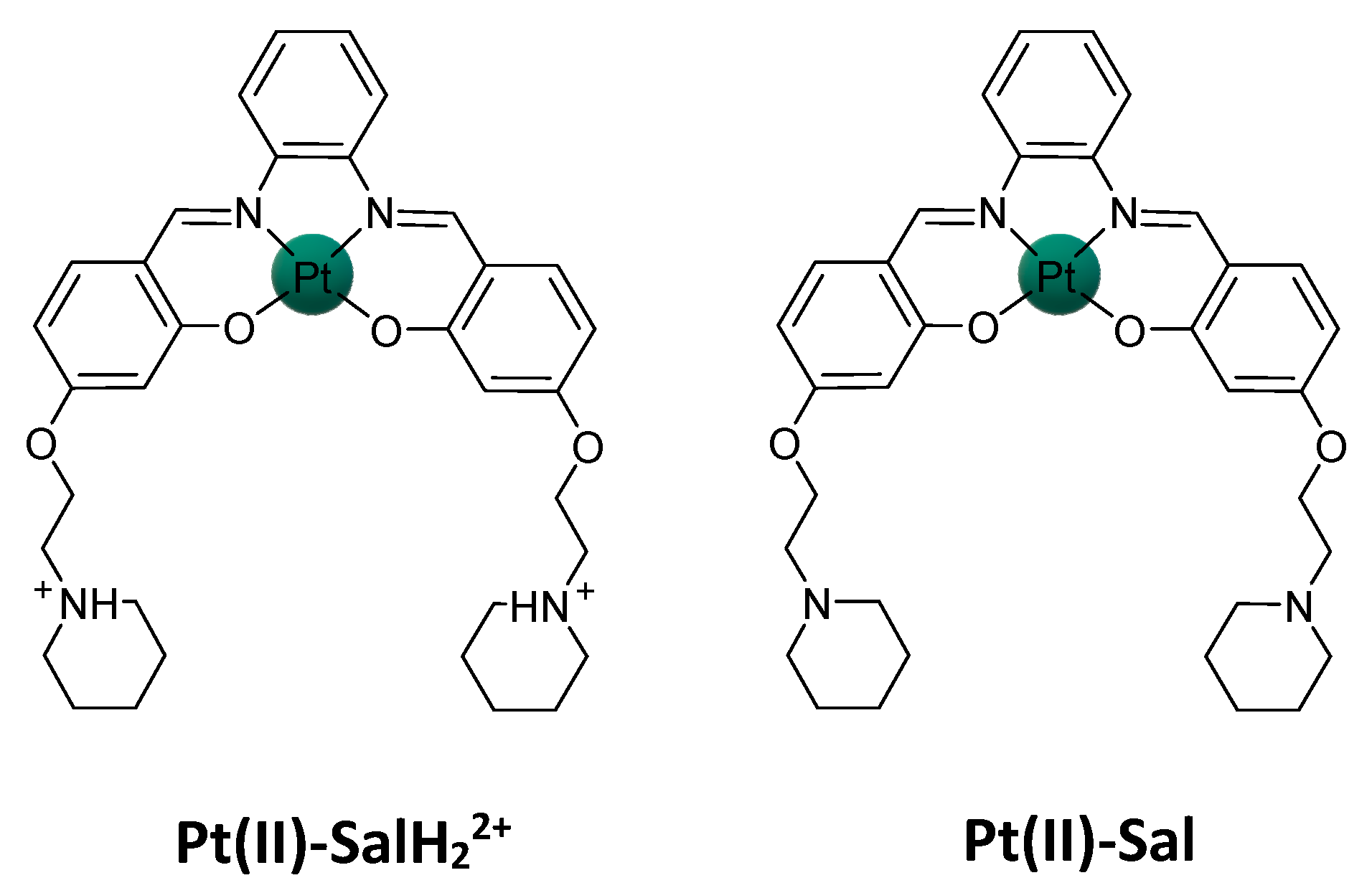{kind=link}
1. Introduction
2. Results
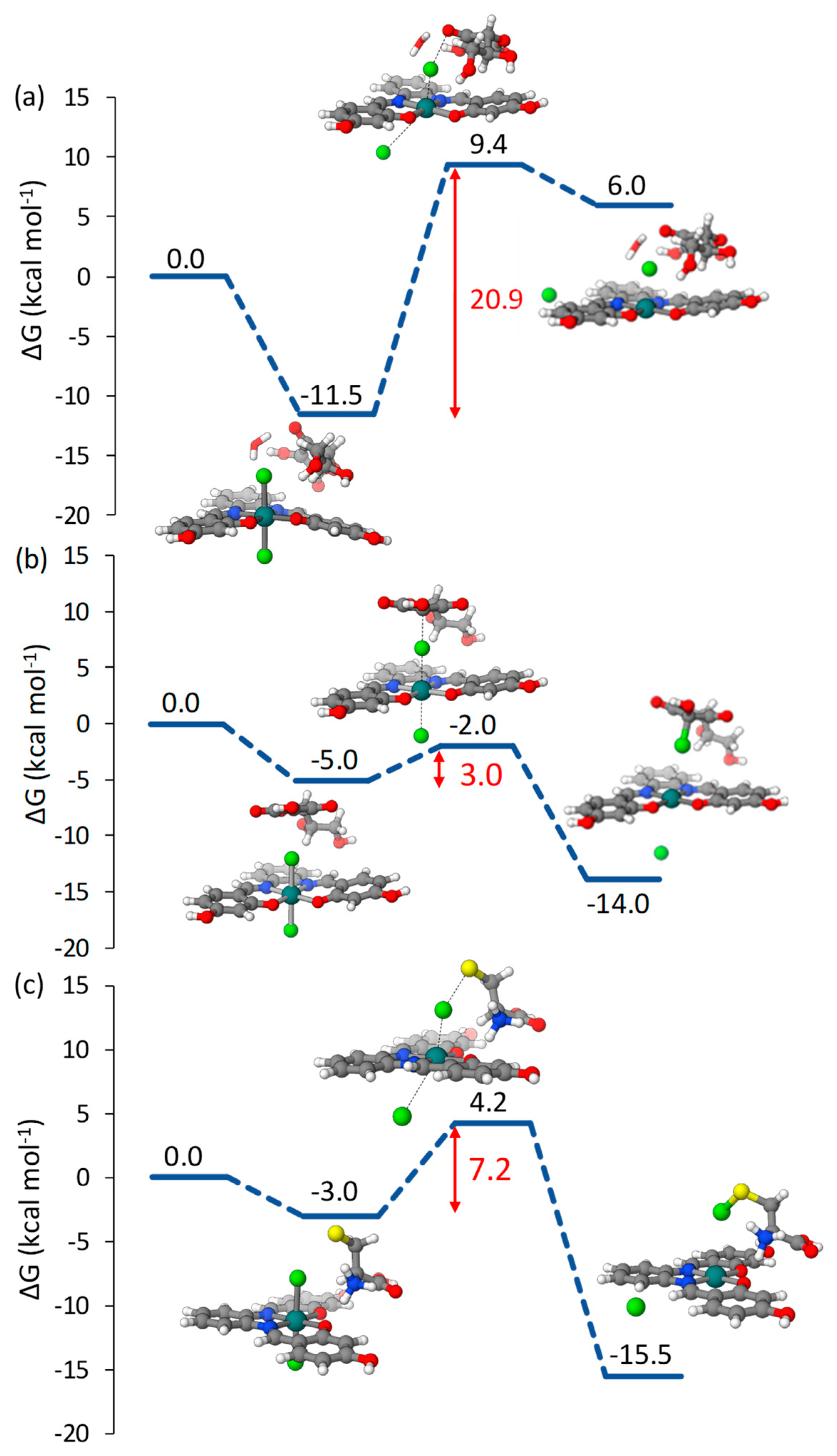
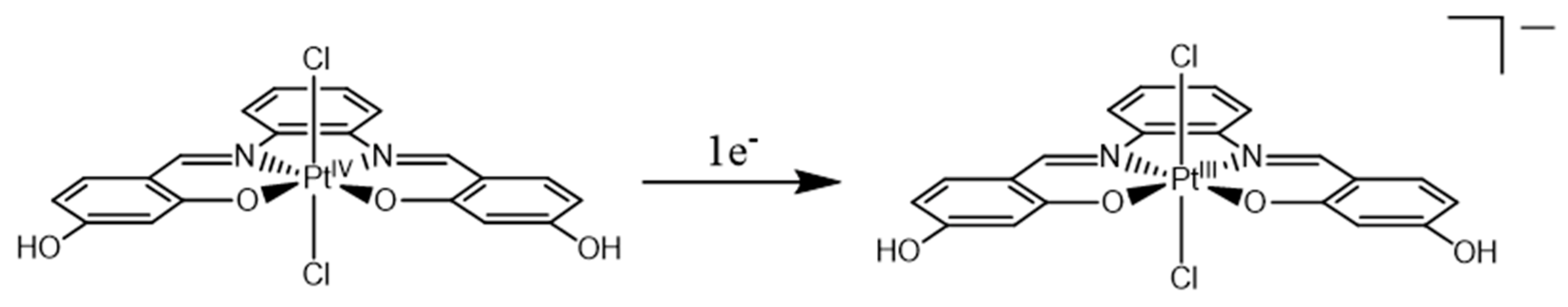
2.1. Pt(IV)-Sal Complex Reduction Mechanism
2.1.1. Pt(IV)-Sal Inner-Sphere Reduction Mechanism
2.1.2. Pt(IV)-Sal Outer-Sphere Reduction
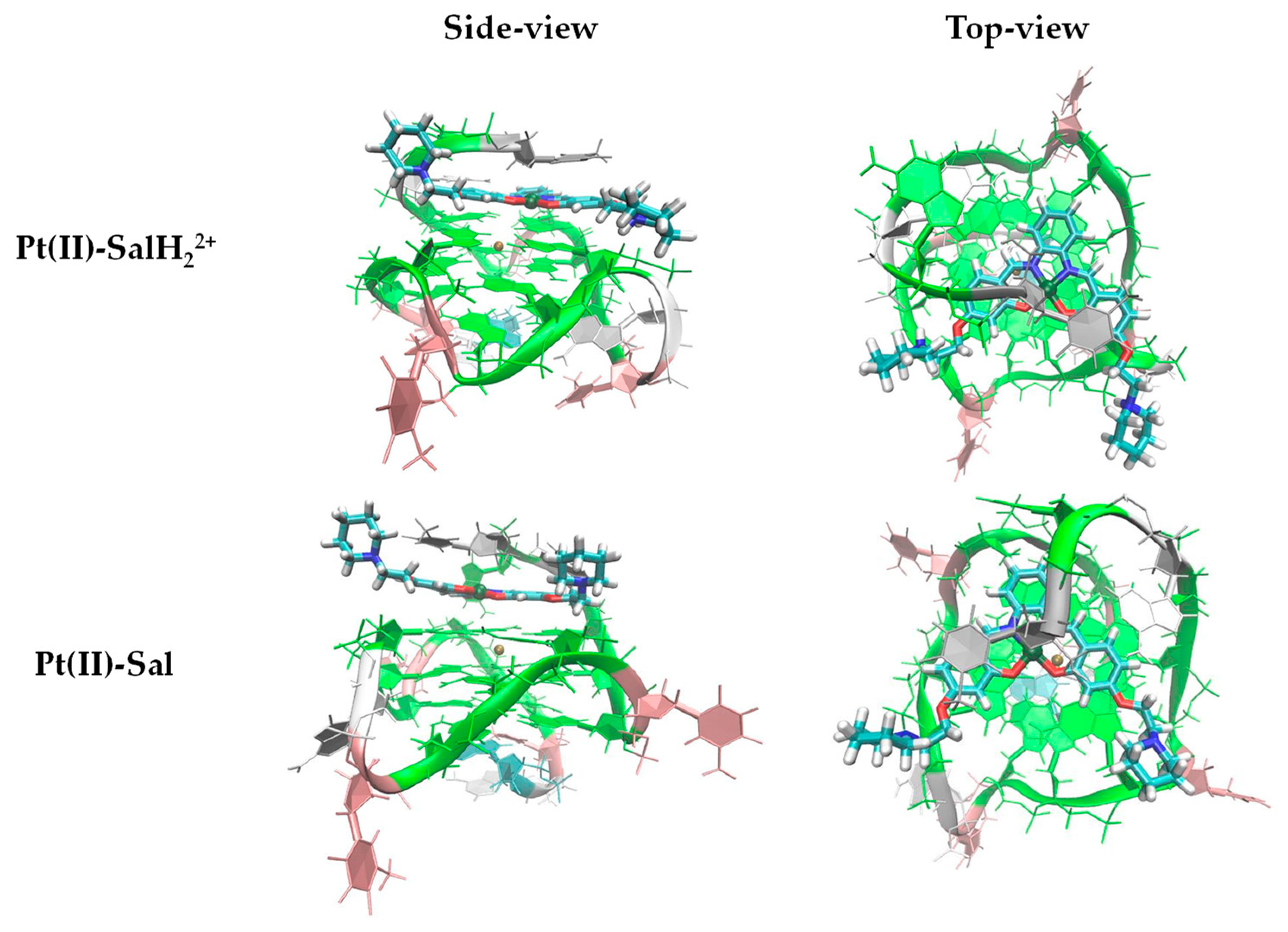
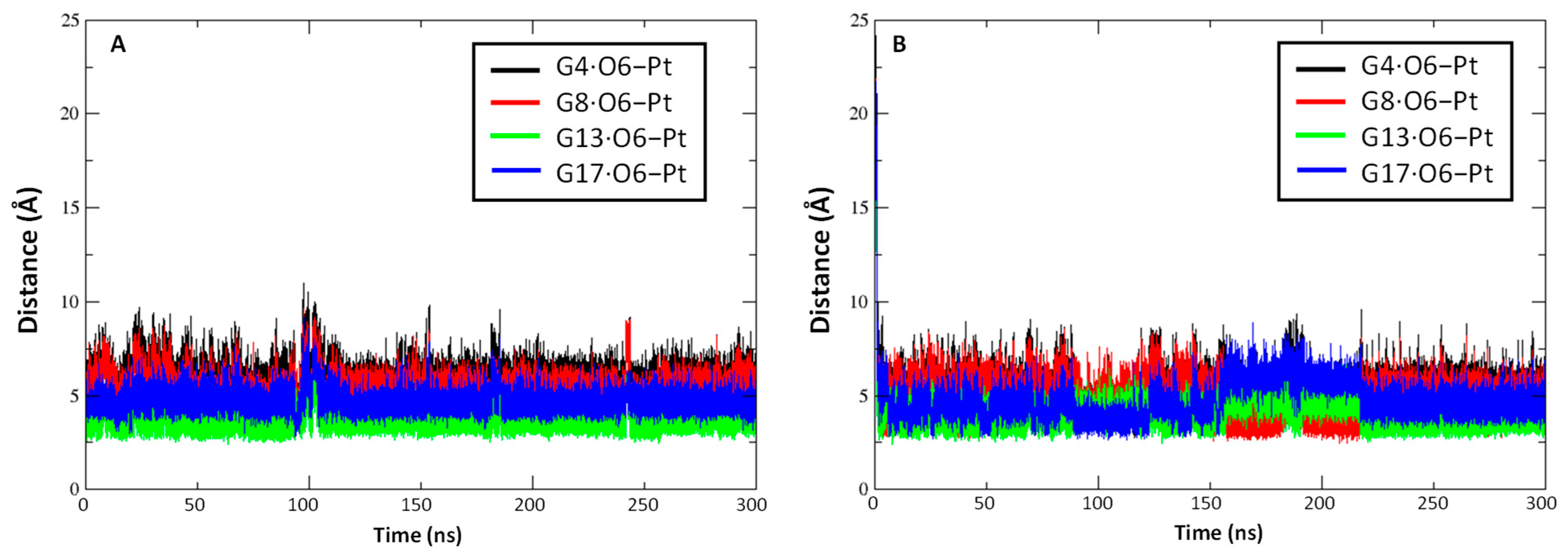
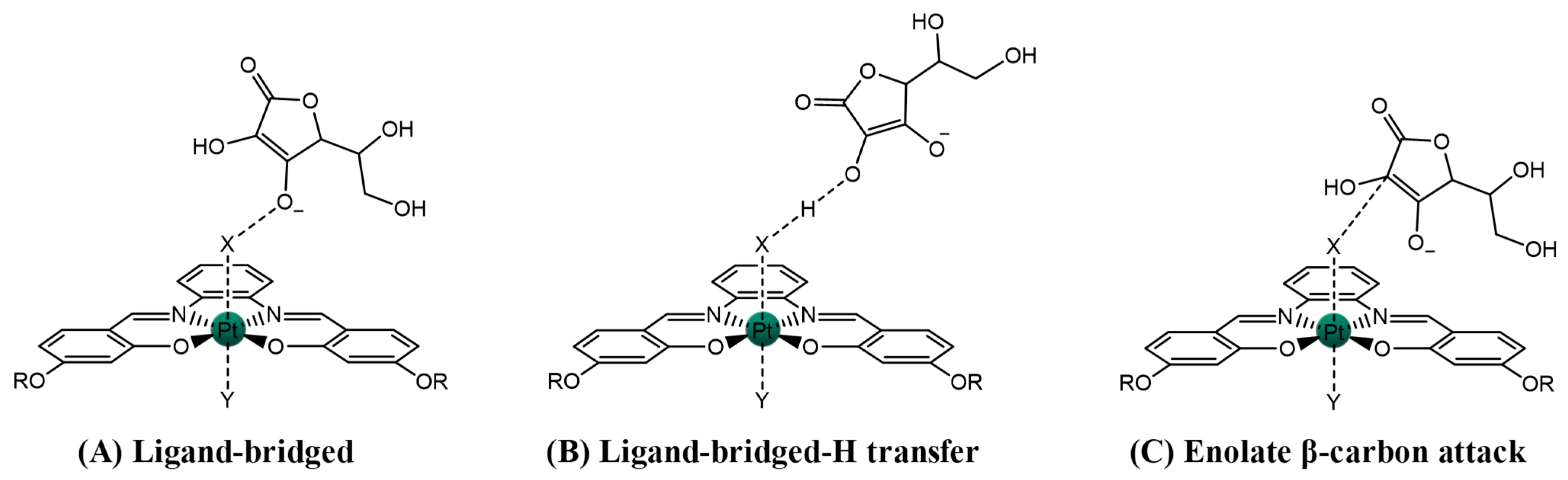
2.2. Pt(IV)-Sal Complex Binding
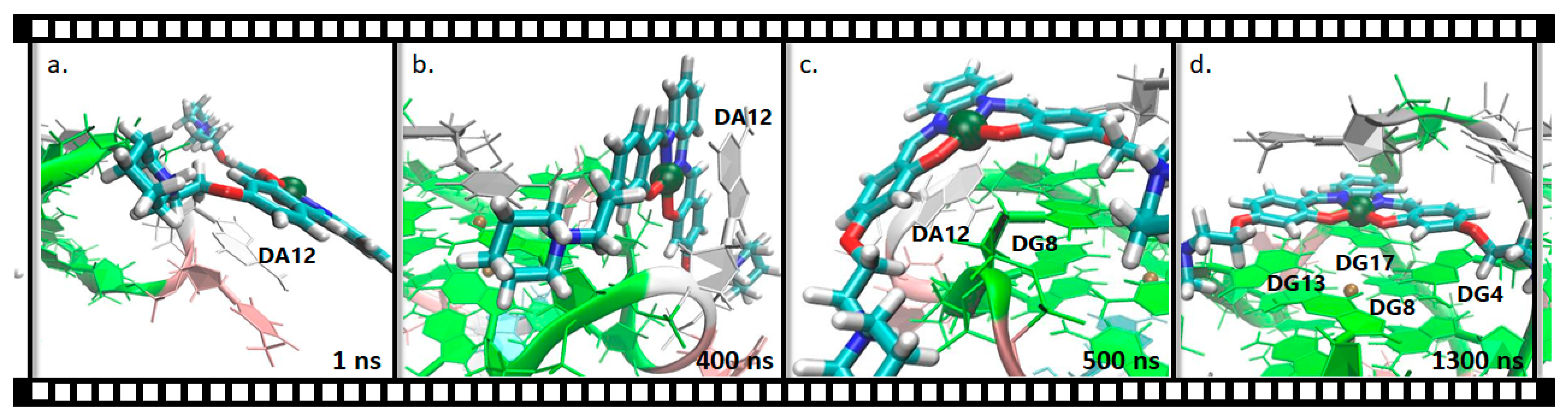
2.2.1. Pt(II)-Sal Binding Interactions
2.2.2. Pt(II)-SalH22+ Targeting Binding Site
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kelland, L. The Resurgence of Platinum-Based Cancer Chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Reedijk, J. New Clues for Platinum Antitumor Chemistry: Kinetically Controlled Metal Binding to DNA. Proc. Natl. Acad. Sci. USA 2003, 100, 3611–3616. [Google Scholar] [CrossRef]
- Jung, Y.; Lippard, S.J. Direct Cellular Responses to Platinum-Induced DNA Damage. Chem. Rev. 2007, 107, 1387–1407. [Google Scholar] [CrossRef] [PubMed]
- Heffeter, P.; Jungwirth, U.; Jakupec, M.; Hartinger, C.; Galanski, M.S.; Elbling, L.; Micksche, M.; Keppler, B.; Berger, W. Resistance against Novel Anticancer Metal Compounds: Differences and Similarities. Drug Resist. Updates 2008, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Panichpisal, K.; Kurtzman, N.; Nugent, K. Cisplatin Nephrotoxicity: A Review. Am. J. Med. Sci. 2007, 334, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Bruijnincx, P.C.; Sadler, P.J. New Trends for Metal Complexes with Anticancer Activity. Curr. Opin. Chem. Biol. 2008, 12, 197–206. [Google Scholar] [CrossRef]
- Ho, Y.-P.; Au-Yeung, S.C.F.; To, K.K.W. Platinum-Based Anticancer Agents: Innovative Design Strategies and Biological Perspectives. Med. Res. Rev. 2003, 23, 633–655. [Google Scholar] [CrossRef]
- Gellert, M.; Lipsett, M.N.; Davies, D.R. Helix Formation by Guanylic Acid. Proc. Natl. Acad. Sci. USA 1962, 48, 2013–2018. [Google Scholar] [CrossRef]
- Patel, D.J.; Phan, A.T.; Kuryavyi, V. Human Telomere, Oncogenic Promoter and 5’-UTR G-Quadruplexes: Diverse Higher Order DNA and RNA Targets for Cancer Therapeutics. Nucleic Acids Res. 2007, 35, 7429–7455. [Google Scholar] [CrossRef]
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, Topology and Structure. Nucleic Acids Res. 2006, 34, 5402–5415. [Google Scholar] [CrossRef]
- Hänsel-Hertsch, R.; di Antonio, M.; Balasubramanian, S. DNA G-Quadruplexes in the Human Genome: Detection, Functions and Therapeutic Potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [Google Scholar] [CrossRef]
- Müller, S.; Rodriguez, R. G-Quadruplex Interacting Small Molecules and Drugs: From Bench toward Bedside. Expert Rev. Clin. Pharmacol. 2014, 7, 663–679. [Google Scholar] [CrossRef]
- Fletcher, T.M.; Sun, D.; Salazar, M.; Hurley, L.H. Effect of DNA Secondary Structure on Human Telomerase Activity. Biochemistry 1998, 37, 5536–5541. [Google Scholar] [CrossRef]
- Campbell, N.; Collie, G.W.; Neidle, S. Crystallography of DNA and RNA G-Quadruplex Nucleic Acids and Their Ligand Complexes. Curr. Protoc. Nucleic Acid Chem. 2012, 50, 17.6.1–17.6.22. [Google Scholar] [CrossRef]
- Georgiades, S.N.; Abd Karim, N.H.; Suntharalingam, K.; Vilar, R. Interaction of Metal Complexes with G-Quadruplex DNA. Angew. Chem. Int. Ed. 2010, 49, 4020–4034. [Google Scholar] [CrossRef]
- Campbell, N.H.; Karim, N.H.A.; Parkinson, G.N.; Gunaratnam, M.; Petrucci, V.; Todd, A.K.; Vilar, R.; Neidle, S. Molecular Basis of Structure–Activity Relationships between Salphen Metal Complexes and Human Telomeric DNA Quadruplexes. J. Med. Chem. 2012, 55, 209–222. [Google Scholar] [CrossRef]
- Reed, J.E.; Arnal, A.A.; Neidle, S.; Vilar, R. Stabilization of G-Quadruplex DNA and Inhibition of Telomerase Activity by Square-Planar Nickel(II) Complexes. J. Am. Chem. Soc. 2006, 128, 5992–5993. [Google Scholar] [CrossRef]
- Arola-Arnal, A.; Benet-Buchholz, J.; Neidle, S.; Vilar, R. Effects of Metal Coordination Geometry on Stabilization of Human Telomeric Quadruplex DNA by Square-Planar and Square-Pyramidal Metal Complexes. Inorg. Chem. 2008, 47, 11910–11919. [Google Scholar] [CrossRef]
- Wu, P.; Ma, D.-L.; Leung, C.-H.; Yan, S.-C.; Zhu, N.; Abagyan, R.; Che, C.-M. Stabilization of G-Quadruplex DNA with Platinum(II) Schiff Base Complexes: Luminescent Probe and Down-Regulation of c- Myc Oncogene Expression. Chem. A Eur. J. 2009, 15, 13008–13021. [Google Scholar] [CrossRef]
- Bandeira, S.; Gonzalez-Garcia, J.; Pensa, E.; Albrecht, T.; Vilar, R. A Redox-Activated G-Quadruplex DNA Binder Based on a Platinum(IV)-Salphen Complex. Angew. Chem. Int. Ed. 2018, 57, 310–313. [Google Scholar] [CrossRef]
- Butler, J.S.; Sadler, P.J. Targeted Delivery of Platinum-Based Anticancer Complexes. Curr. Opin. Chem. Biol. 2013, 17, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Pathak, R.K.; Marrache, S.; Choi, J.H.; Berding, T.B.; Dhar, S. The Prodrug Platin- A: Simultaneous Release of Cisplatin and Aspirin. Angew. Chem. Int. Ed. 2014, 53, 1963–1967. [Google Scholar] [CrossRef] [PubMed]
- Graf, N.; Lippard, S.J. Redox Activation of Metal-Based Prodrugs as a Strategy for Drug Delivery. Adv. Drug Deliv. Rev. 2012, 64, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Bradáč, O.; Zimmermann, T.; Burda, J.v. Can Satraplatin Be Hydrated before the Reduction Process Occurs? The DFT Computational Study. J. Mol. Model. 2013, 19, 4669–4680. [Google Scholar] [CrossRef] [PubMed]
- Ritacco, I.; Mazzone, G.; Russo, N.; Sicilia, E. Investigation of the Inertness to Hydrolysis of Platinum(IV) Prodrugs. Inorg. Chem. 2016, 55, 1580–1586. [Google Scholar] [CrossRef]
- Wexselblatt, E.; Yavin, E.; Gibson, D. Platinum(IV) Prodrugs with Haloacetato Ligands in the Axial Positions Can Undergo Hydrolysis under Biologically Relevant Conditions. Angew. Chem. Int. Ed. 2013, 52, 6059–6062. [Google Scholar] [CrossRef]
- Dabbish, E.; Ponte, F.; Russo, N.; Sicilia, E. Antitumor Platinium(IV) Prodrugs: A Systematic Computational Exploration of Their Reduction Mechanism by L-Ascorbic Acid. Inorg. Chem. 2019, 58, 3851–3860. [Google Scholar] [CrossRef]
- Ejehi, Z.; Ariafard, A. A Computational Mechanistic Investigation into the Reduction of Pt(IV) Prodrugs with Two Axial Chlorides by Biological Reductants. Chem. Commun. 2017, 53, 1413–1416. [Google Scholar] [CrossRef]
- McCormick, M.C.; Keijzer, K.; Polavarapu, A.; Schultz, F.A.; Baik, M.-H. Understanding Intrinsically Irreversible, Non-Nernstian, Two-Electron Redox Processes: A Combined Experimental and Computational Study of the Electrochemical Activation of Platinum(IV) Antitumor Prodrugs. J. Am. Chem. Soc. 2014, 136, 8992–9000. [Google Scholar] [CrossRef]
- Scoditti, S.; Mazzone, G.; Sanna, N.; Sicilia, E. Computational Exploration of the Synergistic Anticancer Effect of a Multi-Action Ru(II)–Pt(IV) Conjugate. Inorg. Chem. 2022, 61, 12903–12912. [Google Scholar] [CrossRef]
- Scoditti, S.; Vigna, V.; Dabbish, E.; Sicilia, E. Iodido Equatorial Ligands Influence on the Mechanism of Action of Pt(IV) and Pt(II) Anti-cancer Complexes: A DFT Computational Study. J. Comput. Chem. 2021, 42, 608–619. [Google Scholar] [CrossRef]
- Ponte, F.; Russo, N.; Sicilia, E. Insights from Computations on the Mechanism of Reduction by Ascorbic Acid of Pt IV Prodrugs with Asplatin and Its Chlorido and Bromido Analogues as Model Systems. Chem. A Eur. J. 2018, 24, 9572–9580. [Google Scholar] [CrossRef]
- Gramatica, P.; Papa, E.; Luini, M.; Monti, E.; Gariboldi, M.B.; Ravera, M.; Gabano, E.; Gaviglio, L.; Osella, D. Antiproliferative Pt(IV) Complexes: Synthesis, Biological Activity, and Quantitative Structure–Activity Relationship Modeling. JBIC J. Biol. Inorg. Chem. 2010, 15, 1157–1169. [Google Scholar] [CrossRef]
- Wexselblatt, E.; Raveendran, R.; Salameh, S.; Friedman-Ezra, A.; Yavin, E.; Gibson, D. On the Stability of Pt IV Pro-Drugs with Haloacetato Ligands in the Axial Positions. Chem. A Eur. J. 2015, 21, 3108–3114. [Google Scholar] [CrossRef]
- Ambrus, A.; Chen, D.; Dai, J.; Jones, R.A.; Yang, D. Solution Structure of the Biologically Relevant G-Quadruplex Element in the Human c-MYC Promoter. Implications for G-Quadruplex Stabilization. Biochemistry 2005, 44, 2048–2058. [Google Scholar] [CrossRef]
- Mulliri, S.; Laaksonen, A.; Spanu, P.; Farris, R.; Farci, M.; Mingoia, F.; Roviello, G.N.; Mocci, F. Spectroscopic and In Silico Studies on the Interaction of Substituted Pyrazolo [1,2-a]Benzo [1–4]Tetrazine-3-One Derivatives with c-Myc G4-DNA. Int. J. Mol. Sci. 2021, 22, 6028. [Google Scholar] [CrossRef]
- Bonsignore, R.; Terenzi, A.; Spinello, A.; Martorana, A.; Lauria, A.; Almerico, A.M.; Keppler, B.K.; Barone, G. G-Quadruplex vs. Duplex-DNA Binding of Nickel(II) and Zinc(II) Schiff Base Complexes. J. Inorg. Biochem. 2016, 161, 115–121. [Google Scholar] [CrossRef]
- Bonsignore, R.; Russo, F.; Terenzi, A.; Spinello, A.; Lauria, A.; Gennaro, G.; Almerico, A.M.; Keppler, B.K.; Barone, G. The Interaction of Schiff Base Complexes of Nickel(II) and Zinc(II) with Duplex and G-Quadruplex DNA. J. Inorg. Biochem. 2018, 178, 106–114. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Shan, Y.; Kim, E.T.; Eastwood, M.P.; Dror, R.O.; Seeliger, M.A.; Shaw, D.E. How Does a Drug Molecule Find Its Target Binding Site? J. Am. Chem. Soc. 2011, 133, 9181–9183. [Google Scholar] [CrossRef]
- Decherchi, S.; Berteotti, A.; Bottegoni, G.; Rocchia, W.; Cavalli, A. The Ligand Binding Mechanism to Purine Nucleoside Phosphorylase Elucidated via Molecular Dynamics and Machine Learning. Nat. Commun. 2015, 6, 6155. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, M.P.; Haldar, S.; Morales, J.C.; Mulholland, A.J.; Galan, M.C. Enhanced Sampling Molecular Dynamics Simulations Correctly Predict the Diverse Activities of a Series of Stiff-Stilbene G-Quadruplex DNA Ligands. Chem. Sci. 2021, 12, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- Castelli, M.; Serapian, S.A.; Marchetti, F.; Triveri, A.; Pirota, V.; Torielli, L.; Collina, S.; Doria, F.; Freccero, M.; Colombo, G. New Perspectives in Cancer Drug Development: Computational Advances with an Eye to Design. RSC Med. Chem. 2021, 12, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Spinello, A.; Barone, G.; Grunenberg, J. Molecular Recognition of Naphthalene Diimide Ligands by Telomeric Quadruplex-DNA: The Importance of the Protonation State and Mediated Hydrogen Bonds. Phys. Chem. Chem. Phys. 2016, 18, 2871–2877. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. G16_C01, Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Andrae, D.; Häussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-Adjustedab Initio Pseudopotentials for the Second and Third Row Transition Elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Miertuš, S.; Tomasi, J. Approximate Evaluations of the Electrostatic Free Energy and Internal Energy Changes in Solution Processes. Chem. Phys. 1982, 65, 239–245. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic Interaction of a Solute with a Continuum. A Direct Utilizaion of AB Initio Molecular Potentials for the Prevision of Solvent Effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Fukui, K. A Formulation of the Reaction Coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A Software Program for PK a Prediction and Protonation State Generation for Drug-like Molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Li, P.; Merz, K.M. MCPB.Py: A Python Based Metal Center Parameter Builder. J. Chem. Inf. Model. 2016, 56, 599–604. [Google Scholar] [CrossRef]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A Refined Force Field for DNA Simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Mahoney, M.W.; Jorgensen, W.L. Diffusion Constant of the TIP5P Model of Liquid Water. J. Chem. Phys. 2001, 114, 363. [Google Scholar] [CrossRef]
- Mahoney, M.W.; Jorgensen, W.L. A Five-Site Model for Liquid Water and the Reproduction of the Density Anomaly by Rigid, Nonpolarizable Potential Functions. J. Chem. Phys. 2000, 112, 8910–8922. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vigna, V.; Scoditti, S.; Spinello, A.; Mazzone, G.; Sicilia, E. Anticancer Activity, Reduction Mechanism and G-Quadruplex DNA Binding of a Redox-Activated Platinum(IV)–Salphen Complex. Int. J. Mol. Sci. 2022, 23, 15579. https://doi.org/10.3390/ijms232415579
Vigna V, Scoditti S, Spinello A, Mazzone G, Sicilia E. Anticancer Activity, Reduction Mechanism and G-Quadruplex DNA Binding of a Redox-Activated Platinum(IV)–Salphen Complex. International Journal of Molecular Sciences. 2022; 23(24):15579. https://doi.org/10.3390/ijms232415579
Chicago/Turabian StyleVigna, Vincenzo, Stefano Scoditti, Angelo Spinello, Gloria Mazzone, and Emilia Sicilia. 2022. "Anticancer Activity, Reduction Mechanism and G-Quadruplex DNA Binding of a Redox-Activated Platinum(IV)–Salphen Complex" International Journal of Molecular Sciences 23, no. 24: 15579. https://doi.org/10.3390/ijms232415579
APA StyleVigna, V., Scoditti, S., Spinello, A., Mazzone, G., & Sicilia, E. (2022). Anticancer Activity, Reduction Mechanism and G-Quadruplex DNA Binding of a Redox-Activated Platinum(IV)–Salphen Complex. International Journal of Molecular Sciences, 23(24), 15579. https://doi.org/10.3390/ijms232415579