

Escherichia coli as a New Platform for the Fast Production of Vault-like Nanoparticles: An Optimized Protocol

 , , and
, , and
Abstract
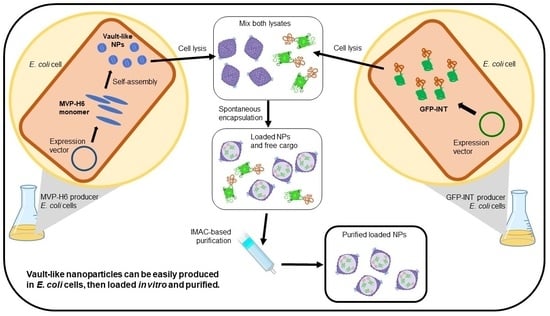
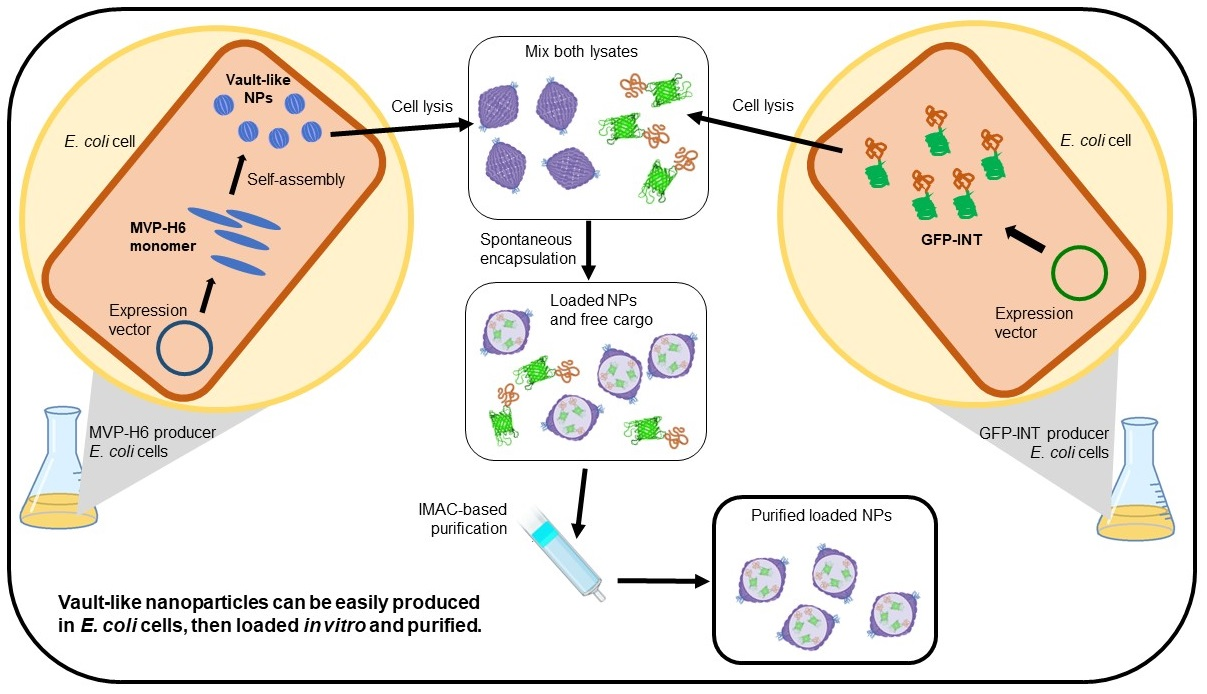{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
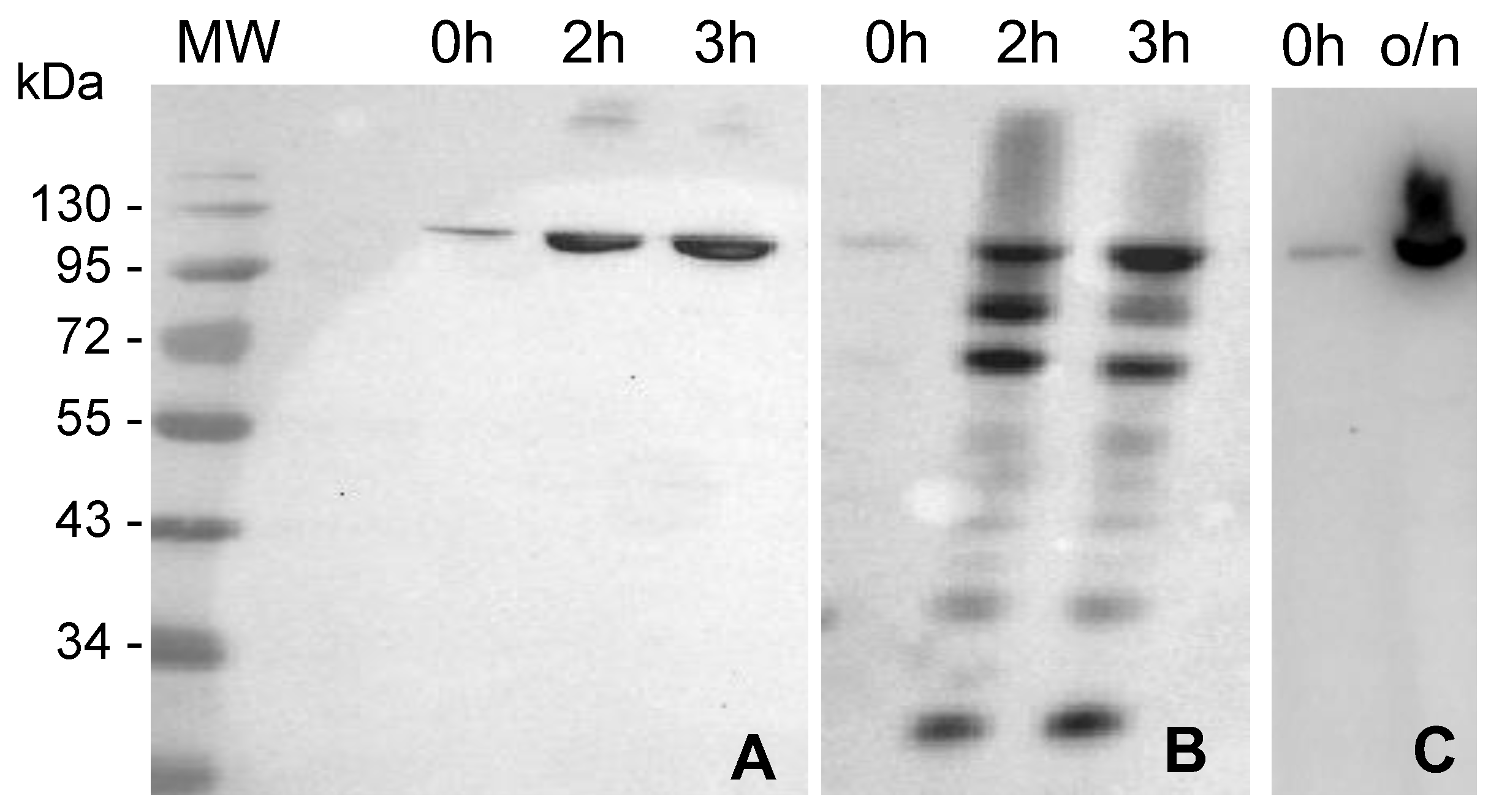
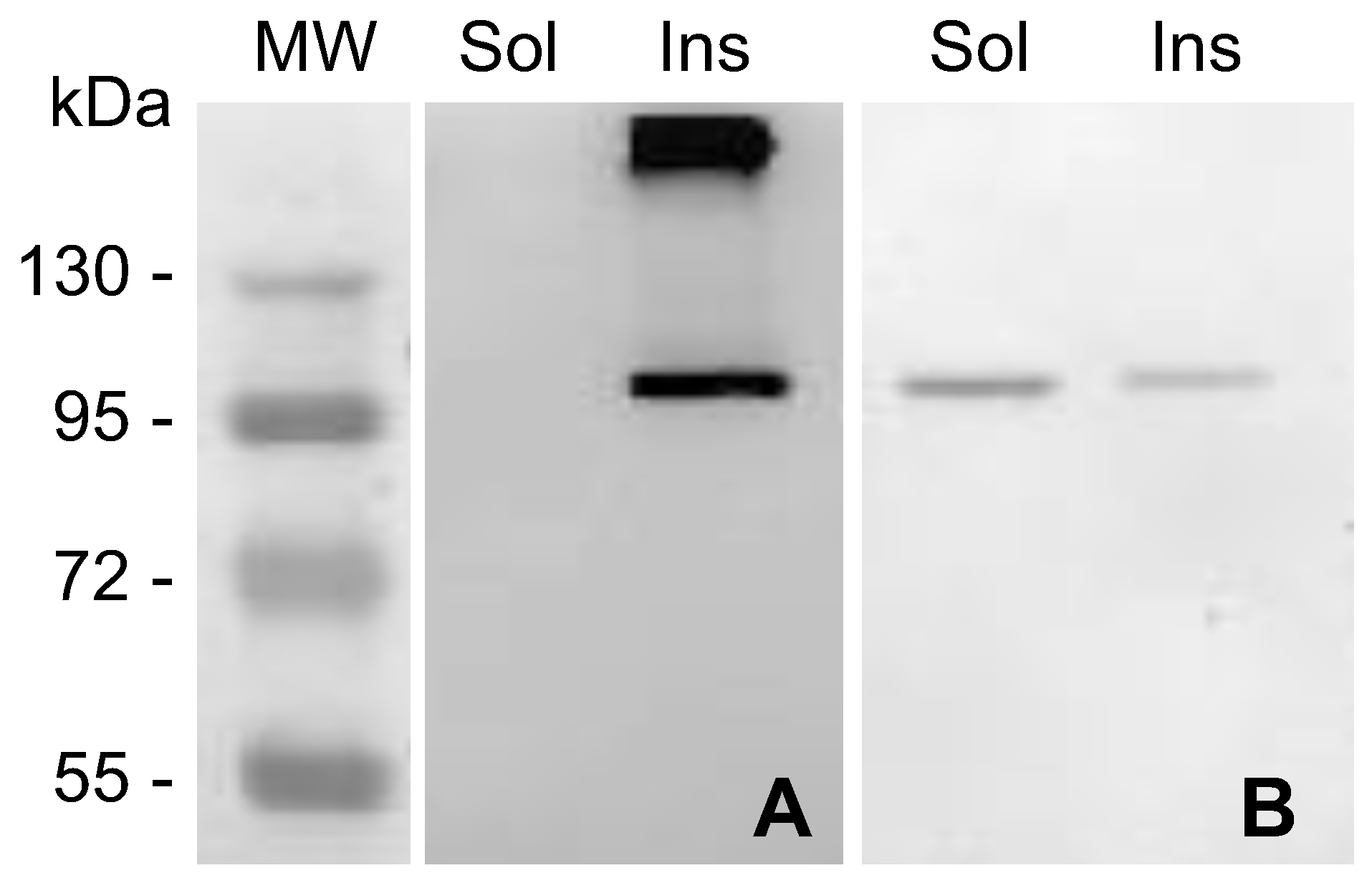
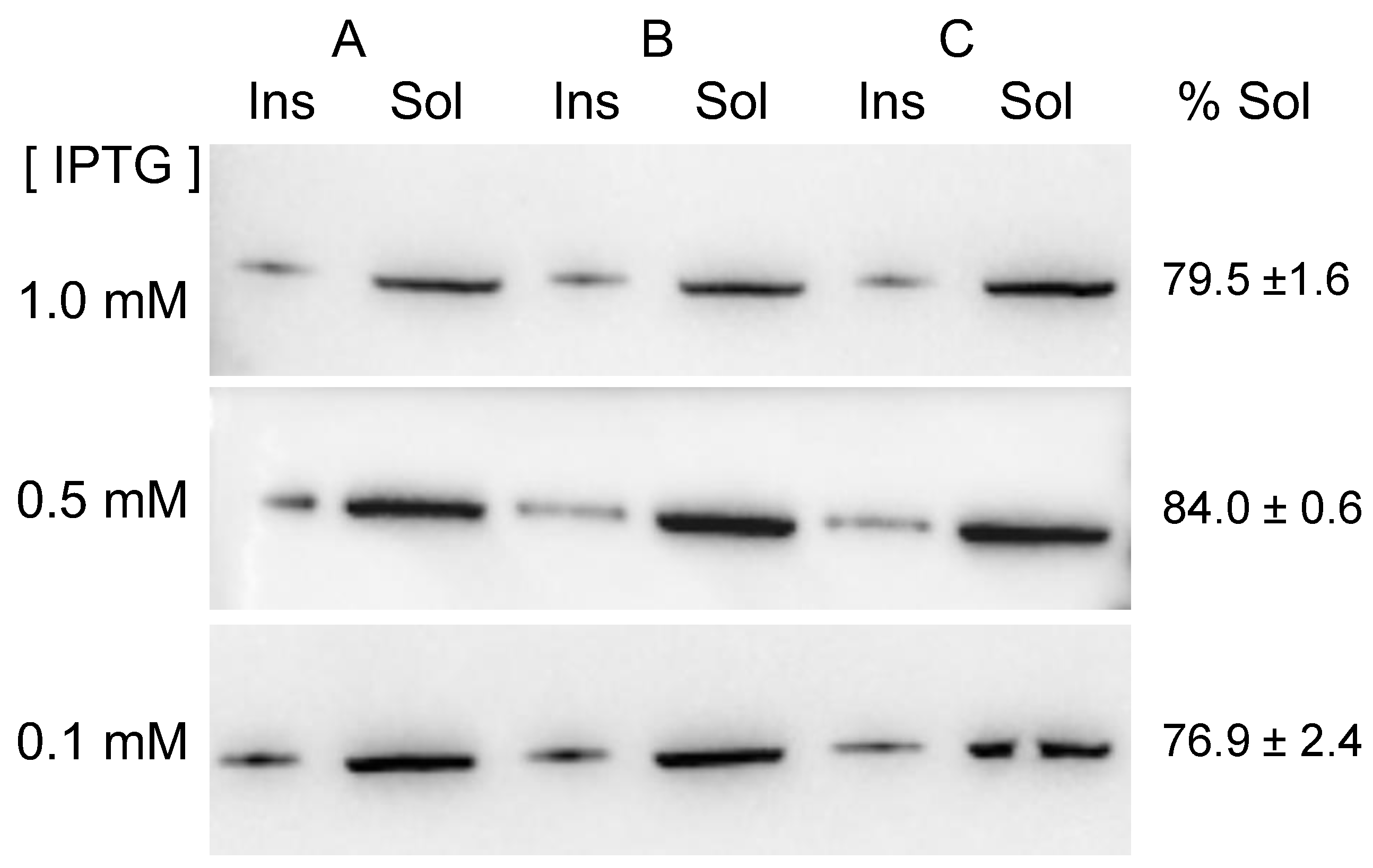
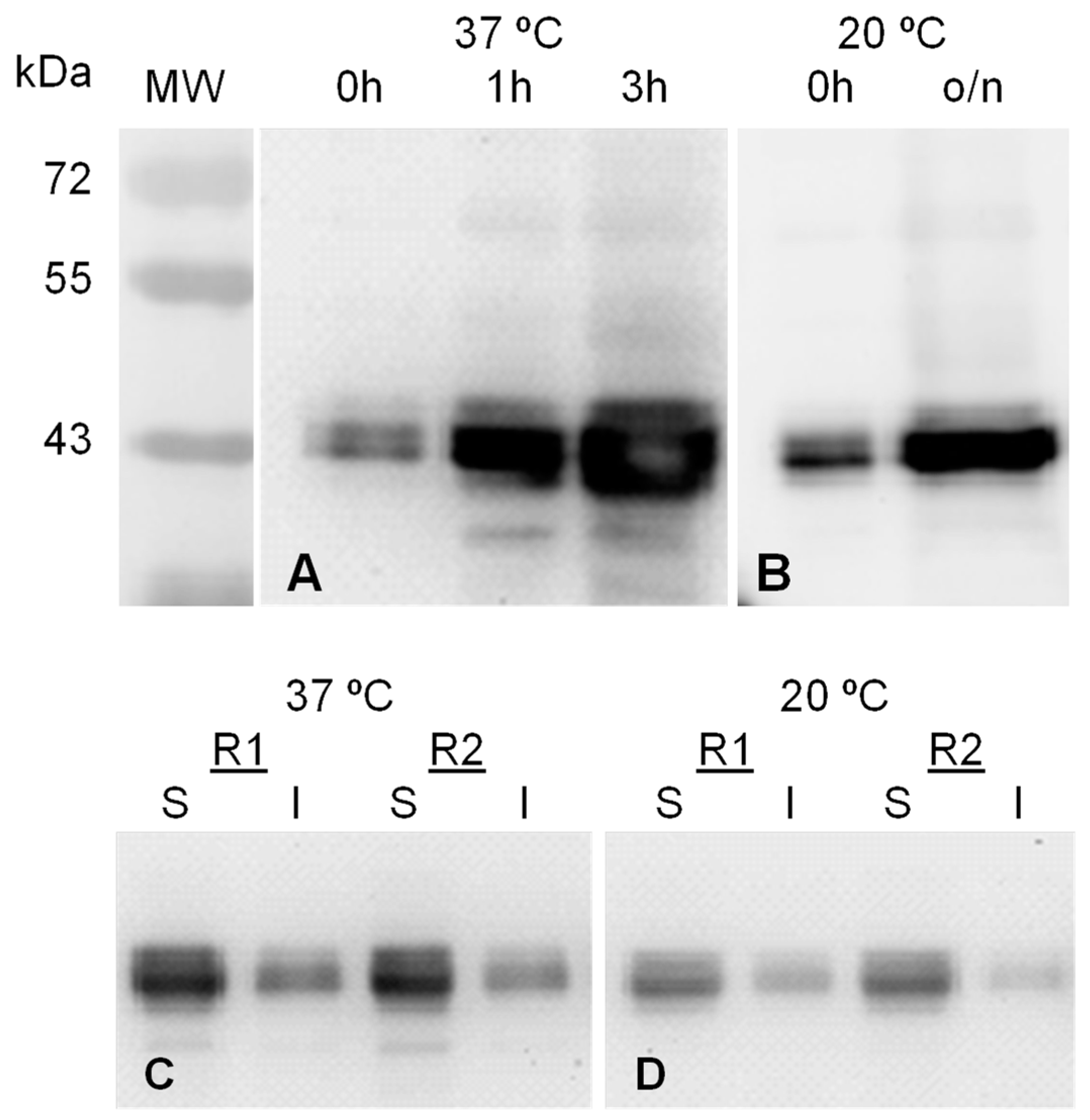
2.1. MVP-H6 Protein Expression and Solubility
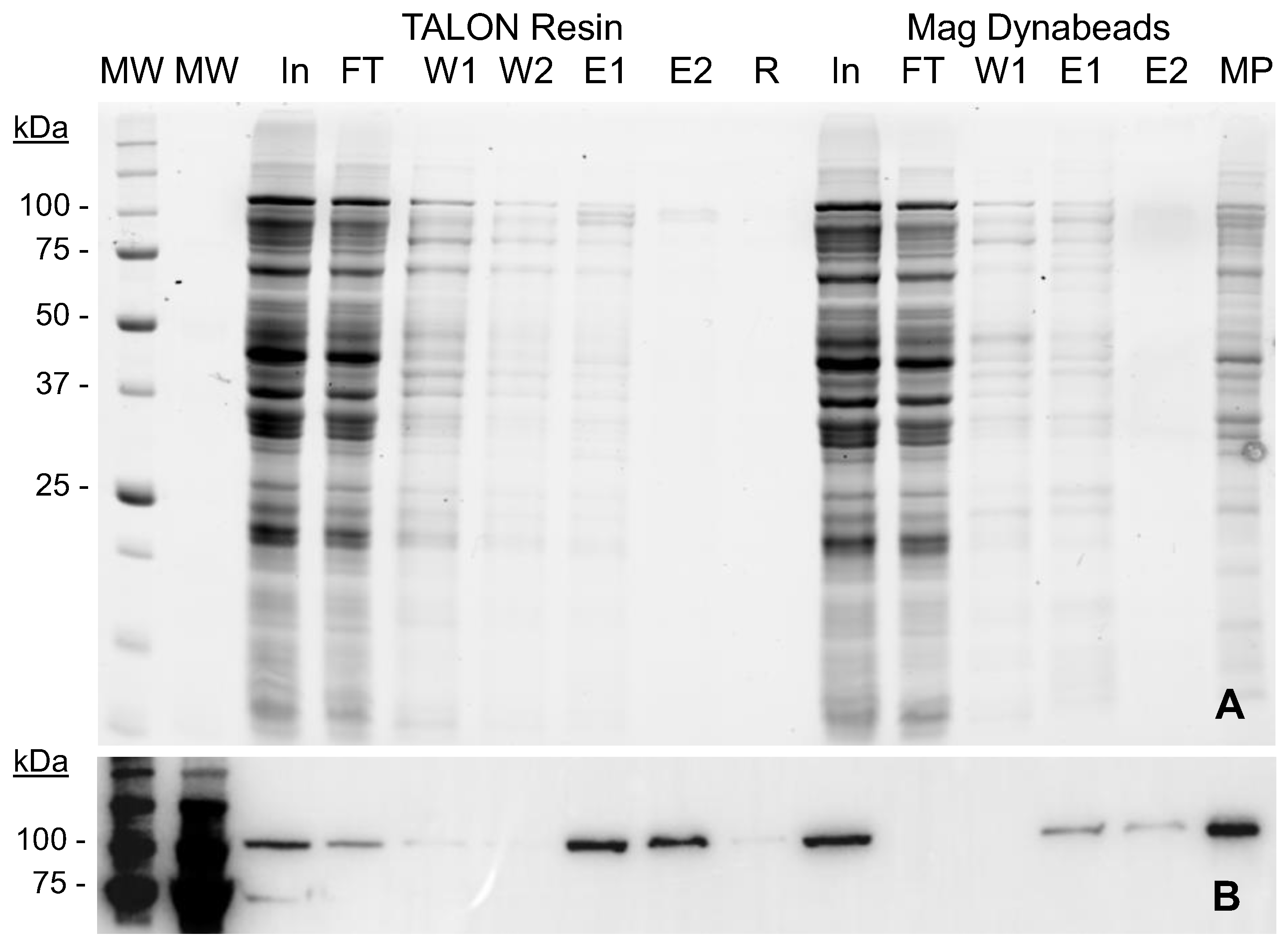
2.2. MVP-H6 Protein Purification
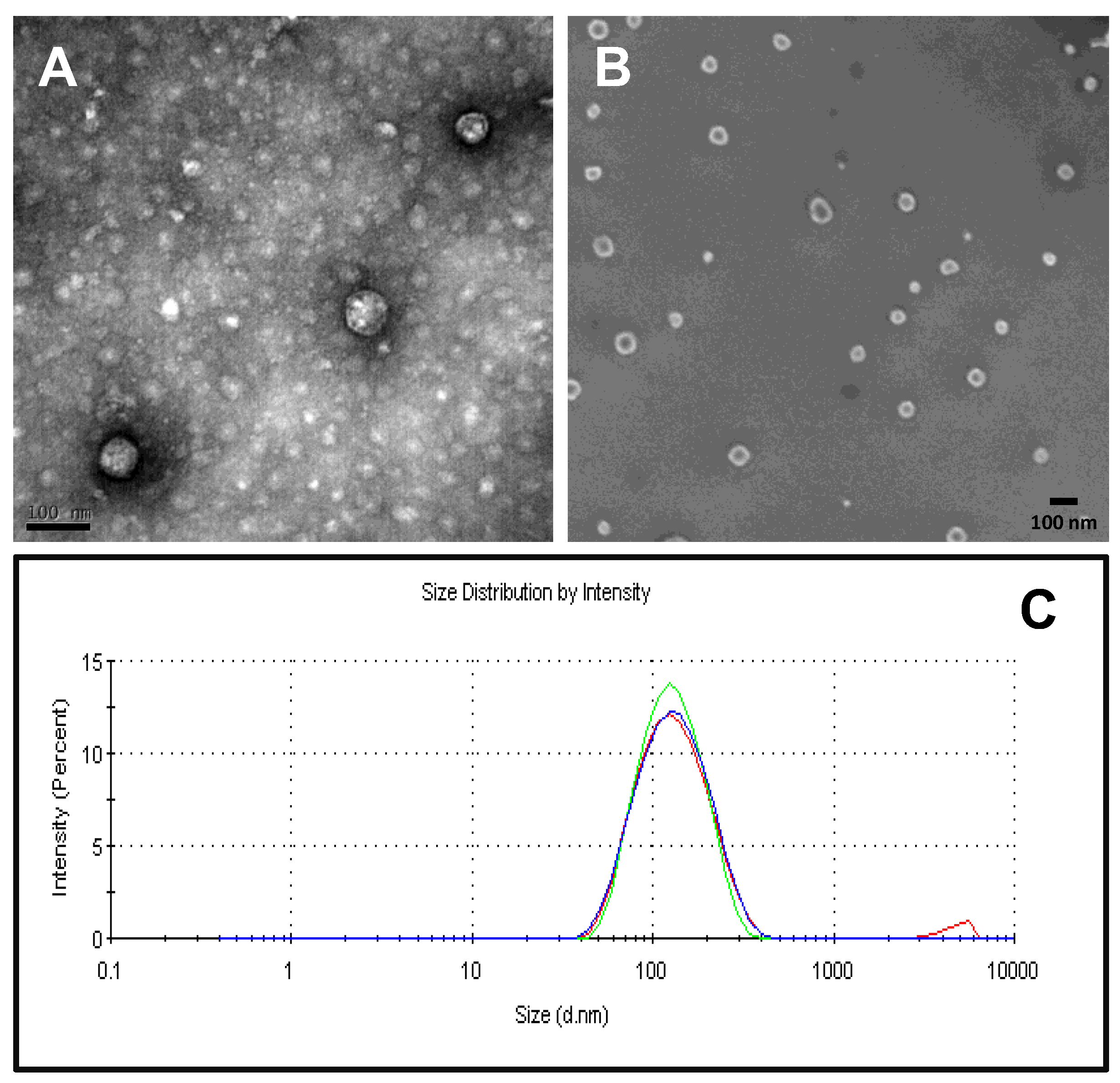
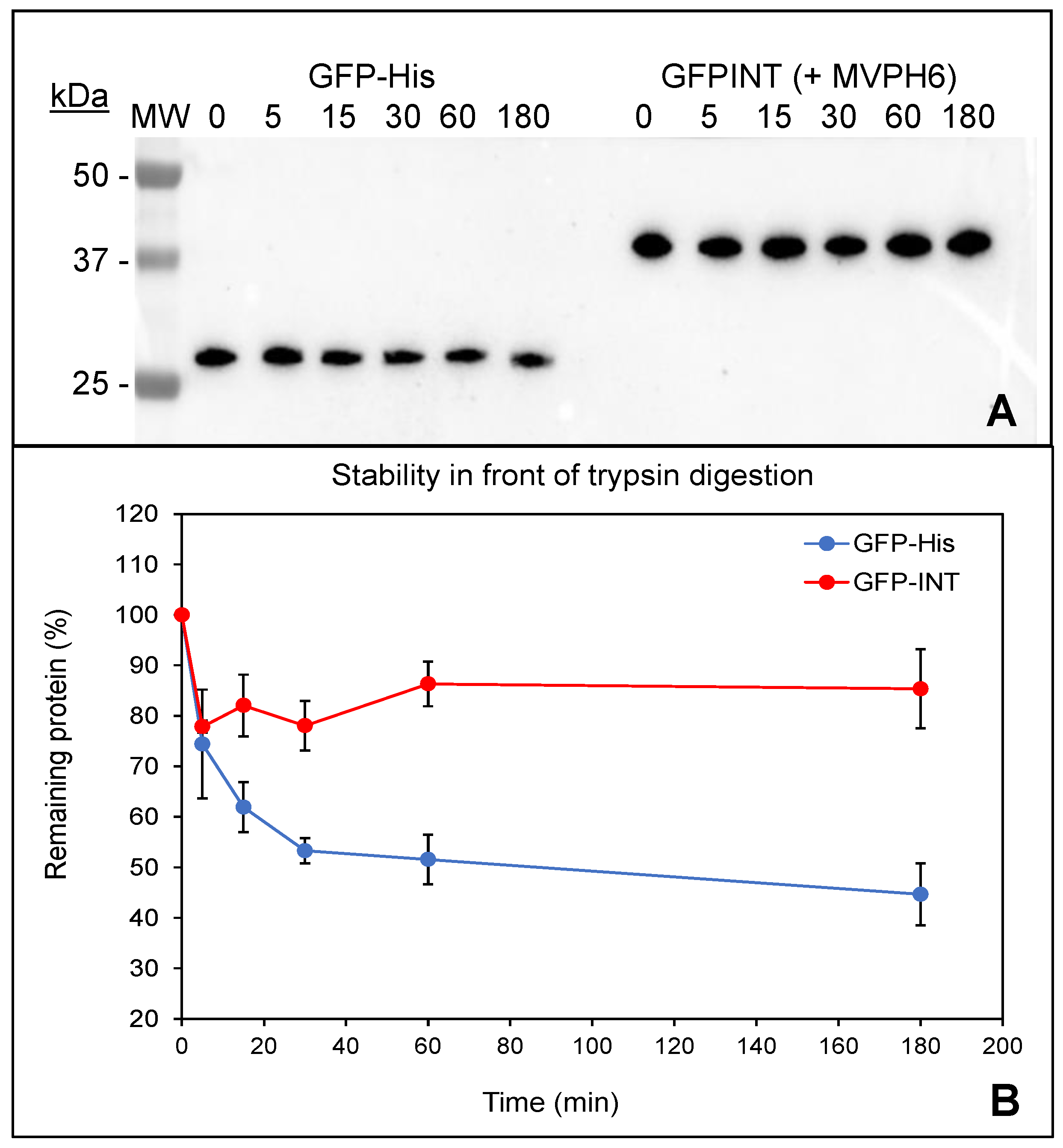
2.3. Physico-Chemical Characterization of Recombinant Nanoparticles
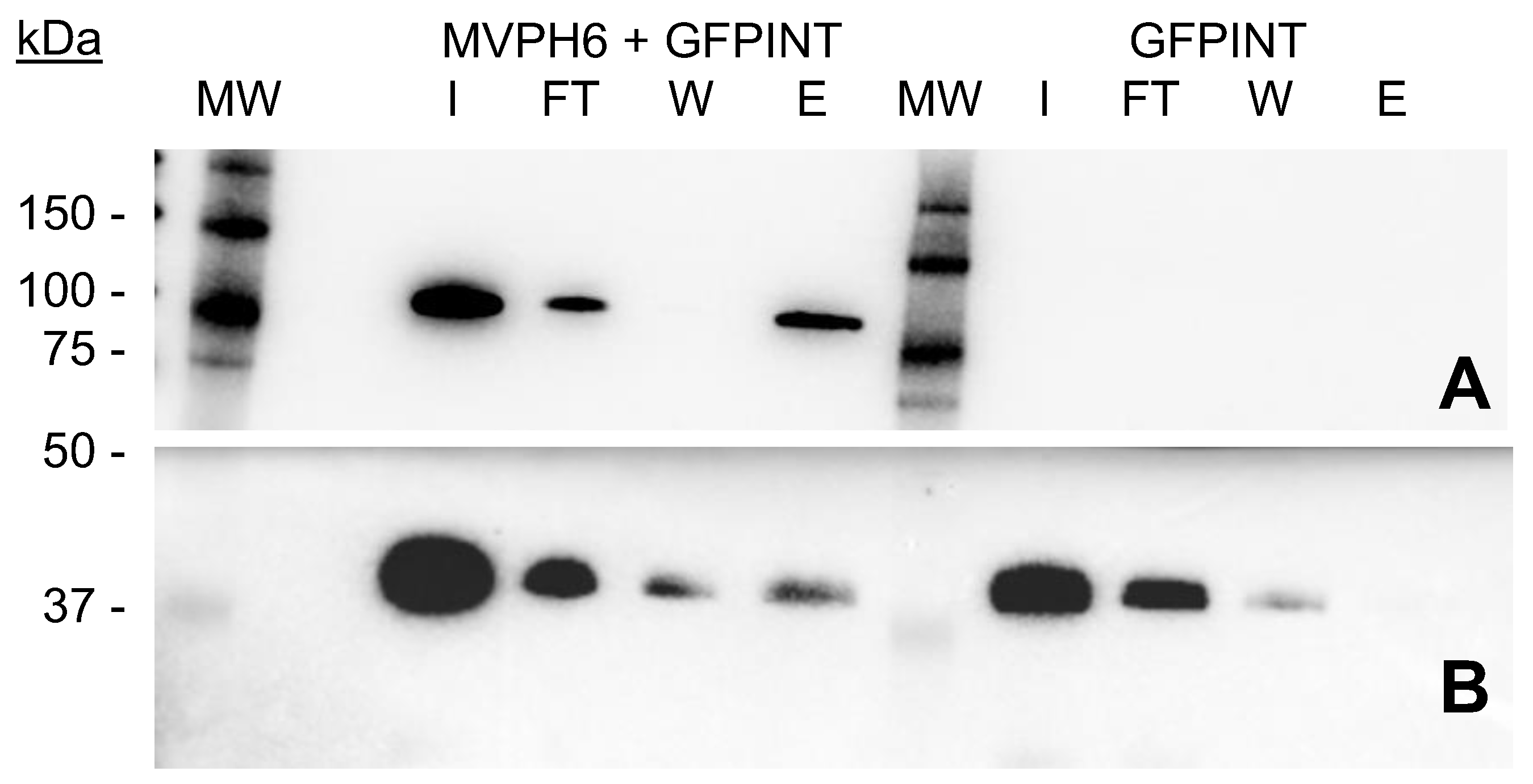
2.4. GFP-INT Protein Expression and Solubility
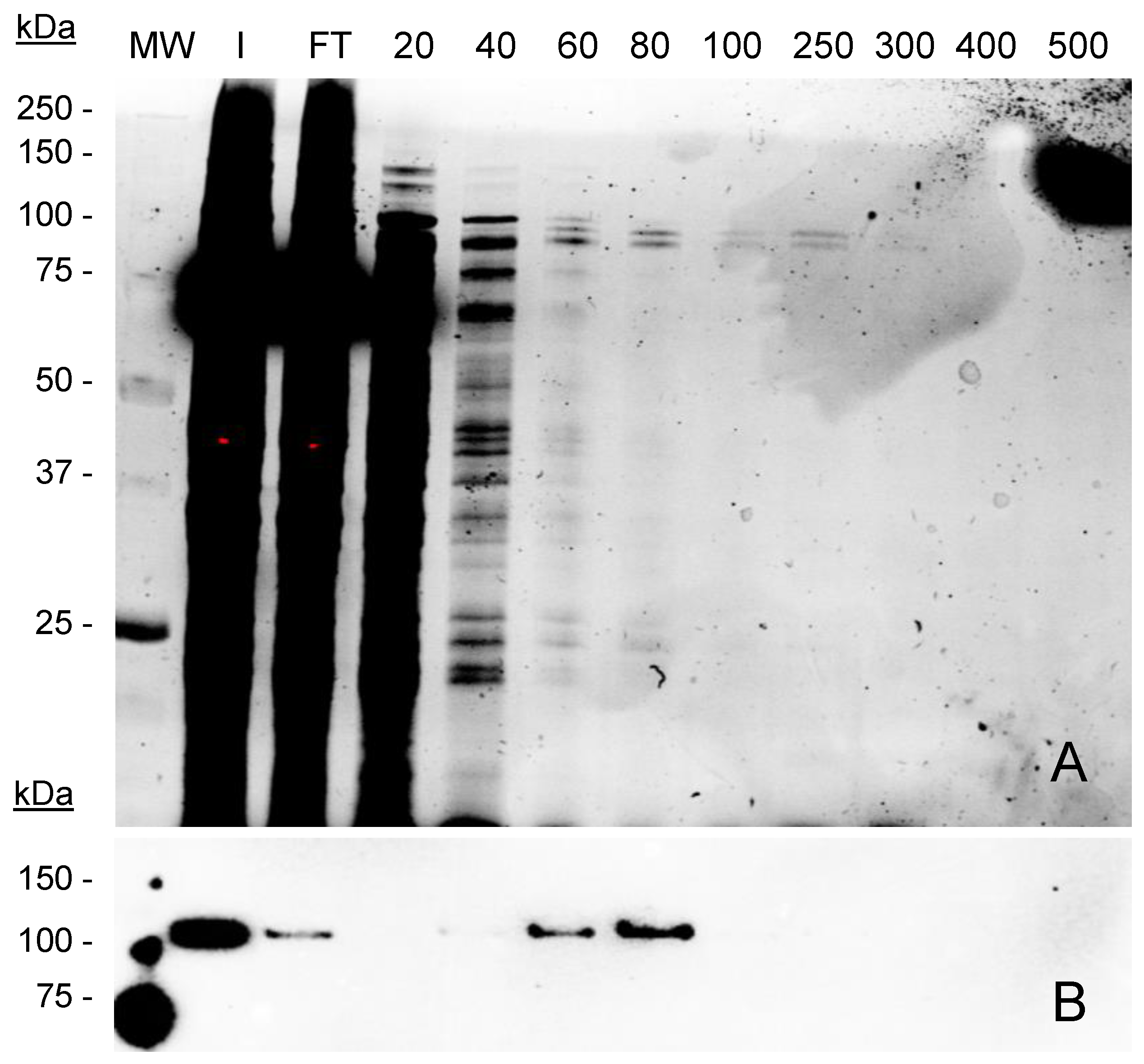
2.5. Encapsulation and Purification of GFP-INT
3. Discussion
4. Materials and Methods
4.1. Synthesis and Cloning of Recombinant Genes
4.2. Escherichia coli Strains and Expression Plasmids
4.3. Recombinant Protein Expression
4.4. Bacterial Cell Fractionation
4.5. SDS-PAGE and Western Blot Analyses
4.6. Protein Purification
4.7. Physico-Chemical Characterization of Recombinant Vault-like Nanoparticles
4.8. GFP-INT Protein Encapsulation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maham, A.; Tang, Z.; Wu, H.; Wang, J.; Lin, Y. Protein-based nanomedicine platforms for drug delivery. Small 2009, 5, 1706–1721. [Google Scholar] [CrossRef] [PubMed]
- McClements, D.J. Encapsulation, protection, and delivery of bioactive proteins and peptides using nanoparticle and microparticle systems: A review. Adv. Colloid Interface Sci. 2018, 253, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Du, J.; Gu, Z.; Liang, M.; Hu, Y.; Zhang, W.; Priceman, S.; Wu, L.; Zhou, Z.H.; Liu, Z.; et al. A novel intracellular protein delivery platform based on single-protein nanocapsules. Nat. Nanotechnol. 2010, 5, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Miralles, N.; Rodríguez-Carmona, E.; Corchero, J.L.; García-Fruitós, E.; Vázquez, E.; Villaverde, A. Engineering protein self-assembling in protein-based nanomedicines for drug delivery and gene therapy. Crit. Rev. Biotechnol. 2015, 35, 209–221. [Google Scholar] [CrossRef]
- Lee, L.A.; Wang, Q. Adaptations of nanoscale viruses and other protein cages for medical applications. Nanomedicine 2006, 2, 137–149. [Google Scholar] [CrossRef]
- Flenniken, M.L.; Uchida, M.; Liepold, L.O.; Kang, S.; Young, M.J.; Douglas, T. A library of protein cage architectures as nanomaterials. Curr. Top. Microbiol. Immunol. 2009, 327, 71–93. [Google Scholar] [CrossRef]
- Uchida, M.; Klem, M.T.; Allen, M.; Suci, P.; Flenniken, M.; Gillitzer, E.; Varpness, Z.; Liepold, L.O.; Young, M.; Douglas, T. Biological Containers: Protein Cages as Multifunctional Nanoplatforms. Adv. Mater. 2007, 19, 1025–1042. [Google Scholar] [CrossRef]
- Molino, N.M.; Wang, S.-W. Caged protein nanoparticles for drug delivery. Curr. Opin. Biotechnol. 2014, 28, 75–82. [Google Scholar] [CrossRef]
- Schoonen, L.; van Hest, J.C.M. Functionalization of protein-based nanocages for drug delivery applications. Nanoscale 2014, 6, 7124–7141. [Google Scholar] [CrossRef]
- Bhaskar, S.; Lim, S. Engineering protein nanocages as carriers for biomedical applications. NPG Asia Mater. 2017, 9, e371. [Google Scholar] [CrossRef]
- Li, F.; Wang, Q. Fabrication of nanoarchitectures templated by virus-based nanoparticles: Strategies and applications. Small 2014, 10, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Giessen, T.W. Encapsulins: Microbial nanocompartments with applications in biomedicine, nanobiotechnology and materials science. Curr. Opin. Chem. Biol. 2016, 34, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Lee, J.; Min, J.; Kang, S. Developing genetically engineered encapsulin protein cage nanoparticles as a targeted delivery nanoplatform. Biomacromolecules 2014, 15, 3794–3801. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Lee, N.K.; Kim, I.-S. Bioengineered protein-based nanocage for drug delivery. Adv. Drug Deliv. Rev. 2016, 106, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Raeeszadeh-Sarmazdeh, M.; Hartzell, E.; Price, J.V.; Chen, W. Protein Nanoparticles as Multifunctional Biocatalysts and Health Assessment Sensors. Curr. Opin. Chem. Eng. 2016, 13, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Juan, A.; Carreño, A.; Mendoza, R.; Corchero, J.L. Latest Advances in the Development of Eukaryotic Vaults as Targeted Drug Delivery Systems. Pharmaceutics 2019, 11, 300. [Google Scholar] [CrossRef]
- Tanaka, H.; Kato, K.; Yamashita, E.; Sumizawa, T.; Zhou, Y.; Yao, M.; Iwasaki, K.; Yoshimura, M.; Tsukihara, T. The structure of rat liver vault at 3.5 angstrom resolution. Science 2009, 323, 384–388. [Google Scholar] [CrossRef]
- Tanaka, H.; Tsukihara, T. Structural studies of large nucleoprotein particles, vaults. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 416–433. [Google Scholar] [CrossRef]
- Stephen, A.G.; Raval-Fernandes, S.; Huynh, T.; Torres, M.; Kickhoefer, V.A.; Rome, L.H. Assembly of vault-like particles in insect cells expressing only the major vault protein. J. Biol. Chem. 2001, 276, 23217–23220. [Google Scholar] [CrossRef]
- Kickhoefer, V.A.; Garcia, Y.; Mikyas, Y.; Johansson, E.; Zhou, J.C.; Raval-Fernandes, S.; Minoofar, P.; Zink, J.I.; Dunn, B.; Stewart, P.L.; et al. Engineering of vault nanocapsules with enzymatic and fluorescent properties. Proc. Natl. Acad. Sci. USA 2005, 102, 4348–4352. [Google Scholar] [CrossRef]
- Poderycki, M.J.; Kickhoefer, V.A.; Kaddis, C.S.; Raval-Fernandes, S.; Johansson, E.; Zink, J.I.; Loo, J.A.; Rome, L.H. The vault exterior shell is a dynamic structure that allows incorporation of vault-associated proteins into its interior. Biochemistry 2006, 45, 12184–12193. [Google Scholar] [CrossRef] [PubMed]
- Kickhoefer, V.A.; Han, M.; Raval-Fernandes, S.; Poderycki, M.J.; Moniz, R.J.; Vaccari, D.; Silvestry, M.; Stewart, P.L.; Kelly, K.A.; Rome, L.H. Targeting vault nanoparticles to specific cell surface receptors. ACS Nano 2009, 3, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-Y.; Wiethoff, C.M.; Kickhoefer, V.A.; Rome, L.H.; Nemerow, G.R. Vault nanoparticles containing an adenovirus-derived membrane lytic protein facilitate toxin and gene transfer. ACS Nano 2009, 3, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Kar, U.K.; Jiang, J.; Champion, C.I.; Salehi, S.; Srivastava, M.; Sharma, S.; Rabizadeh, S.; Niazi, K.; Kickhoefer, V.; Rome, L.H.; et al. Vault nanocapsules as adjuvants favor cell-mediated over antibody-mediated immune responses following immunization of mice. PLoS ONE 2012, 7, e38553. [Google Scholar] [CrossRef]
- Kar, U.K.; Srivastava, M.K.; Andersson, A.; Baratelli, F.; Huang, M.; Kickhoefer, V.A.; Dubinett, S.M.; Rome, L.H.; Sharma, S. Novel CCL21-vault nanocapsule intratumoral delivery inhibits lung cancer growth. PLoS ONE 2011, 6, e18758. [Google Scholar] [CrossRef]
- Champion, C.I.; Kickhoefer, V.A.; Liu, G.; Moniz, R.J.; Freed, A.S.; Bergmann, L.L.; Vaccari, D.; Raval-Fernandes, S.; Chan, A.M.; Rome, L.H.; et al. A vault nanoparticle vaccine induces protective mucosal immunity. PLoS ONE 2009, 4, e5409. [Google Scholar] [CrossRef]
- Wang, M.; Abad, D.; Kickhoefer, V.A.; Rome, L.H.; Mahendra, S. Vault Nanoparticles Packaged with Enzymes as an Efficient Pollutant Biodegradation Technology. ACS Nano 2015, 9, 10931–10940. [Google Scholar] [CrossRef]
- Voth, B.L.; Pelargos, P.E.; Barnette, N.E.; Bhatt, N.S.; Chen, C.H.J.; Lagman, C.; Chung, L.K.; Nguyen, T.; Sheppard, J.P.; Romiyo, P.; et al. Intratumor injection of CCL21-coupled vault nanoparticles is associated with reduction in tumor volume in an in vivo model of glioma. J. Neurooncol. 2020, 147, 599–605. [Google Scholar] [CrossRef]
- Mrazek, J.; Toso, D.; Ryazantsev, S.; Zhang, X.; Zhou, Z.H.; Fernandez, B.C.; Kickhoefer, V.A.; Rome, L.H. Polyribosomes are molecular 3D nanoprinters that orchestrate the assembly of vault particles. ACS Nano 2014, 8, 11552–11559. [Google Scholar] [CrossRef]
- Matsumoto, N.M.; Buchman, G.W.; Rome, L.H.; Maynard, H.D. Dual pH- and Temperature-Responsive Protein Nanoparticles. Eur. Polym. J. 2015, 69, 532–539. [Google Scholar] [CrossRef]
- Wang, M.; Kickhoefer, V.A.; Rome, L.H.; Foellmer, O.K.; Mahendra, S. Synthesis and assembly of human vault particles in yeast. Biotechnol. Bioeng. 2018, 115, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Walsh, G.; Jefferis, R. Post-translational modifications in the context of therapeutic proteins. Nat. Biotechnol. 2006, 24, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Terpe, K. Overview of bacterial expression systems for heterologous protein production: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2006, 72, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-J.; Lin, H.; Yang, X. Industrial production of recombinant therapeutics in Escherichia coli and its recent advancements. J. Ind. Microbiol. Biotechnol. 2012, 39, 383–399. [Google Scholar] [CrossRef]
- Blattner, F.R.; Plunkett, G., 3rd; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Walsh, G. Biopharmaceutical benchmarks 2014. Nat. Biotechnol. 2014, 32, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Martín, F.; Carreño, A.; Mendoza, R.; Caruana, P.; Rodriguez, F.; Bravo, M.; Benito, A.; Ferrer-Miralles, N.; Céspedes, M.V.; Corchero, J.L. All-in-one biofabrication and loading of recombinant vaults in human cells. Biofabrication 2022, 14, 025018. [Google Scholar] [CrossRef]
- Ishida, O.; Maruyama, K.; Sasaki, K.; Iwatsuru, M. Size-dependent extravasation and interstitial localization of polyethyleneglycol liposomes in solid tumor-bearing mice. Int. J. Pharm. 1999, 190, 49–56. [Google Scholar] [CrossRef]
- Kong, G.; Braun, R.D.; Dewhirst, M.W. Hyperthermia enables tumor-specific nanoparticle delivery: Effect of particle size. Cancer Res. 2000, 60, 4440–4445. [Google Scholar]
- Vinogradov, S.V.; Bronich, T.K.; Kabanov, A.V. Nanosized cationic hydrogels for drug delivery: Preparation, properties and interactions with cells. Adv. Drug Deliv. Rev. 2002, 54, 135–147. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Stolnik, S.; Illum, L.; Davis, S.S. Long circulating microparticulate drug carriers. Adv. Drug Deliv. Rev. 1995, 16, 195–214. [Google Scholar] [CrossRef]
- Rome, L.H.; Kickhoefer, V.A. Development of the vault particle as a platform technology. ACS Nano 2013, 7, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Deo, V.K.; Tsuji, Y.; Yasuda, T.; Kato, T.; Sakamoto, N.; Suzuki, H.; Park, E.Y. Expression of an RSV-gag virus-like particle in insect cell lines and silkworm larvae. J. Virol. Methods 2011, 177, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.-C.; Huang, J.-H.; Lai, C.-W.; Sheng, H.-C.; Shih, S.-R.; Ho, M.-S.; Hu, Y.-C. Expression, purification and characterization of enterovirus-71 virus-like particles. World J. Gastroenterol. 2006, 12, 921–927. [Google Scholar] [CrossRef]
- Morenweiser, R. Downstream processing of viral vectors and vaccines. Gene Ther. 2005, 12 (Suppl. S1), S103–S110. [Google Scholar] [CrossRef]
- Yang, J.; Srinivasan, A.; Sun, Y.; Mrazek, J.; Shu, Z.; Kickhoefer, V.A.; Rome, L.H. Vault nanoparticles engineered with the protein transduction domain, TAT48, enhances cellular uptake. Integr. Biol. 2013, 5, 151–158. [Google Scholar] [CrossRef]
- Galbiati, E.; Avvakumova, S.; La Rocca, A.; Pozzi, M.; Messali, S.; Magnaghi, P.; Colombo, M.; Prosperi, D.; Tortora, P. A fast and straightforward procedure for vault nanoparticle purification and the characterization of its endocytic uptake. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2254–2260. [Google Scholar] [CrossRef]
- Franzreb, M.; Siemann-Herzberg, M.; Hobley, T.J.; Thomas, O.R.T. Protein purification using magnetic adsorbent particles. Appl. Microbiol. Biotechnol. 2006, 70, 505–516. [Google Scholar] [CrossRef]
- Peter, J.F.; Otto, A.M. Magnetic particles as powerful purification tool for high sensitive mass spectrometric screening procedures. Proteomics 2010, 10, 628–633. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández, R.; Carreño, A.; Mendoza, R.; Benito, A.; Ferrer-Miralles, N.; Céspedes, M.V.; Corchero, J.L. Escherichia coli as a New Platform for the Fast Production of Vault-like Nanoparticles: An Optimized Protocol. Int. J. Mol. Sci. 2022, 23, 15543. https://doi.org/10.3390/ijms232415543
Fernández R, Carreño A, Mendoza R, Benito A, Ferrer-Miralles N, Céspedes MV, Corchero JL. Escherichia coli as a New Platform for the Fast Production of Vault-like Nanoparticles: An Optimized Protocol. International Journal of Molecular Sciences. 2022; 23(24):15543. https://doi.org/10.3390/ijms232415543
Chicago/Turabian StyleFernández, Roger, Aida Carreño, Rosa Mendoza, Antoni Benito, Neus Ferrer-Miralles, María Virtudes Céspedes, and José Luis Corchero. 2022. "Escherichia coli as a New Platform for the Fast Production of Vault-like Nanoparticles: An Optimized Protocol" International Journal of Molecular Sciences 23, no. 24: 15543. https://doi.org/10.3390/ijms232415543
APA StyleFernández, R., Carreño, A., Mendoza, R., Benito, A., Ferrer-Miralles, N., Céspedes, M. V., & Corchero, J. L. (2022). Escherichia coli as a New Platform for the Fast Production of Vault-like Nanoparticles: An Optimized Protocol. International Journal of Molecular Sciences, 23(24), 15543. https://doi.org/10.3390/ijms232415543