
Therapeutic Targeting of Glutaminolysis as a Novel Strategy to Combat Cancer Stem Cells
 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Catabolism of Glutamine
3. Cancer Glutaminolysis
3.1. Alteration of Systemic Glutamine Utilization by Cancer
3.2. The Significance of Glutamine Metabolism for Cancer Survival
3.3. Determinants of Glutamine Dependence in Cancer
4. Development of Anti-Glutaminolysis Drugs
4.1. Glutamine Depletion
4.2. Inhibitors of Glutamine Uptake
4.3. Antagonists of Glutamine
4.4. Glutaminase Inhibitors
4.4.1. BPTES
4.4.2. CB-839
4.4.3. Compound 968
4.4.4. C9.22
4.4.5. Combination Therapies
5. Glutaminolysis in CSCs
5.1. Glutamine Dependence in CSCs
5.2. Oncogenotypes That Drive Glutamine Dependence, Stemness, and Treatment Resistance in CSCs
5.2.1. c-MYC
5.2.2. KRAS
5.2.3. TAp73
5.3. GLS1 in CSCs
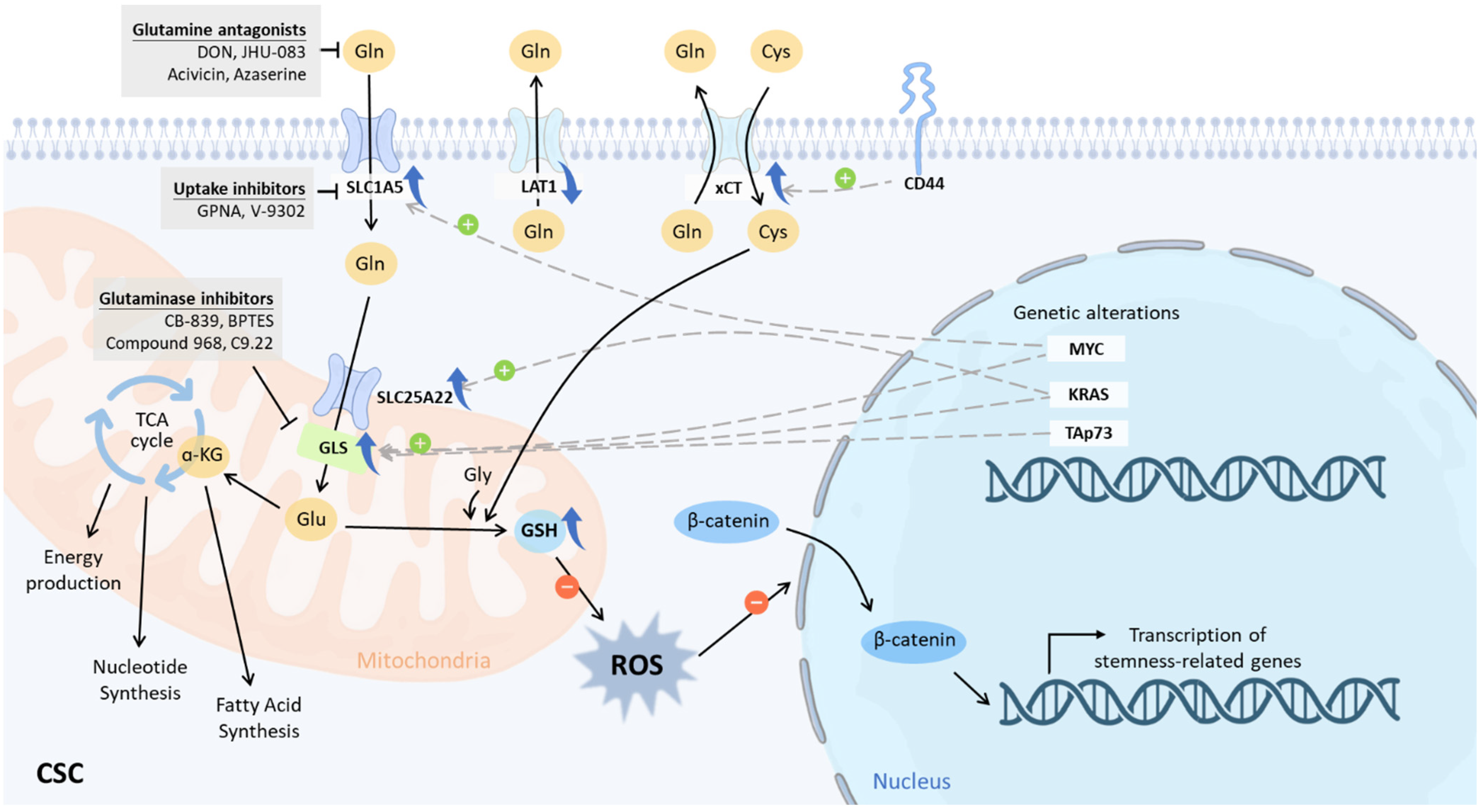
6. Glutaminolysis-Specific Intervention for CSCs
7. Future Perspectives
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [Green Version]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Ghisolfi, L.; Keates, A.C.; Zhang, J.; Xiang, S.; Lee, D.K.; Li, C.J. Induction of cancer cell stemness by chemotherapy. Cell Cycle 2012, 11, 2691–2698. [Google Scholar] [CrossRef] [Green Version]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Touil, Y.; Igoudjil, W.; Corvaisier, M.; Dessein, A.F.; Vandomme, J.; Monte, D.; Stechly, L.; Skrypek, N.; Langlois, C.; Grard, G.; et al. Colon cancer cells escape 5FU chemotherapy-induced cell death by entering stemness and quiescence associated with the c-Yes/YAP axis. Clin. Cancer Res. 2014, 20, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Rogoff, H.A.; Keates, S.; Gao, Y.; Murikipudi, S.; Mikule, K.; Leggett, D.; Li, W.; Pardee, A.B.; Li, C.J. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc. Natl. Acad. Sci. USA 2015, 112, 1839–1844. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Le, A. Glutamine Metabolism in Cancer. Adv. Exp. Med. Biol. 2018, 1063, 13–32. [Google Scholar] [PubMed]
- Mukha, A.; Kahya, U.; Dubrovska, A. Targeting glutamine metabolism and autophagy: The combination for prostate cancer radiosensitization. Autophagy 2021, 17, 3879–3881. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.K.; Lau, P.M.; Kwan, Y.W.; Kong, S.K. Mitochondrial Fuel Dependence on Glutamine Drives Chemo-Resistance in the Cancer Stem Cells of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 3315. [Google Scholar] [CrossRef]
- Jagust, P.; Alcalá, S.; Sainz Jr, B.; Heeschen, C.; Sancho, P. Glutathione metabolism is essential for self-renewal and chemoresistance of pancreatic cancer stem cells. World J. Stem Cells 2020, 12, 1410–1428. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Wicha, M.S. Targeting Cancer Stem Cell Redox Metabolism to Enhance Therapy Responses. Semin Radiat. Oncol. 2019, 29, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar]
- Shen, Y.-A.; Hong, J.; Asaka, R.; Asaka, S.; Hsu, F.-C.; Suryo Rahmanto, Y.; Jung, J.-G.; Chen, Y.-W.; Yen, T.-T.; Tomaszewski, A.; et al. Inhibition of the MYC-Regulated Glutaminase Metabolic Axis Is an Effective Synthetic Lethal Approach for Treating Chemoresistant Ovarian Cancers. Cancer Res. 2020, 80, 4514–4526. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, Z.; Wu, H.; Xi, Z.; Liu, Y. Glutathione selectively modulates the binding of platinum drugs to human copper chaperone Cox17. Biochem. J. 2015, 472, 217–223. [Google Scholar] [CrossRef]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef] [PubMed]
- Park, J.K.; Coffey, N.J.; Limoges, A.; Le, A. The Heterogeneity of Lipid Metabolism in Cancer. Adv. Exp. Med. Biol. 2018, 1063, 33–55. [Google Scholar] [PubMed]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Saqcena, M.; Foster, D.A. Synthetic lethality in KRas-driven cancer cells created by glutamine deprivation. Oncoscience 2015, 2, 807–808. [Google Scholar] [CrossRef]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V.; Hamaker, M.; Sun, P.; Le, A.; Gao, P. Therapeutic targeting of cancer cell metabolism. J. Mol. Med. 2011, 89, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Hirschey, M.D.; DeBerardinis, R.J.; Diehl, A.M.E.; Drew, J.E.; Frezza, C.; Green, M.F.; Jones, L.W.; Ko, Y.H.; Le, A.; Lea, M.A.; et al. Dysregulated metabolism contributes to oncogenesis. Semin Cancer Biol 2015, 35, S129–S150. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.K.; Park, K.G. Targeting Glutamine Metabolism for Cancer Treatment. Biomol. Ther. 2018, 26, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Yang, Q.; Wang, H.; Melick, C.H.; Navlani, R.; Frank, A.R.; Jewell, J.L. Glutamine and asparagine activate mTORC1 independently of Rag GTPases. J. Biol. Chem. 2020, 295, 2890–2899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodineau, C.; Tomé, M.; Courtois, S.; Costa, A.S.H.; Sciacovelli, M.; Rousseau, B.; Richard, E.; Vacher, P.; Parejo-Pérez, C.; Bessede, E.; et al. Two parallel pathways connect glutamine metabolism and mTORC1 activity to regulate glutamoptosis. Nat. Commun. 2021, 12, 4814. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Fendt, S.M.; Li, C.; Poulogiannis, G.; Choo, A.Y.; Chapski, D.J.; Jeong, S.M.; Dempsey, J.M.; Parkhitko, A.; Morrison, T.; et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 2013, 153, 840–854. [Google Scholar] [CrossRef] [Green Version]
- Bhutia, Y.D.; Ganapathy, V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim. Biophys. Acta 2016, 1863, 2531–2539. [Google Scholar] [CrossRef]
- van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [Green Version]
- Bolzoni, M.; Chiu, M.; Accardi, F.; Vescovini, R.; Airoldi, I.; Storti, P.; Todoerti, K.; Agnelli, L.; Missale, G.; Andreoli, R.; et al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: A new attractive target. Blood 2016, 128, 667–679. [Google Scholar] [CrossRef]
- Abu Aboud, O.; Habib, S.L.; Trott, J.; Stewart, B.; Liang, S.; Chaudhari, A.J.; Sutcliffe, J.; Weiss, R.H. Glutamine Addiction in Kidney Cancer Suppresses Oxidative Stress and Can Be Exploited for Real-Time Imaging. Cancer Res. 2017, 77, 6746–6758. [Google Scholar] [CrossRef] [Green Version]
- Vanhove, K.; Derveaux, E.; Graulus, G.J.; Mesotten, L.; Thomeer, M.; Noben, J.P.; Guedens, W.; Adriaensens, P. Glutamine Addiction and Therapeutic Strategies in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 252. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.W.; Lee, M.; Lee, Y.S.; Kim, S.H.; Lee, J.C.; Park, J.J.; Nam, H.Y.; Kim, M.R.; Han, M.W.; Kim, S.W.; et al. p53-dependent glutamine usage determines susceptibility to oxidative stress in radioresistant head and neck cancer cells. Cell. Signal. 2021, 77, 109820. [Google Scholar] [CrossRef] [PubMed]
- Lowman, X.H.; Hanse, E.A.; Yang, Y.; Ishak Gabra, M.B.; Tran, T.Q.; Li, H.; Kong, M. p53 Promotes Cancer Cell Adaptation to Glutamine Deprivation by Upregulating Slc7a3 to Increase Arginine Uptake. Cell Rep. 2019, 26, 3051–3060.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef] [Green Version]
- Tajan, M.; Hock, A.K.; Blagih, J.; Robertson, N.A.; Labuschagne, C.F.; Kruiswijk, F.; Humpton, T.J.; Adams, P.D.; Vousden, K.H. A Role for p53 in the Adaptation to Glutamine Starvation through the Expression of SLC1A3. Cell Metab. 2018, 28, 721–736.e6. [Google Scholar] [CrossRef] [Green Version]
- Durán, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, K.; Bhatia, R.; Rauth, S.; Kisling, A.; Atri, P.; Thompson, C.; Vengoji, R.; Ram Krishn, S.; Shinde, D.; Thomas, V.; et al. Mucin 5AC Serves as the Nexus for β-Catenin/c-Myc Interplay to Promote Glutamine Dependency During Pancreatic Cancer Chemoresistance. Gastroenterology 2022, 162, 253–268.e13. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; He, J.; Guo, Z.; He, H.; Yang, S.; Huang, L.; Pan, H.; He, J. Rhophilin-2 Upregulates Glutamine Synthetase by Stabilizing c-Myc Protein and Confers Resistance to Glutamine Deprivation in Lung Cancer. Front. Oncol. 2020, 10, 571384. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cai, B.; Ding, M.; Su, Z.; Liu, Y.; Shen, L. c-Myc Overexpression Promotes Oral Cancer Cell Proliferation and Migration by Enhancing Glutaminase and Glutamine Synthetase Activity. Am. J. Med. Sci. 2019, 358, 235–242. [Google Scholar] [CrossRef]
- Lukey, M.J.; Greene, K.S.; Erickson, J.W.; Wilson, K.F.; Cerione, R.A. The oncogenic transcription factor c-Jun regulates glutaminase expression and sensitizes cells to glutaminase-targeted therapy. Nat. Commun. 2016, 7, 11321. [Google Scholar] [CrossRef] [Green Version]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef] [PubMed]
- Raho, S.; Capobianco, L.; Malivindi, R.; Vozza, A.; Piazzolla, C.; De Leonardis, F.; Gorgoglione, R.; Scarcia, P.; Pezzuto, F.; Agrimi, G.; et al. KRAS-regulated glutamine metabolism requires UCP2-mediated aspartate transport to support pancreatic cancer growth. Nat. Metab. 2020, 2, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, M.R.; Lane, A.N.; Robertson, B.; Kemp, S.; Liu, Y.; Hill, B.G.; Dean, D.C.; Clem, B.F. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 2014, 33, 556–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, R.; Sánchez-Rivera, F.J.; Westcott, P.M.K.; Mercer, K.L.; Bhutkar, A.; Muir, A.; González Robles, T.J.; Lamboy Rodríguez, S.; Liao, L.Z.; Ng, S.R.; et al. Keap1 mutation renders lung adenocarcinomas dependent on Slc33a1. Nat. Cancer 2020, 1, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, M.J.; Bennett, B.D.; Joshi, A.D.; Gao, P.; Thomas, A.G.; Ferraris, D.V.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Rabinowitz, J.D.; et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010, 70, 8981–8987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emadi, A.; Jun, S.A.; Tsukamoto, T.; Fathi, A.T.; Minden, M.D.; Dang, C.V. Inhibition of glutaminase selectively suppresses the growth of primary acute myeloid leukemia cells with IDH mutations. Exp. Hematol. 2014, 42, 247–251. [Google Scholar] [CrossRef]
- Gallipoli, P.; Giotopoulos, G.; Tzelepis, K.; Costa, A.S.H.; Vohra, S.; Medina-Perez, P.; Basheer, F.; Marando, L.; Di Lisio, L.; Dias, J.M.L.; et al. Glutaminolysis is a metabolic dependency in FLT3(ITD) acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood 2018, 131, 1639–1653. [Google Scholar] [CrossRef] [Green Version]
- Daemen, A.; Liu, B.; Song, K.; Kwong, M.; Gao, M.; Hong, R.; Nannini, M.; Peterson, D.; Liederer, B.M.; de la Cruz, C.; et al. Pan-Cancer Metabolic Signature Predicts Co-Dependency on Glutaminase and De Novo Glutathione Synthesis Linked to a High-Mesenchymal Cell State. Cell Metab. 2018, 28, 383–399.e9. [Google Scholar] [CrossRef] [Green Version]
- Muir, A.; Danai, L.V.; Gui, D.Y.; Waingarten, C.Y.; Lewis, C.A.; Vander Heiden, M.G. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife 2017, 6, e27713. [Google Scholar] [CrossRef]
- Yang, W.H.; Qiu, Y.; Stamatatos, O.; Janowitz, T.; Lukey, M.J. Enhancing the Efficacy of Glutamine Metabolism Inhibitors in Cancer Therapy. Trends Cancer 2021, 7, 790–804. [Google Scholar] [CrossRef]
- Christen, S.; Lorendeau, D.; Schmieder, R.; Broekaert, D.; Metzger, K.; Veys, K.; Elia, I.; Buescher, J.M.; Orth, M.F.; Davidson, S.M.; et al. Breast Cancer-Derived Lung Metastases Show Increased Pyruvate Carboxylase-Dependent Anaplerosis. Cell Rep. 2016, 17, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Thibault, A.; Cooper, M.R.; Figg, W.D.; Venzon, D.J.; Sartor, A.O.; Tompkins, A.C.; Weinberger, M.S.; Headlee, D.J.; McCall, N.A.; Samid, D.; et al. A phase I and pharmacokinetic study of intravenous phenylacetate in patients with cancer. Cancer Res. 1994, 54, 1690–1694. [Google Scholar]
- Ghasemian, A.; Al-Marzoqi, A.H.; Al-Abodi, H.R.; Alghanimi, Y.K.; Kadhum, S.A.; Shokouhi Mostafavi, S.K.; Fattahi, A. Bacterial l-asparaginases for cancer therapy: Current knowledge and future perspectives. J. Cell. Physiol. 2019, 234, 19271–19279. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Achreja, A.; Yeung, T.L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Shimizu, K.; Kaira, K.; Tomizawa, Y.; Sunaga, N.; Kawashima, O.; Oriuchi, N.; Tominaga, H.; Nagamori, S.; Kanai, Y.; Yamada, M.; et al. ASC amino-acid transporter 2 (ASCT2) as a novel prognostic marker in non-small cell lung cancer. Br. J. Cancer 2014, 110, 2030–2039. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Ge, M.; Hu, J.; Li, X.; Che, L.; Sun, K.; Cheng, L.; Huang, Y.; Pilo, M.G.; Cigliano, A.; et al. A functional mammalian target of rapamycin complex 1 signaling is indispensable for c-Myc-driven hepatocarcinogenesis. Hepatology 2017, 66, 167–181. [Google Scholar] [CrossRef] [Green Version]
- Bernhardt, S.; Bayerlová, M.; Vetter, M.; Wachter, A.; Mitra, D.; Hanf, V.; Lantzsch, T.; Uleer, C.; Peschel, S.; John, J. Proteomic profiling of breast cancer metabolism identifies SHMT2 and ASCT2 as prognostic factors. Breast Cancer Res. 2017, 19, 112. [Google Scholar] [CrossRef] [Green Version]
- Esslinger, C.S.; Cybulski, K.A.; Rhoderick, J.F. Ngamma-aryl glutamine analogues as probes of the ASCT2 neutral amino acid transporter binding site. Bioorg. Med. Chem. 2005, 13, 1111–1118. [Google Scholar] [CrossRef]
- Chiu, M.; Sabino, C.; Taurino, G.; Bianchi, M.G.; Andreoli, R.; Giuliani, N.; Bussolati, O. GPNA inhibits the sodium-independent transport system L for neutral amino acids. Amino Acids 2017, 49, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Bröer, A.; Rahimi, F.; Bröer, S. Deletion of Amino Acid Transporter ASCT2 (SLC1A5) Reveals an Essential Role for Transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to Sustain Glutaminolysis in Cancer Cells. J. Biol. Chem. 2016, 291, 13194–13205. [Google Scholar] [CrossRef] [PubMed]
- Corti, A.; Dominici, S.; Piaggi, S.; Belcastro, E.; Chiu, M.; Taurino, G.; Pacini, S.; Bussolati, O.; Pompella, A. γ-Glutamyltransferase enzyme activity of cancer cells modulates L-γ-glutamyl-p-nitroanilide (GPNA) cytotoxicity. Sci. Rep. 2019, 9, 891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Bröer, A.; Fairweather, S.; Bröer, S. Disruption of Amino Acid Homeostasis by Novel ASCT2 Inhibitors Involves Multiple Targets. Front. Pharm. 2018, 9, 785. [Google Scholar] [CrossRef] [Green Version]
- Coffey, G.L.; Ehrlich, J.; Fisher, M.W.; Hillegas, A.B.; Kohberger, D.L.; Machamer, H.E.; Rightsel, W.A.; Roegner, F.R. 6-Diazo-5-oxo-L-norleucine, a new tumor-inhibitory substance. I. Biologic studies. Antibiot. Chemother. 1956, 6, 487–497. [Google Scholar]
- Lemberg, K.M.; Vornov, J.J.; Rais, R.; Slusher, B.S. We’re Not “DON” Yet: Optimal Dosing and Prodrug Delivery of 6-Diazo-5-oxo-L-norleucine. Mol. Cancer 2018, 17, 1824–1832. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, M.; Rodriguez, G.; Kuhn, J.G.; Brown, T.; Weiss, G.; MacGovren, J.P.; Von Hoff, D.D.; Rowinsky, E.K. A Phase I and pharmacological study of the glutamine antagonist acivicin with the amino acid solution aminosyn in patients with advanced solid malignancies. Clin. Cancer Res. 1998, 4, 2763–2770. [Google Scholar]
- Lyons, S.D.; Sant, M.E.; Christopherson, R.I. Cytotoxic mechanisms of glutamine antagonists in mouse L1210 leukemia. J. Biol. Chem. 1990, 265, 11377–11381. [Google Scholar] [CrossRef]
- Rais, R.; Jancarik, A.; Tenora, L.; Nedelcovych, M.; Alt, J.; Englert, J.; Rojas, C.; Le, A.; Elgogary, A.; Tan, J.; et al. Discovery of 6-Diazo-5-oxo-l-norleucine (DON) Prodrugs with Enhanced CSF Delivery in Monkeys: A Potential Treatment for Glioblastoma. J. Med. Chem. 2016, 59, 8621–8633. [Google Scholar] [CrossRef]
- Tenora, L.; Alt, J.; Dash, R.P.; Gadiano, A.J.; Novotna, K.; Veeravalli, V.; Lam, J.; Kirkpatrick, Q.R.; Lemberg, K.M.; Majer, P.; et al. Tumor-Targeted Delivery of 6-Diazo-5-oxo-l-norleucine (DON) Using Substituted Acetylated Lysine Prodrugs. J. Med. Chem. 2019, 62, 3524–3538. [Google Scholar] [CrossRef]
- DeBerardinis, R.J. Tumor Microenvironment, Metabolism, and Immunotherapy. N. Engl. J. Med. 2020, 382, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, F.; Fan, N.; Zhou, C.; Li, D.; Macvicar, T.; Dong, Q.; Bruns, C.J.; Zhao, Y. Targeting Glutaminolysis: New Perspectives to Understand Cancer Development and Novel Strategies for Potential Target Therapies. Front. Oncol. 2020, 10, 589508. [Google Scholar] [CrossRef]
- Xu, X.; Meng, Y.; Li, L.; Xu, P.; Wang, J.; Li, Z.; Bian, J. Overview of the Development of Glutaminase Inhibitors: Achievements and Future Directions. J. Med. Chem. 2019, 62, 1096–1115. [Google Scholar] [CrossRef]
- Zimmermann, S.C.; Wolf, E.F.; Luu, A.; Thomas, A.G.; Stathis, M.; Poore, B.; Nguyen, C.; Le, A.; Rojas, C.; Slusher, B.S.; et al. Allosteric Glutaminase Inhibitors Based on a 1,4-Di(5-amino-1,3,4-thiadiazol-2-yl)butane Scaffold. ACS Med. Chem. Lett. 2016, 7, 520–524. [Google Scholar] [CrossRef] [Green Version]
- Duvall, B.; Zimmermann, S.C.; Gao, R.-D.; Thomas, A.G.; Kalčic, F.; Veeravalli, V.; Elgogary, A.; Rais, R.; Rojas, C.; Le, A.; et al. Allosteric kidney-type glutaminase (GLS) inhibitors with a mercaptoethyl linker. Bioorg. Med. Chem. 2020, 28, 115698. [Google Scholar] [CrossRef]
- Shukla, K.; Ferraris, D.V.; Thomas, A.G.; Stathis, M.; Duvall, B.; Delahanty, G.; Alt, J.; Rais, R.; Rojas, C.; Gao, P.; et al. Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J. Med. Chem. 2012, 55, 10551–10563. [Google Scholar] [CrossRef] [Green Version]
- Finlay, M.R.V.; Anderton, M.; Bailey, A.; Boyd, S.; Brookfield, J.; Cairnduff, C.; Charles, M.; Cheasty, A.; Critchlow, S.E.; Culshaw, J.; et al. Discovery of a Thiadiazole-Pyridazine-Based Allosteric Glutaminase 1 Inhibitor Series That Demonstrates Oral Bioavailability and Activity in Tumor Xenograft Models. J. Med. Chem. 2019, 62, 6540–6560. [Google Scholar] [CrossRef]
- Elgogary, A.; Xu, Q.; Poore, B.; Alt, J.; Zimmermann, S.C.; Zhao, L.; Fu, J.; Chen, B.; Xia, S.; Liu, Y.; et al. Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E5328–E5336. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.; Dias, S.M.; Dang, C.V.; et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, E.R.K.; Rodrigues, C.T.; Campos, H.J.C.; Paradela, L.S.; Dias, M.M.; Novaes da Silva, B.; de Valega Negrao, C.V.; Gonçalves, K.D.; Ascenção, C.F.; Adamoski, D.; et al. High-Throughput Screening Reveals New Glutaminase Inhibitor Molecules. ACS Pharm. Transl. Sci. 2021, 4, 1849–1866. [Google Scholar] [CrossRef] [PubMed]
- Soth, M.J.; Le, K.; Di Francesco, M.E.; Hamilton, M.M.; Liu, G.; Burke, J.P.; Carroll, C.L.; Kovacs, J.J.; Bardenhagen, J.P.; Bristow, C.A.; et al. Discovery of IPN60090, a Clinical Stage Selective Glutaminase-1 (GLS-1) Inhibitor with Excellent Pharmacokinetic and Physicochemical Properties. J. Med. Chem. 2020, 63, 12957–12977. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.Z.; Yang, C.W.; Chang, H.Y.; Hsu, H.Y.; Chen, I.S.; Chang, H.S.; Lee, C.H.; Lee, J.C.; Kumar, C.R.; Qiu, Y.Q.; et al. Discovery of selective inhibitors of Glutaminase-2, which inhibit mTORC1, activate autophagy and inhibit proliferation in cancer cells. Oncotarget 2014, 5, 6087–6101. [Google Scholar] [CrossRef]
- Yeh, T.K.; Kuo, C.C.; Lee, Y.Z.; Ke, Y.Y.; Chu, K.F.; Hsu, H.Y.; Chang, H.Y.; Liu, Y.W.; Song, J.S.; Yang, C.W.; et al. Design, Synthesis, and Evaluation of Thiazolidine-2,4-dione Derivatives as a Novel Class of Glutaminase Inhibitors. J. Med. Chem. 2017, 60, 5599–5612. [Google Scholar] [CrossRef]
- DeLaBarre, B.; Gross, S.; Fang, C.; Gao, Y.; Jha, A.; Jiang, F.; Song, J.J.; Wei, W.; Hurov, J.B. Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry 2011, 50, 10764–10770. [Google Scholar] [CrossRef]
- Thangavelu, K.; Pan, C.Q.; Karlberg, T.; Balaji, G.; Uttamchandani, M.; Suresh, V.; Schuler, H.; Low, B.C.; Sivaraman, J. Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, 7705–7710. [Google Scholar] [CrossRef] [Green Version]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer 2014, 13, 890–901. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Feng, X.; Chen, Y.; Selfridge, J.E.; Gorityala, S.; Du, Z.; Wang, J.M.; Hao, Y.; Cioffi, G.; Conlon, R.A.; et al. 5-Fluorouracil Enhances the Antitumor Activity of the Glutaminase Inhibitor CB-839 against PIK3CA-Mutant Colorectal Cancers. Cancer Res. 2020, 80, 4815–4827. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Tannir, N.M.; Iliopoulos, O.; Lee, R.J.; Telli, M.L.; Fan, A.C.; DeMichele, A.; Haas, N.B.; Patel, M.R.; Harding, J.J.; et al. Telaglenastat Plus Cabozantinib or Everolimus for Advanced or Metastatic Renal Cell Carcinoma: An Open-Label Phase I Trial. Clin. Cancer Res. 2022, 28, 1540–1548. [Google Scholar] [CrossRef]
- Lee, C.H.; Motzer, R.; Emamekhoo, H.; Matrana, M.; Percent, I.; Hsieh, J.J.; Hussain, A.; Vaishampayan, U.; Liu, S.; McCune, S.; et al. Telaglenastat plus Everolimus in Advanced Renal Cell Carcinoma: A Randomized, Double-Blinded, Placebo-Controlled, Phase II ENTRATA Trial. Clin. Cancer Res. 2022, 28, 3248–3255. [Google Scholar] [CrossRef] [PubMed]
- Tannir, N.M.; Agarwal, N.; Porta, C.; Lawrence, N.J.; Motzer, R.; McGregor, B.; Lee, R.J.; Jain, R.K.; Davis, N.; Appleman, L.J.; et al. Efficacy and Safety of Telaglenastat Plus Cabozantinib vs Placebo Plus Cabozantinib in Patients With Advanced Renal Cell Carcinoma: The CANTATA Randomized Clinical Trial. JAMA Oncol. 2022, 8, 1411–1418. [Google Scholar] [CrossRef] [PubMed]
- Stalnecker, C.A.; Ulrich, S.M.; Li, Y.; Ramachandran, S.; McBrayer, M.K.; DeBerardinis, R.J.; Cerione, R.A.; Erickson, J.W. Mechanism by which a recently discovered allosteric inhibitor blocks glutamine metabolism in transformed cells. Proc. Natl. Acad. Sci. USA 2015, 112, 394–399. [Google Scholar] [CrossRef] [Green Version]
- Lukey, M.J.; Cluntun, A.A.; Katt, W.P.; Lin, M.J.; Druso, J.E.; Ramachandran, S.; Erickson, J.W.; Le, H.H.; Wang, Z.E.; Blank, B.; et al. Liver-Type Glutaminase GLS2 Is a Druggable Metabolic Node in Luminal-Subtype Breast Cancer. Cell Rep. 2019, 29, 76–88.e7. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Li, W.; Pan, G.; Wang, C.; Li, D.; Liu, N.; Sheng, X.; Yuan, L. The Glutaminase Inhibitor Compound 968 Exhibits Potent in vitro and in vivo Anti-tumor Effects in Endometrial Cancer. Anticancer Agents Med. Chem. 2022. [Google Scholar] [CrossRef]
- Yuan, L.; Sheng, X.; Clark, L.H.; Zhang, L.; Guo, H.; Jones, H.M.; Willson, A.K.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V.L. Glutaminase inhibitor compound 968 inhibits cell proliferation and sensitizes paclitaxel in ovarian cancer. Am. J. Transl. Res. 2016, 8, 4265–4277. [Google Scholar]
- Wang, J.J.; Siu, M.K.; Jiang, Y.X.; Leung, T.H.; Chan, D.W.; Wang, H.G.; Ngan, H.Y.; Chan, K.K. A Combination of Glutaminase Inhibitor 968 and PD-L1 Blockade Boosts the Immune Response against Ovarian Cancer. Biomolecules 2021, 11, 1749. [Google Scholar] [CrossRef]
- Wang, D.; Meng, G.; Zheng, M.; Zhang, Y.; Chen, A.; Wu, J.; Wei, J. The Glutaminase-1 Inhibitor 968 Enhances Dihydroartemisinin-Mediated Antitumor Efficacy in Hepatocellular Carcinoma Cells. PLoS ONE 2016, 11, e0166423. [Google Scholar] [CrossRef] [Green Version]
- Xi, J.; Sun, Y.; Zhang, M.; Fa, Z.; Wan, Y.; Min, Z.; Xu, H.; Xu, C.; Tang, J. GLS1 promotes proliferation in hepatocellular carcinoma cells via AKT/GSK3β/CyclinD1 pathway. Exp Cell Res 2019, 381, 1–9. [Google Scholar] [CrossRef]
- Xie, C.; Jin, J.; Bao, X.; Zhan, W.H.; Han, T.Y.; Gan, M.; Zhang, C.; Wang, J. Inhibition of mitochondrial glutaminase activity reverses acquired erlotinib resistance in non-small cell lung cancer. Oncotarget 2016, 7, 610–621. [Google Scholar] [CrossRef] [Green Version]
- Effenberger, M.; Bommert, K.S.; Kunz, V.; Kruk, J.; Leich, E.; Rudelius, M.; Bargou, R.; Bommert, K. Glutaminase inhibition in multiple myeloma induces apoptosis via MYC degradation. Oncotarget 2017, 8, 85858–85867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamarudin, M.N.A.; Sarker, M.M.R.; Zhou, J.R.; Parhar, I. Metformin in colorectal cancer: Molecular mechanism, preclinical and clinical aspects. J. Exp. Clin. Cancer Res. 2019, 38, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Lee, K.J.; Seo, Y.; Kwon, J.H.; Yoon, J.P.; Kang, J.Y.; Lee, H.J.; Park, S.J.; Hong, S.P.; Cheon, J.H.; et al. Effects of metformin on colorectal cancer stem cells depend on alterations in glutamine metabolism. Sci. Rep. 2018, 8, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udupa, S.; Nguyen, S.; Hoang, G.; Nguyen, T.; Quinones, A.; Pham, K.; Asaka, R.; Nguyen, K.; Zhang, C.; Elgogary, A.; et al. Upregulation of the Glutaminase II Pathway Contributes to Glutamate Production upon Glutaminase 1 Inhibition in Pancreatic Cancer. Proteomics 2019, 19, e1800451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Kirsch, B.J.; Asaka, R.; Nabi, K.; Quinones, A.; Tan, J.; Antonio, M.J.; Camelo, F.; Li, T.; Nguyen, S.; et al. Uncovering the Role of N-Acetyl-Aspartyl-Glutamate as a Glutamate Reservoir in Cancer. Cell Rep. 2019, 27, 491–501.e6. [Google Scholar] [CrossRef]
- Best, S.A.; Gubser, P.M.; Sethumadhavan, S.; Kersbergen, A.; Negrón Abril, Y.L.; Goldford, J.; Sellers, K.; Abeysekera, W.; Garnham, A.L.; McDonald, J.A.; et al. Glutaminase inhibition impairs CD8 T cell activation in STK11-/Lkb1-deficient lung cancer. Cell Metab. 2022, 34, 874–887.e6. [Google Scholar] [CrossRef]
- Johnson, M.O.; Wolf, M.M.; Madden, M.Z.; Andrejeva, G.; Sugiura, A.; Contreras, D.C.; Maseda, D.; Liberti, M.V.; Paz, K.; Kishton, R.J.; et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 2018, 175, 1780–1795.e19. [Google Scholar] [CrossRef] [Green Version]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Kawana, K.; Adachi, K.; Fujimoto, A.; Yoshida, M.; Nakamura, H.; Nishida, H.; Inoue, T.; Taguchi, A.; Takahashi, J.; et al. Spheroid cancer stem cells display reprogrammed metabolism and obtain energy by actively running the tricarboxylic acid (TCA) cycle. Oncotarget 2016, 7, 33297–33305. [Google Scholar] [CrossRef]
- Li, D.; Fu, Z.; Chen, R.; Zhao, X.; Zhou, Y.; Zeng, B.; Yu, M.; Zhou, Q.; Lin, Q.; Gao, W.; et al. Inhibition of glutamine metabolism counteracts pancreatic cancer stem cell features and sensitizes cells to radiotherapy. Oncotarget 2015, 6, 31151–31163. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Cao, Y.; Meng, G.; Qian, L.; Xu, T.; Yan, C.; Luo, O.; Wang, S.; Wei, J.; Ding, Y.; et al. Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBioMedicine 2019, 39, 239–254. [Google Scholar] [CrossRef] [Green Version]
- Mukha, A.; Kahya, U.; Linge, A.; Chen, O.; Löck, S.; Lukiyanchuk, V.; Richter, S.; Alves, T.C.; Peitzsch, M.; Telychko, V.; et al. GLS-driven glutamine catabolism contributes to prostate cancer radiosensitivity by regulating the redox state, stemness and ATG5-mediated autophagy. Theranostics 2021, 11, 7844–7868. [Google Scholar] [CrossRef]
- Sato, M.; Kawana, K.; Adachi, K.; Fujimoto, A.; Taguchi, A.; Fujikawa, T.; Yoshida, M.; Nakamura, H.; Nishida, H.; Inoue, T.; et al. Low uptake of fluorodeoxyglucose in positron emission tomography/computed tomography in ovarian clear cell carcinoma may reflect glutaminolysis of its cancer stem cell-like properties. Oncol. Rep. 2017, 37, 1883–1888. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.; Kang, J.; Kim, T.I.; Joo, C.G. MRI assessment of glutamine uptake correlates with the distribution of glutamine transporters and cancer stem cell markers. Sci. Rep. 2022, 12, 5511. [Google Scholar] [CrossRef]
- Timmerman, L.A.; Holton, T.; Yuneva, M.; Louie, R.J.; Padró, M.; Daemen, A.; Hu, M.; Chan, D.A.; Ethier, S.P.; van ‘t Veer, L.J.; et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 2013, 24, 450–465. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.A.; Pan, S.C.; Chu, I.; Lai, R.Y.; Wei, Y.H. Targeting cancer stem cells from a metabolic perspective. Exp. Biol. Med. 2020, 245, 465–476. [Google Scholar] [CrossRef] [Green Version]
- Sharif, T.; Dai, C.; Martell, E.; Ghassemi-Rad, M.S.; Hanes, M.R.; Murphy, P.J.; Kennedy, B.E.; Venugopal, C.; Subapanditha, M.; Giacomantonio, C.A.; et al. TAp73 Modifies Metabolism and Positively Regulates Growth of Cancer Stem-Like Cells in a Redox-Sensitive Manner. Clin. Cancer Res. 2019, 25, 2001–2017. [Google Scholar] [CrossRef]
- Lee, S.Y.; Ju, M.K.; Jeon, H.M.; Lee, Y.J.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Oncogenic Metabolism Acts as a Prerequisite Step for Induction of Cancer Metastasis and Cancer Stem Cell Phenotype. Oxid. Med. Cell. Longev. 2018, 2018, 1027453. [Google Scholar] [CrossRef] [Green Version]
- Le Grand, M.; Mukha, A.; Püschel, J.; Valli, E.; Kamili, A.; Vittorio, O.; Dubrovska, A.; Kavallaris, M. Interplay between MycN and c-Myc regulates radioresistance and cancer stem cell phenotype in neuroblastoma upon glutamine deprivation. Theranostics 2020, 10, 6411–6429. [Google Scholar] [CrossRef]
- Janssen, K.P.; Alberici, P.; Fsihi, H.; Gaspar, C.; Breukel, C.; Franken, P.; Rosty, C.; Abal, M.; El Marjou, F.; Smits, R.; et al. APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology 2006, 131, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Xu, J.; Bian, X.; Wu, J.L.; Kang, W.; Qian, Y.; Li, W.; Chen, H.; Gou, H.; Liu, D.; et al. In Colorectal Cancer Cells With Mutant KRAS, SLC25A22-Mediated Glutaminolysis Reduces DNA Demethylation to Increase WNT Signaling, Stemness, and Drug Resistance. Gastroenterology 2020, 159, 2163–2180.e6. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Du, W.; Yang, X. A critical role of glucose-6-phosphate dehydrogenase in TAp73-mediated cell proliferation. Cell Cycle 2013, 12, 3720–3726. [Google Scholar] [CrossRef] [Green Version]
- Du, W.; Jiang, P.; Mancuso, A.; Stonestrom, A.; Brewer, M.D.; Minn, A.J.; Mak, T.W.; Wu, M.; Yang, X. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat Cell Biol. 2013, 15, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Folmes, C.D.; Terzic, A. Energy metabolism in the acquisition and maintenance of stemness. Semin. Cell Dev. Biol. 2016, 52, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem cells and the impact of ROS signaling. Development 2014, 141, 4206–4218. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Wang, S.; Zaal, E.A.; Wang, C.; Wu, H.; Bosma, A.; Jochems, F.; Isima, N.; Jin, G.; Lieftink, C.; et al. A powerful drug combination strategy targeting glutamine addiction for the treatment of human liver cancer. Elife 2020, 9, e56749. [Google Scholar] [CrossRef]
- Saha, S.K.; Islam, S.M.R.; Abdullah-Al-Wadud, M.; Islam, S.; Ali, F.; Park, K.S. Multiomics Analysis Reveals that GLS and GLS2 Differentially Modulate the Clinical Outcomes of Cancer. J. Clin. Med. 2019, 8, 355. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Liu, P.P.; Hou, G.; Shao, J.; Yang, J.; Liu, K.; Lu, W.; Wen, S.; Hu, Y.; Huang, P. Regulation of stem-like cancer cells by glutamine through β-catenin pathway mediated by redox signaling. Mol. Cancer 2017, 16, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamarajan, P.; Rajendiran, T.M.; Kinchen, J.; Bermúdez, M.; Danciu, T.; Kapila, Y.L. Head and Neck Squamous Cell Carcinoma Metabolism Draws on Glutaminolysis, and Stemness Is Specifically Regulated by Glutaminolysis via Aldehyde Dehydrogenase. J. Proteome Res. 2017, 16, 1315–1326. [Google Scholar] [CrossRef] [Green Version]
- Sekine, S.; Lan, B.Y.; Bedolli, M.; Feng, S.; Hebrok, M. Liver-specific loss of beta-catenin blocks glutamine synthesis pathway activity and cytochrome p450 expression in mice. Hepatology 2006, 43, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Cadoret, A.; Ovejero, C.; Terris, B.; Souil, E.; Lévy, L.; Lamers, W.H.; Kitajewski, J.; Kahn, A.; Perret, C. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 2002, 21, 8293–8301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jariyal, H.; Gupta, C.; Andhale, S.; Gadge, S.; Srivastava, A. Comparative stemness and differentiation of luminal and basal breast cancer stem cell type under glutamine-deprivation. J. Cell Commun. Signal. 2021, 15, 207–222. [Google Scholar] [CrossRef]
- Huang, T.; Song, X.; Xu, D.; Tiek, D.; Goenka, A.; Wu, B.; Sastry, N.; Hu, B.; Cheng, S.Y. Stem cell programs in cancer initiation, progression, and therapy resistance. Theranostics 2020, 10, 8721–8743. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mayea, Y.; Mir, C.; Masson, F.; Paciucci, R.; ME, L.L. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin. Cancer Biol. 2020, 60, 166–180. [Google Scholar] [CrossRef]
- Koch, K.; Hartmann, R.; Tsiampali, J.; Uhlmann, C.; Nickel, A.C.; He, X.; Kamp, M.A.; Sabel, M.; Barker, R.A.; Steiger, H.J.; et al. A comparative pharmaco-metabolomic study of glutaminase inhibitors in glioma stem-like cells confirms biological effectiveness but reveals differences in target-specificity. Cell Death Discov. 2020, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Restall, I.J.; Cseh, O.; Richards, L.M.; Pugh, T.J.; Luchman, H.A.; Weiss, S. Brain Tumor Stem Cell Dependence on Glutaminase Reveals a Metabolic Vulnerability through the Amino Acid Deprivation Response Pathway. Cancer Res. 2020, 80, 5478–5490. [Google Scholar] [CrossRef]
- Poonaki, E.; Nickel, A.C.; Shafiee Ardestani, M.; Rademacher, L.; Kaul, M.; Apartsin, E.; Meuth, S.G.; Gorji, A.; Janiak, C.; Kahlert, U.D. CD133-Functionalized Gold Nanoparticles as a Carrier Platform for Telaglenastat (CB-839) against Tumor Stem Cells. Int. J. Mol. Sci. 2022, 23, 5479. [Google Scholar] [CrossRef]
- Jaggupilli, A.; Ly, S.; Nguyen, K.; Anand, V.; Yuan, B.; El-Dana, F.; Yan, Y.; Arvanitis, Z.; Piyarathna, D.W.B.; Putluri, N.; et al. Metabolic stress induces GD2(+) cancer stem cell-like phenotype in triple-negative breast cancer. Br. J. Cancer 2022, 126, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Mou, J.; Shao, B.; Wei, Y.; Liang, H.; Takano, N.; Semenza, G.L.; Xie, G. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. 2019, 10, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kao, T.-W.; Chuang, Y.-C.; Lee, H.-L.; Kuo, C.-C.; Shen, Y.-A. Therapeutic Targeting of Glutaminolysis as a Novel Strategy to Combat Cancer Stem Cells. Int. J. Mol. Sci. 2022, 23, 15296. https://doi.org/10.3390/ijms232315296
Kao T-W, Chuang Y-C, Lee H-L, Kuo C-C, Shen Y-A. Therapeutic Targeting of Glutaminolysis as a Novel Strategy to Combat Cancer Stem Cells. International Journal of Molecular Sciences. 2022; 23(23):15296. https://doi.org/10.3390/ijms232315296
Chicago/Turabian StyleKao, Ting-Wan, Yao-Chen Chuang, Hsin-Lun Lee, Chia-Chun Kuo, and Yao-An Shen. 2022. "Therapeutic Targeting of Glutaminolysis as a Novel Strategy to Combat Cancer Stem Cells" International Journal of Molecular Sciences 23, no. 23: 15296. https://doi.org/10.3390/ijms232315296
APA StyleKao, T.-W., Chuang, Y.-C., Lee, H.-L., Kuo, C.-C., & Shen, Y.-A. (2022). Therapeutic Targeting of Glutaminolysis as a Novel Strategy to Combat Cancer Stem Cells. International Journal of Molecular Sciences, 23(23), 15296. https://doi.org/10.3390/ijms232315296