
Synthesis, Biological Evaluation and Computational Studies of New Hydrazide Derivatives Containing 1,3,4-Oxadiazole as Antitubercular Agents
 , ,
, ,  ,
,
Abstract
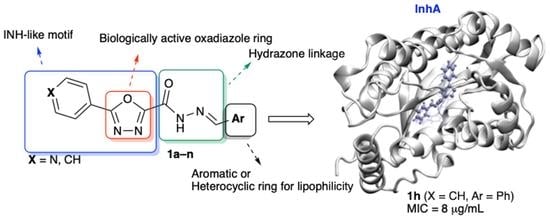
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Antimycobacterial Activity
2.3. Cytotoxicity Study
2.4. Molecular Modelling
2.5. In Silico Pharmacokinetic Parameters
3. Materials and Methods
3.1. Chemistry
3.1.1. Chemical Reagents and Instruments
3.1.2. General Procedure for the Synthesis of Intermediates 2a,b
3.1.3. General Procedure for the Synthesis of Intermediates 3a,b
3.1.4. General Procedure for the Synthesis of Final Compounds 1a–n
3.2. Antimycobacterial, Antifungal and Cytotoxicity Studies
3.2.1. Cultures Preparation
3.2.2. Staging of the Solutions of the Test Compounds
3.2.3. Determination of Antimycobacterial Activity
3.2.4. Determination of Antifungal Activity
3.2.5. Cytotoxicity
3.3. Computational Methods
3.3.1. Docking
3.3.2. Pocket Analysis
3.3.3. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Global Tuberculosis Report 2020. Available online: https://www.who.int/publications/i/item/9789240013131 (accessed on 18 October 2022).
- World Health Organization (WHO). Global Tuberculosis Report 2021. Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2021 (accessed on 18 October 2022).
- Milligan, D.L.; Tran, S.L.; Strych, U.; Cook, G.M.; Krause, K.L. The alanine racemase of Mycobacterium smegmatis is essential for growth in the absence of D-alanine. J. Bacteriol. 2007, 189, 8381–8386. [Google Scholar] [CrossRef] [PubMed]
- Olaleye, O.; Raghunand, T.R.; Bhat, S.; He, J.; Tyagi, S.; Lamichhan, E.G.; Gu, P.; Zhou, S.; Grosset, J.; Bishai, W.R.; et al. Methionine aminopeptidases from Mycobacterium tuberculosis as novel antimycobacterial targets. Chem. Biol. 2010, 17, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Krátký, M.; Vinsová, J.; Novotná, E.; Mandíková, J.; Wsól, W.; Trejtnar, F.; Ulmann, V.; Stolarikova, J.; Fernandes, S.; Bhat, S.; et al. Salicylanilide derivatives block Mycobacterium tuberculosis through inhibition of isocitrate lyase and methionine aminopeptidase. Tuberculosis 2012, 92, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, N.; Zhang, H.; Knudson, S.E.; Slayden, R.A.; Tonge, P.J. Synthesis and SAR studies of 1,4-benzoxazine MenB inhibitors: Novel antibacterial agents against Mycobacterium tuberculosis. Bioorg. Med. Chem Lett. 2010, 20, 6306–6309. [Google Scholar] [CrossRef][Green Version]
- Makarov, V.; Manina, G.; Mikusova, K.; Möllmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones Kill Mycobacterium tuberculosis by Blocking Arabinan Synthesis. Science 2009, 324, 801–804. [Google Scholar] [CrossRef]
- Vilcheze, C.; Morbidoni, H.R.; Weisbrod, T.R.; Iwamoto, H.; Kuo, M.; Sacchettini, J.C.; Jacobs, W.R., Jr. Inactivation of the inhA-Encoded Fatty Acid Synthase II (FASII) Enoyl-Acyl Carrier Protein Reductase Induces Accumulation of the FASI End Products and Cell Lysis of Mycobacterium smegmatis. J. Bacteriol. 2000, 182, 4059–4067. [Google Scholar] [CrossRef]
- Beruve, G. An overview of molecular hybrids in drug discovery. Expert Opin. Drug Discov. 2016, 11, 281–305. [Google Scholar] [CrossRef]
- Zampieri, D.; Mamolo, M.G. Hybridization Approach to Drug Discovery Inhibiting Mycobacterium tuberculosis. An Overview. Curr. Top. Med. Chem. 2021, 21, 777–788. [Google Scholar] [CrossRef]
- Ziyaev, A.A.; Ismailova, D.S. Biological activity of 5-(2,3,4-Pyridyl)-1,3,4-oxadiazol-2-thiones and their derivatives. World J. Pharm. Res. 2017, 6, 5–57. [Google Scholar] [CrossRef]
- Shah, S.R.; Katariya, K.D. 1,3-Oxazole-isoniazid hybrids: Synthesis, antitubercular activity, and their docking studies. J. Het. Chem. 2020, 57, 1682–1691. [Google Scholar] [CrossRef]
- Mamolo, M.G.; Falagiani, V.; Zampieri, D.; Vio, L.; Banfi, E. Synthesis and antimycobacterial activity of [5-(pyridin-2-yl)-1,3,4-thiadiazol-2-ylthio]acetic acid arylidene-hydrazide derivatives. Il Farm. 2001, 56, 587–592. [Google Scholar] [CrossRef]
- Desai, N.C.; Somani, H.; Trivedi, A.; Bhatt, K.; Nawale, L.; Khedkar, V.M.; Jha, P.C.; Sarkar, D. Synthesis, biological evaluation and molecular docking study of some novel indole and pyridine based 1,3,4-oxadiazole derivatives as potential antitubercular agents. Bioorg. Med. Chem. Lett. 2016, 26, 1776–1783. [Google Scholar] [CrossRef]
- Vosatka, R.; Kratký, M.; Svarcova, M.; Janousek, J.; Stolarkova, J.; Madacki, J.; Huszar, S.; Mikusova, K.; Kordulakova, J.; Trejtnar, F.; et al. New lipophilic isoniazid derivatives and their 1,3,4-oxadiazole analogues: Synthesis, antimycobacterial activity and investigation of their mechanism of action. Eur. J. Med. Chem. 2018, 151, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Elma, F. In silico identification of novel chemical compounds with antituberculosis activity for the inhibition of InhA and EthR proteins from Mycobacterium tuberculosis. J. Clin. Tuberc. Other Mycobact. Dis. 2021, 24, 100246. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Dost, J.; Heschel, M.; Stein, J. Preparation of 1,3,4-oxadiazole-2-carboxylic acid derivatives. J. Prak. Chem. 1985, 327, 109–116. [Google Scholar] [CrossRef]
- Palomino, J.C.; Martin, A.; Camacho, M.; Guerra, H.; Swings, J.; Portales, F. Resazurin microtiter assay plate: Simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2002, 46, 2720–2722. [Google Scholar] [CrossRef]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef]
- Uzarski, J.S.; DiVito, M.D.; Wertheim, J.A.; Miller, W.M. Essential design considerations for the resazurin reduction assay to noninvasively quantify cell expansion within perfused extracellular matrix scaffolds. Biomaterials 2017, 129, 163–175. [Google Scholar] [CrossRef]
- Taneja, N.K.; Tyagi, J.S. Resazurin reduction assays for screening of anti-tubercular compounds against dormant and actively growing Mycobacterium tuberculosis, Mycobacterium bovis BCG and Mycobacterium smegmatis. J. Antimicrob. Chemother. 2007, 60, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, D.; Mamolo, M.G.; Filingeri, J.; Fortuna, S.; De Logu, A.; Sanna, A.; Zanon, D. Design, synthesis and antimycobacterial activity of benzoxazinone derivatives and open-ring analogues: Preliminary data and computational analysis. Bioorg. Med. Chem. Lett. 2019, 29, 2468–2474. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J. MOPAC—A semiempirical molecular-orbital program. J. Comput. Aided Mol. Des. 1990, 4, 1–103. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Dias, M.V.B.; Vasconcelos, I.B.; Prado, A.M.X.; Fadel, V.; Basso, L.A.; Filgueira de Azevedo, W., Jr.; Santos, D.S. Crystallographic studies on the binding of isonicotinyl-NAD adduct to wild-type and isoniazid resistant 2-trans-enoyl-ACP (CoA) reductase from Mycobacterium tuberculosis. J. Struct. Biol. 2007, 159, 369–380. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, S.; Hu, Q.; Gao, S.; Ma, X.; Zhang, W.; Shen, Y.; Chen, F.; Lai, L.; Pei, J. CavityPlus: A web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res. 2018, 46, W374–W379. [Google Scholar] [CrossRef]
- Yuan, Y.; Pei, J.; Lai, L. Binding site detection and druggability prediction of protein targets for structure-based drug design. Curr. Pharm. Des. 2013, 19, 2326–2333. [Google Scholar] [CrossRef]
- Chen, J.; Lai, L. Pocket v. 2: Further developments on receptor-based pharmacophore modelling. J. Chem. Inf. Mod. 2006, 46, 2684–2691. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; Van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, K.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef] [PubMed]
- Hess, B. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open-source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Cmpd | X | Ar | MIC μg/mL (μM) | CC50 a (μM) |
| 1a | N | Ph | 64 (218) | nt |
| 1b | N | 4-ClPh | >64 (-) | nt |
| 1c | N | 4-CH3Ph | 64 (195) | nt |
| 1d | N | 2-Pyrrolyl | 64 (226) | nt |
| 1e | N | 2-Pyridyl | 32 (108) | nt |
| 1f | N | 4-Pyridyl | 64 (217) | nt |
| 1g | N | 2-Furyl | 64 (226) | nt |
| 1h | CH | Ph | 8 (27.4) | >>100 |
| 1i | CH | 4-ClPh | 16 (48.9) | nt |
| 1j | CH | 4-CH3Ph | 16 (52.2) | nt |
| 1k | CH | 2-Pyrrolyl | 8 (28.3) | 50 ± 6 |
| 1l | CH | 2-Pyridyl | 8 (27.4) | 100 ± 12 |
| 1m | CH | 4-Pyridyl | 8 (27.4) | 128 ± 16 |
| 1n | CH | 2-Furyl | 8 (28.3) | >>100 |
| INH | - | - | 0.25 (1.8) | nt |
| Cpd | M. tuberculosis Strain | ||||
|---|---|---|---|---|---|
| Rifa-R a | INH-R b | Pyr-R c | Str-R d | H37Rv | |
| MIC μg/mL (μM) | |||||
| 1a | >64 | >64 | >64 | >64 | >64 |
| 1b | >64 | >64 | >64 | >64 | >64 |
| 1c | >64 | >64 | >64 | >64 | >64 |
| 1d | >64 | >64 | >64 | >64 | >64 |
| 1e | 32 (108) | >64 | 4 (13.6) | 32 (108) | 16 (54.0) |
| 1f | >64 | >64 | >64 | >64 | >64 |
| 1g | >64 | >64 | >64 | >64 | >64 |
| 1h | >64 | >64 | 64 (196) | >64 | >64 |
| 1i | >64 | >64 | 32 (97.8) | >64 | >64 |
| 1j | >64 | >64 | 16 (52.2) | >64 | >64 |
| 1k | >64 | >64 | 4 (14.1) | >64 | 8 (28.3) |
| 1l | 32 (54.4) | >64 | 4 (13.6) | 64 (109) | 8 (27.1) |
| 1m | >64 | >64 | 16 (54.8) | >64 | >64 |
| 1n | 32 (103) | >64 | 16 (56.6) | 32 (103) | 16 (56.6) |
| INH | 0.25 (1.8) | >64 | 0.25 (1.8) | 0.25 (1.8) | 0.25 (1.8) |
| RIF | >64 | 0.25 (0.3) | 0.25 (0.3) | 0.25 (0.3) | 0.5 (0.6) |
| Cmpd | MW b | HBA c | HBD d | clogP e | clogS f (mol/L) | TPSA g (Å < 140) | RO5 Violation | BBB Permeant | GI abs. | |
|---|---|---|---|---|---|---|---|---|---|---|
| RO5 a | <500 | ≤10 | ≤5 | ≤5 | ≤5 | - | ≤1 | - | - | |
| 1h | 292.29 | 5 | 1 | 2.45 | −3.57 | 80.38 | 0 | No | High | |
| 1k | 281.27 | 5 | 2 | 1.71 | −2.94 | 96.17 | 0 | No | High | |
| 1l | 293.28 | 6 | 1 | 1.84 | −3.11 | 93.27 | 0 | No | High | |
| 1m | 293.28 | 6 | 1 | 1.72 | −2.90 | 93.27 | 0 | No | High | |
| 1n | 282.25 | 6 | 1 | 1.86 | −3.12 | 93.52 | 0 | No | High | |
| INH | 137.14 | 3 | 2 | −0.35 | −0.56 | 68.01 | 0 | No | High |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zampieri, D.; Fortuna, S.; Romano, M.; De Logu, A.; Cabiddu, G.; Sanna, A.; Mamolo, M.G. Synthesis, Biological Evaluation and Computational Studies of New Hydrazide Derivatives Containing 1,3,4-Oxadiazole as Antitubercular Agents. Int. J. Mol. Sci. 2022, 23, 15295. https://doi.org/10.3390/ijms232315295
Zampieri D, Fortuna S, Romano M, De Logu A, Cabiddu G, Sanna A, Mamolo MG. Synthesis, Biological Evaluation and Computational Studies of New Hydrazide Derivatives Containing 1,3,4-Oxadiazole as Antitubercular Agents. International Journal of Molecular Sciences. 2022; 23(23):15295. https://doi.org/10.3390/ijms232315295
Chicago/Turabian StyleZampieri, Daniele, Sara Fortuna, Maurizio Romano, Alessandro De Logu, Gianluigi Cabiddu, Adriana Sanna, and Maria Grazia Mamolo. 2022. "Synthesis, Biological Evaluation and Computational Studies of New Hydrazide Derivatives Containing 1,3,4-Oxadiazole as Antitubercular Agents" International Journal of Molecular Sciences 23, no. 23: 15295. https://doi.org/10.3390/ijms232315295
APA StyleZampieri, D., Fortuna, S., Romano, M., De Logu, A., Cabiddu, G., Sanna, A., & Mamolo, M. G. (2022). Synthesis, Biological Evaluation and Computational Studies of New Hydrazide Derivatives Containing 1,3,4-Oxadiazole as Antitubercular Agents. International Journal of Molecular Sciences, 23(23), 15295. https://doi.org/10.3390/ijms232315295