
RNA-Interference-Mediated miR-122-Based Gene Regulation in Colon Cancer, a Structural In Silico Analysis
 , and
, and
Abstract
1. Introduction
2. Results
2.1. miRNA-122-Associated Genes
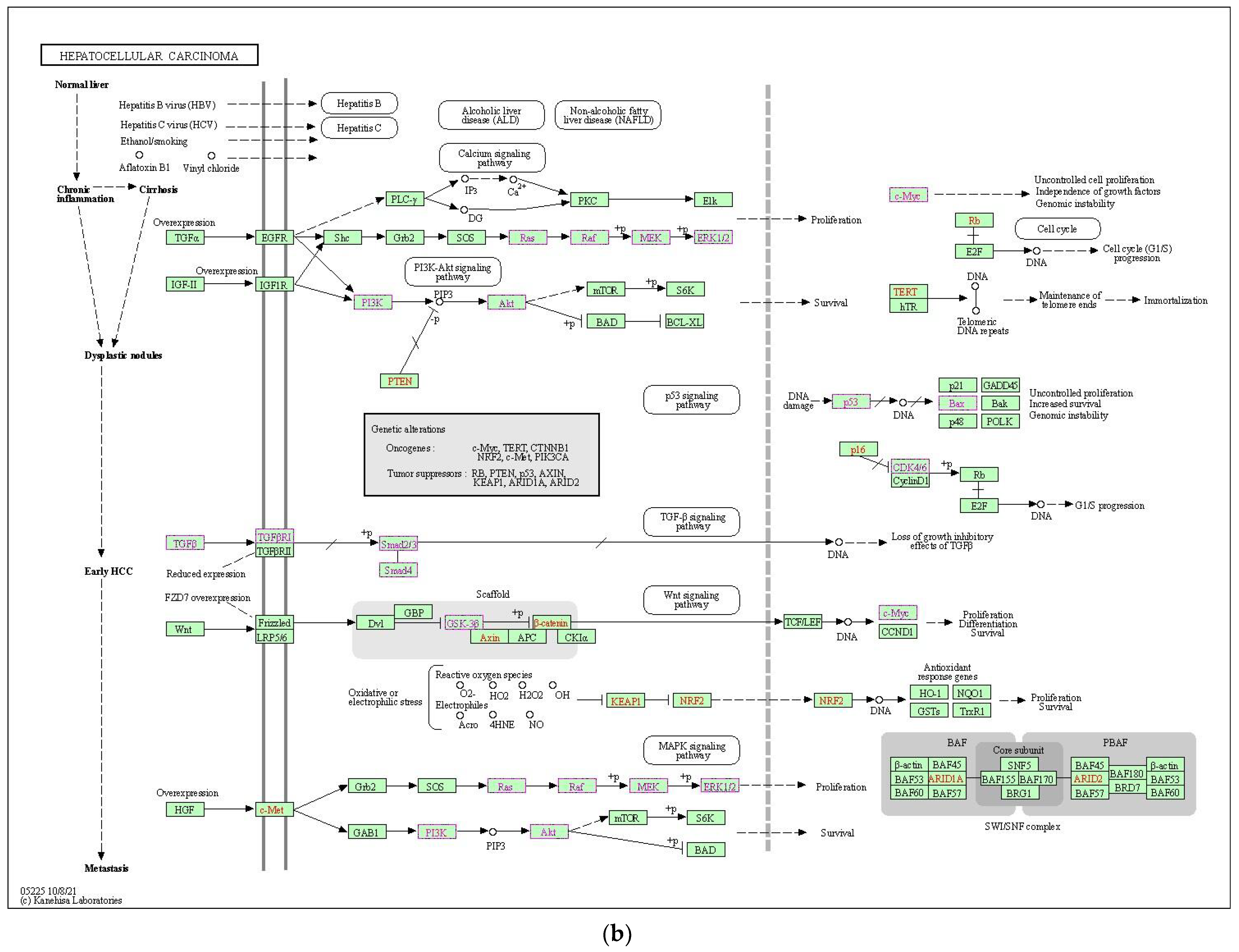
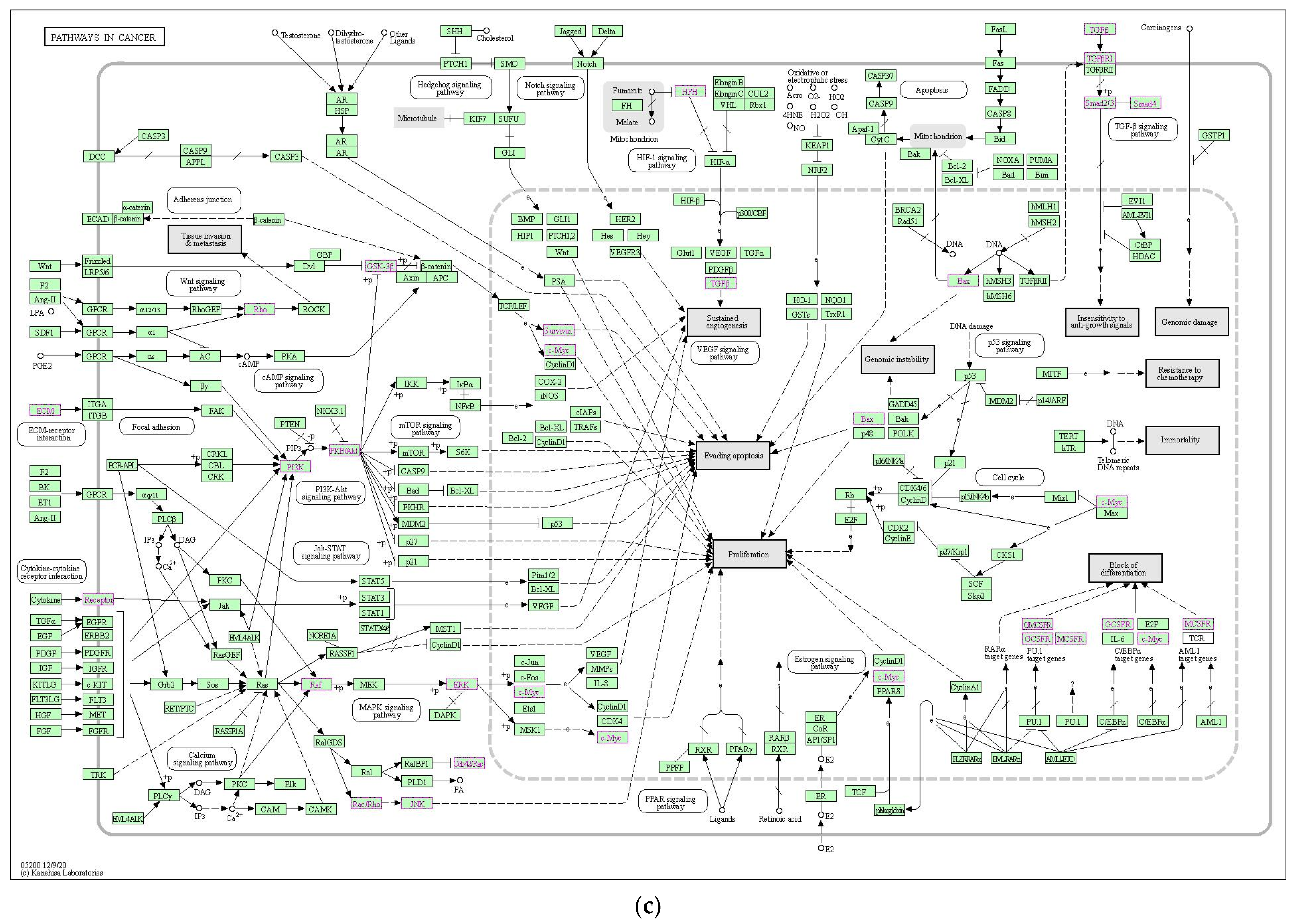
2.2. Functional Analysis and Pathway Enrichment Assessment
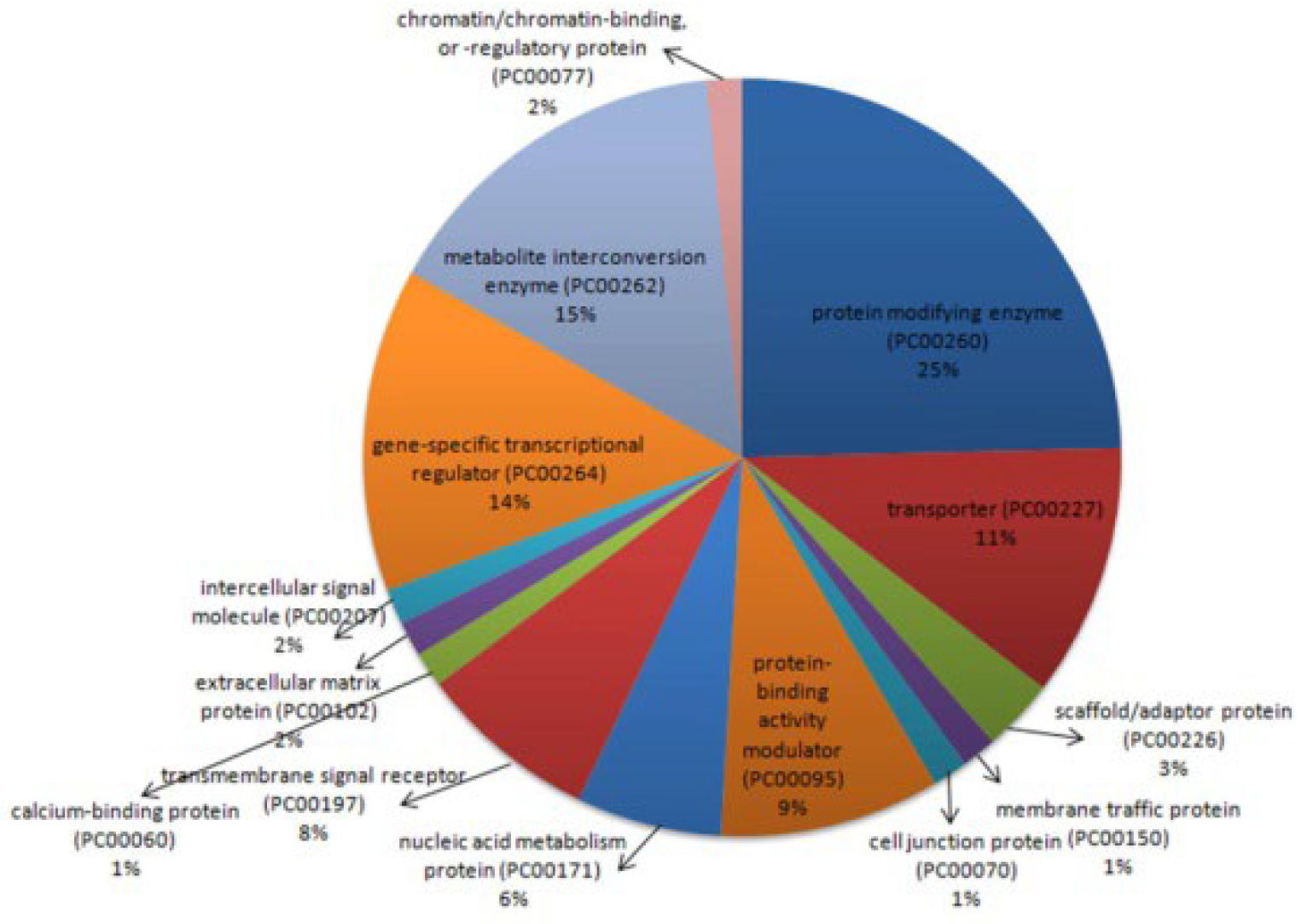
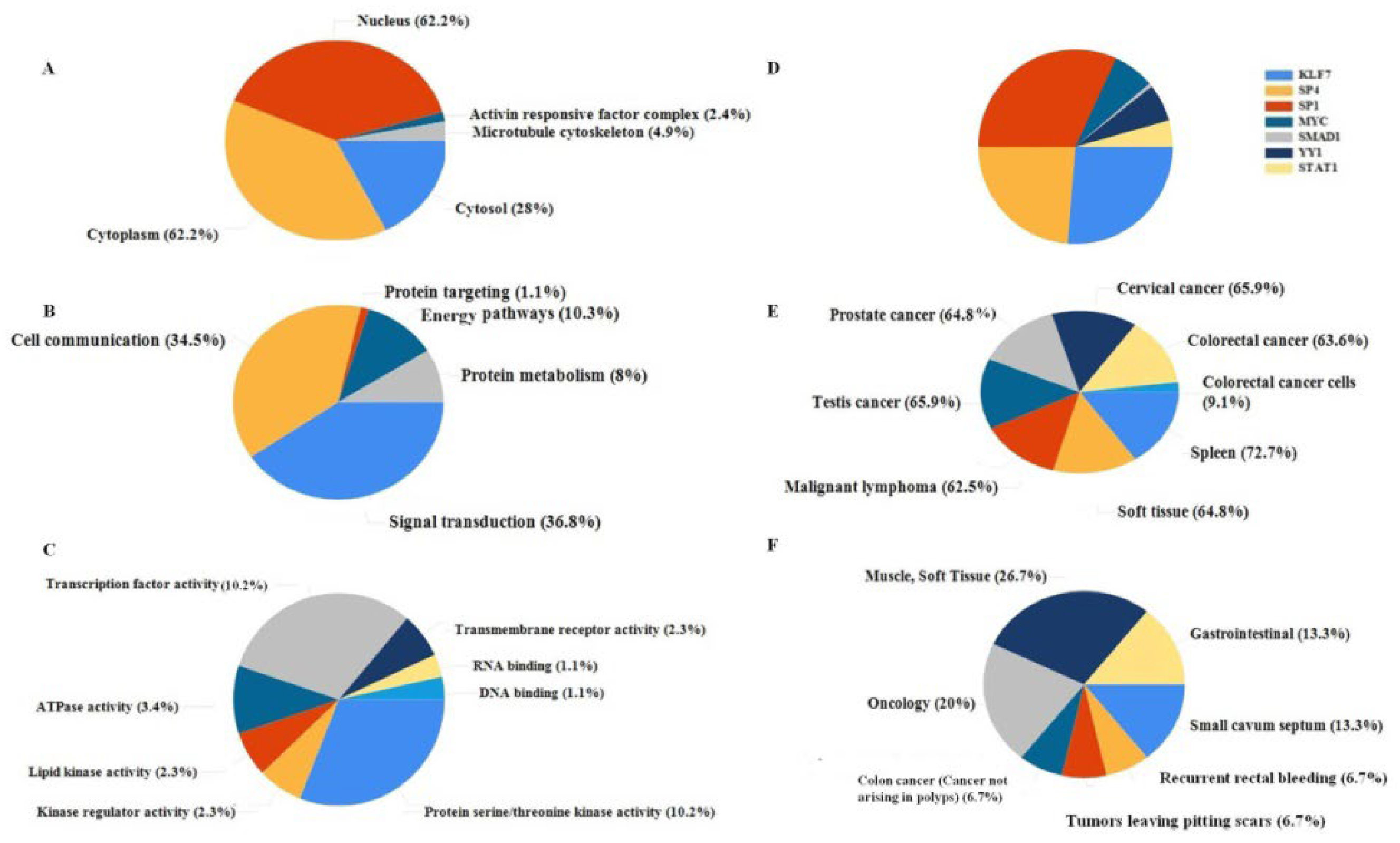
2.2.1. Functional Classification
2.2.2. Functional Annotation
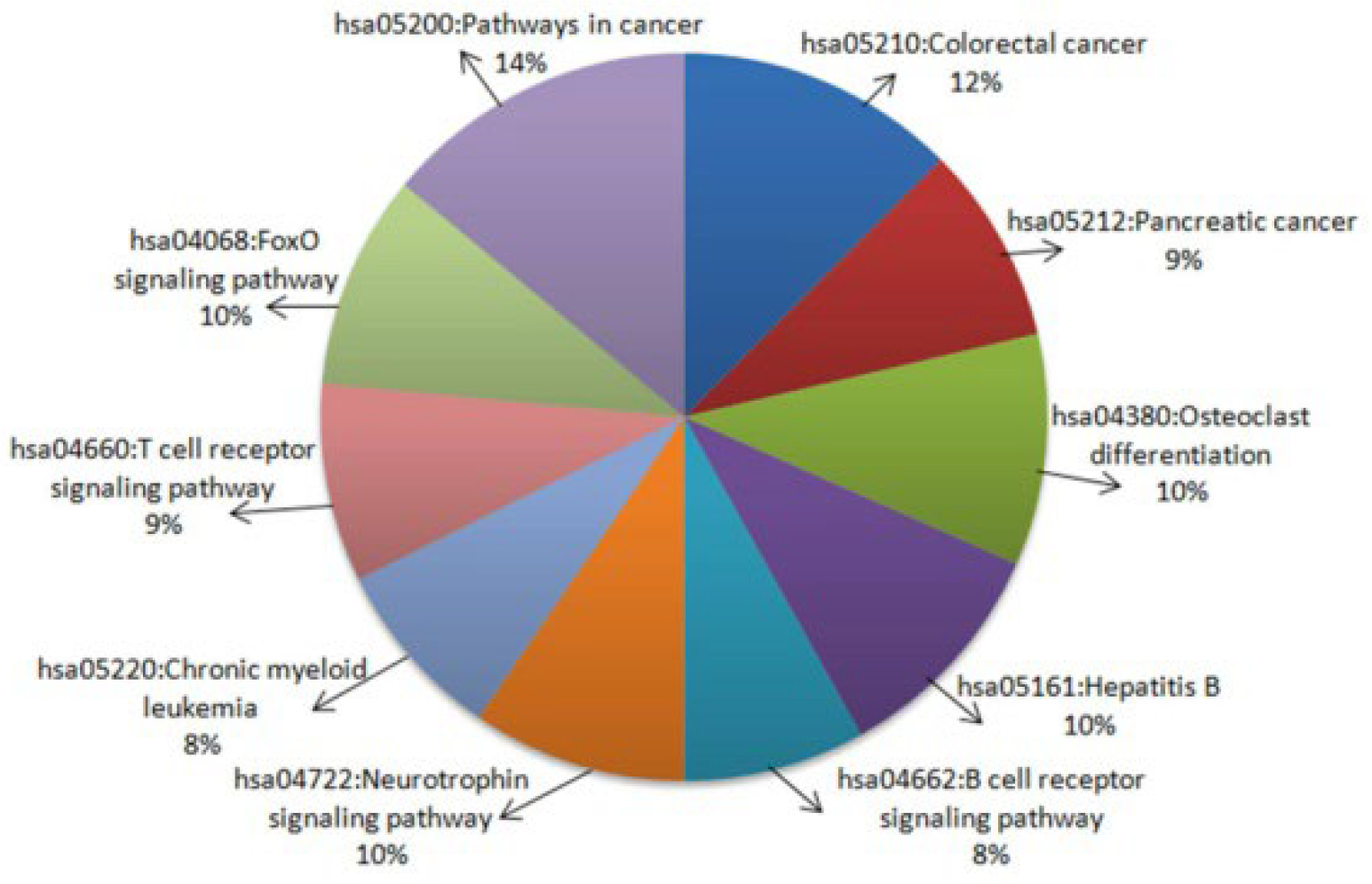
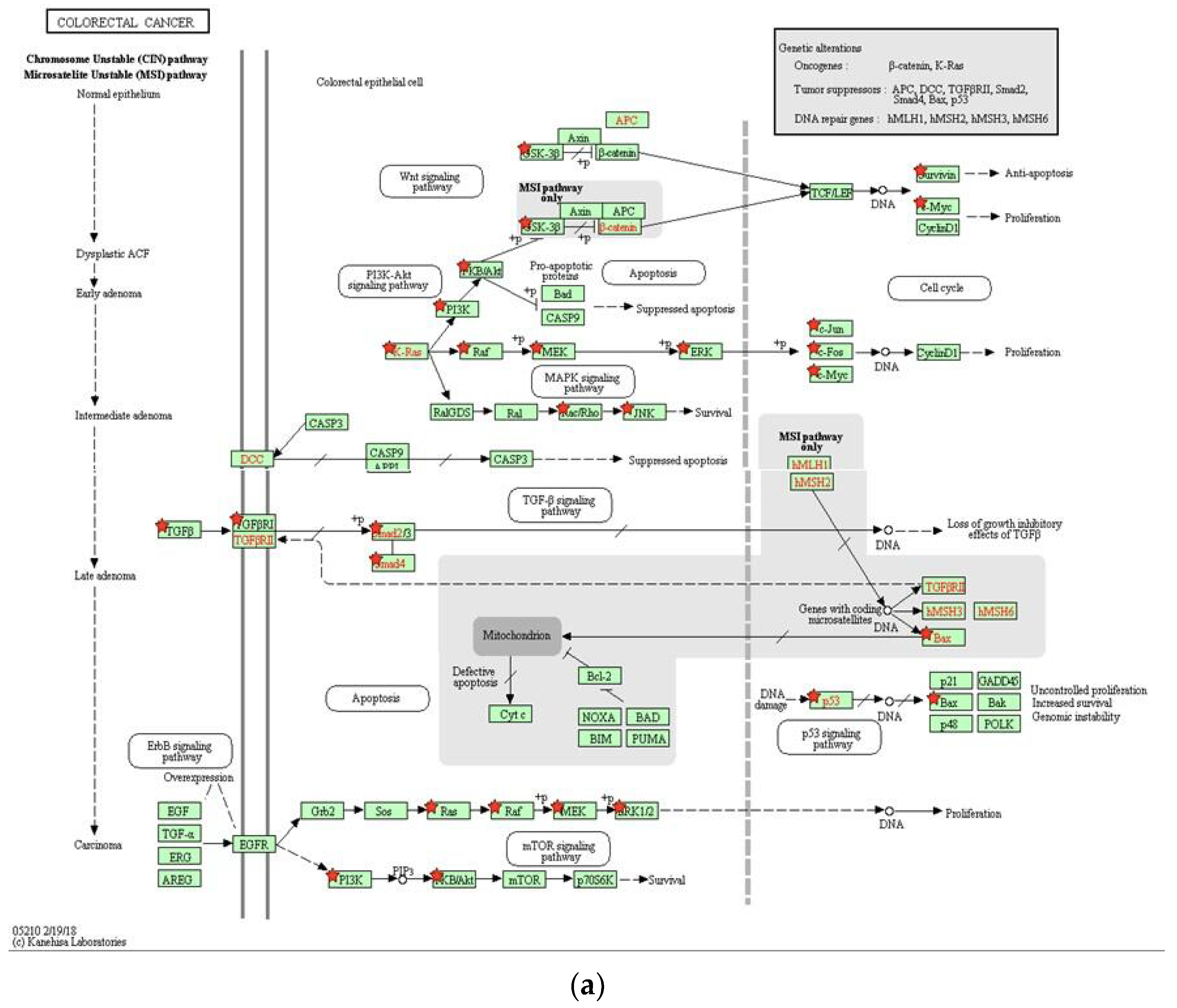
2.2.3. Functional Enrichment
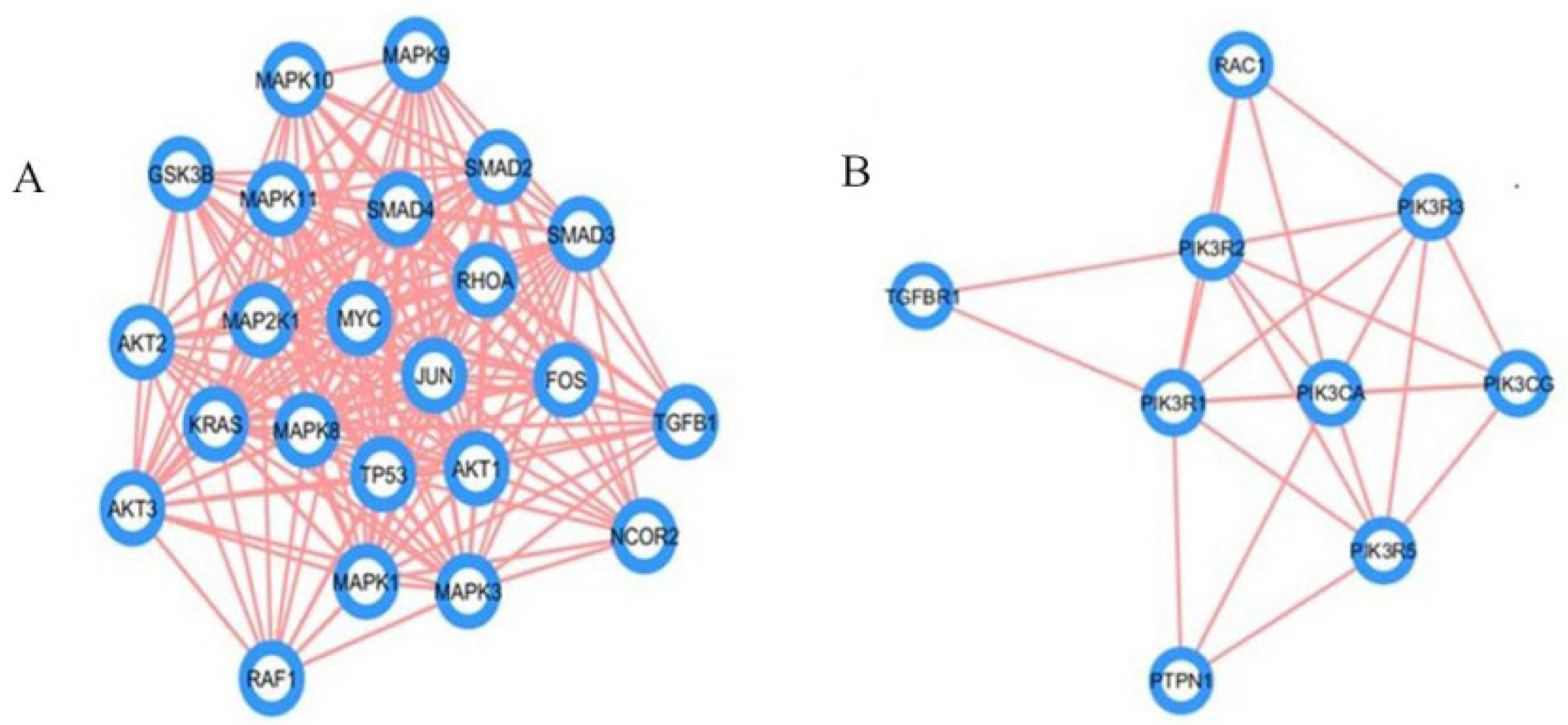
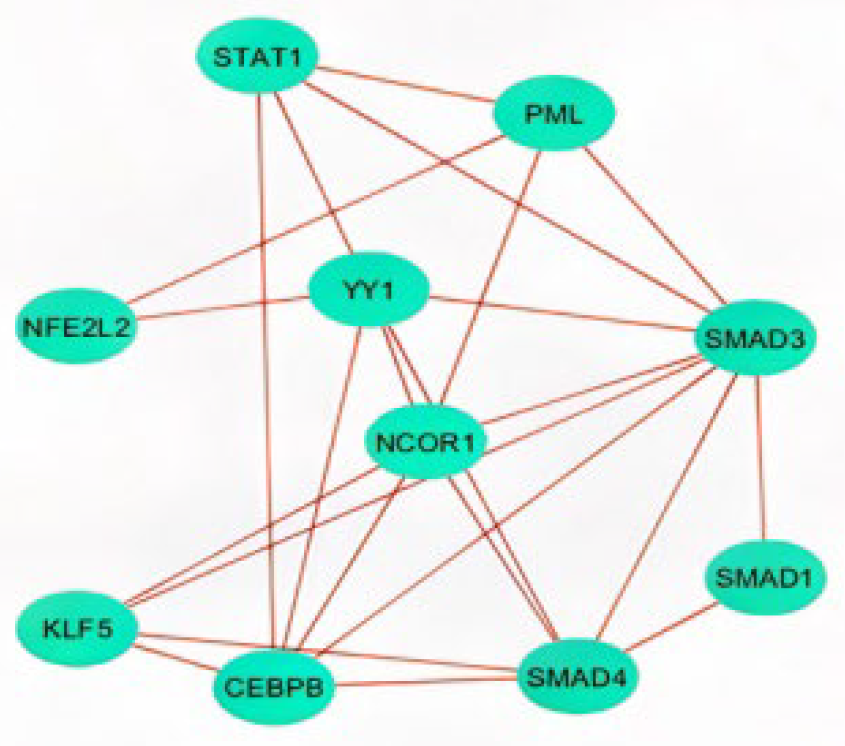
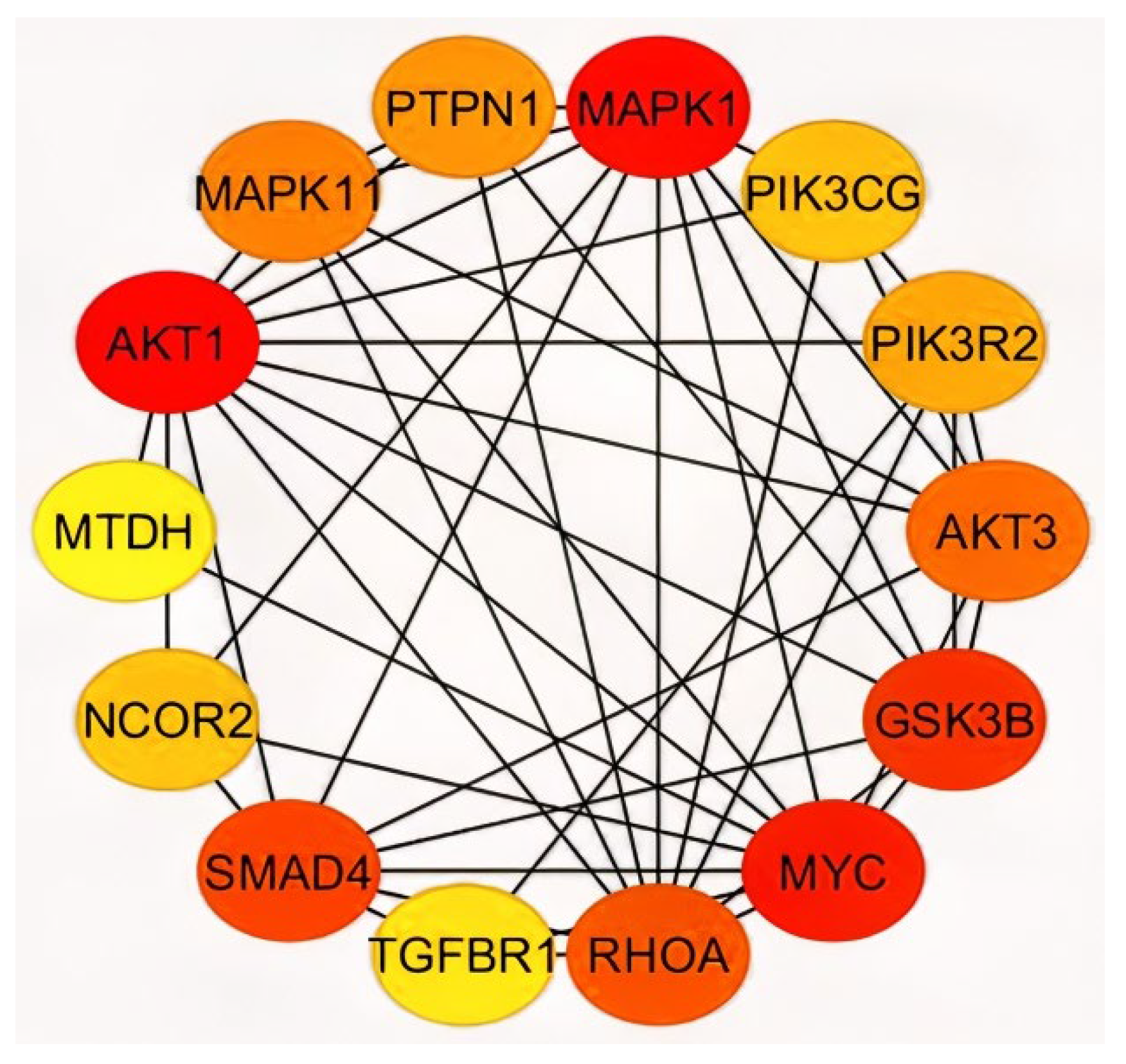
2.3. Analysis of Hub Genes and Binding Targets
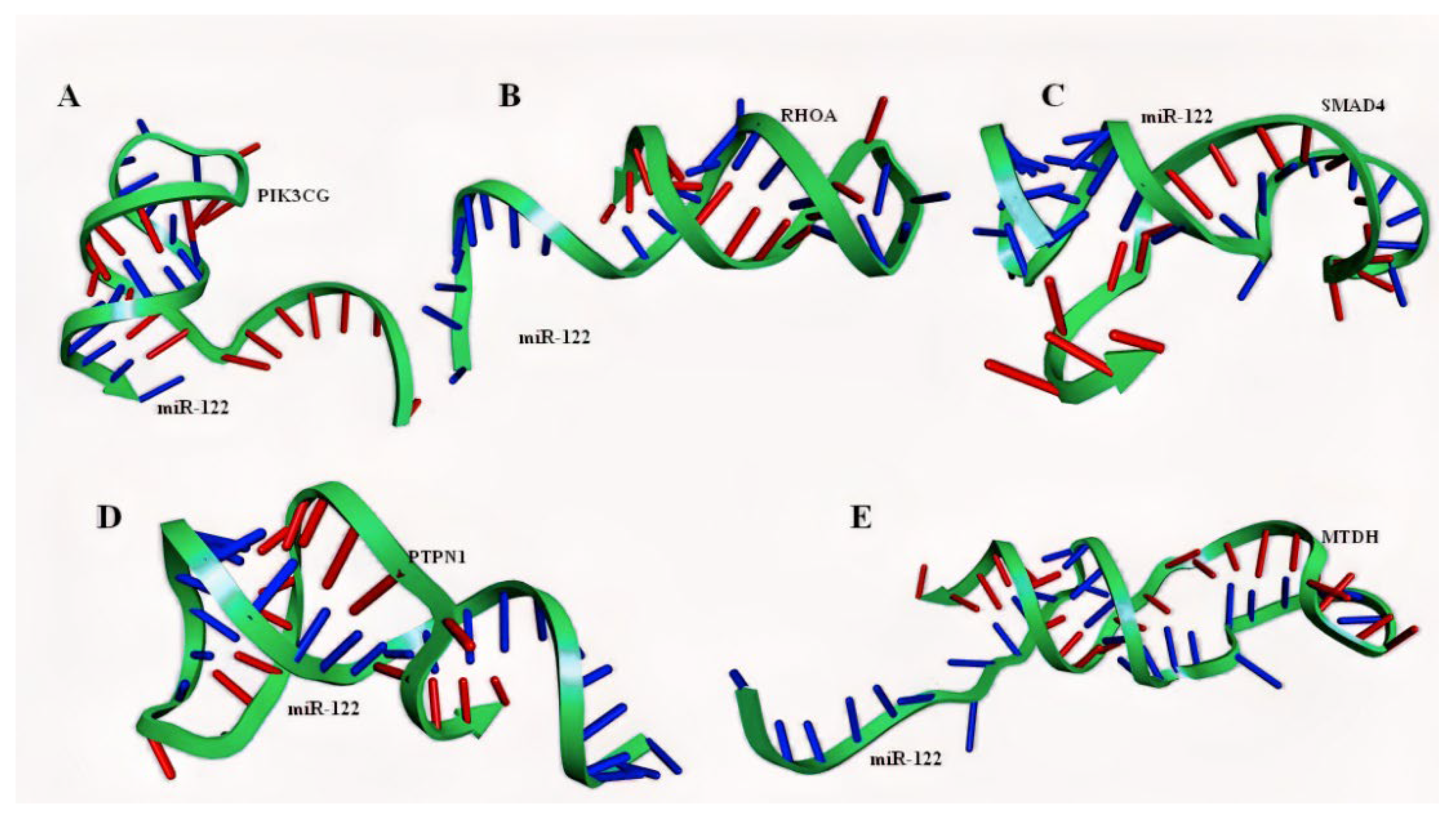
2.4. Molecular Modelling of miRNA–mRNA Duplex
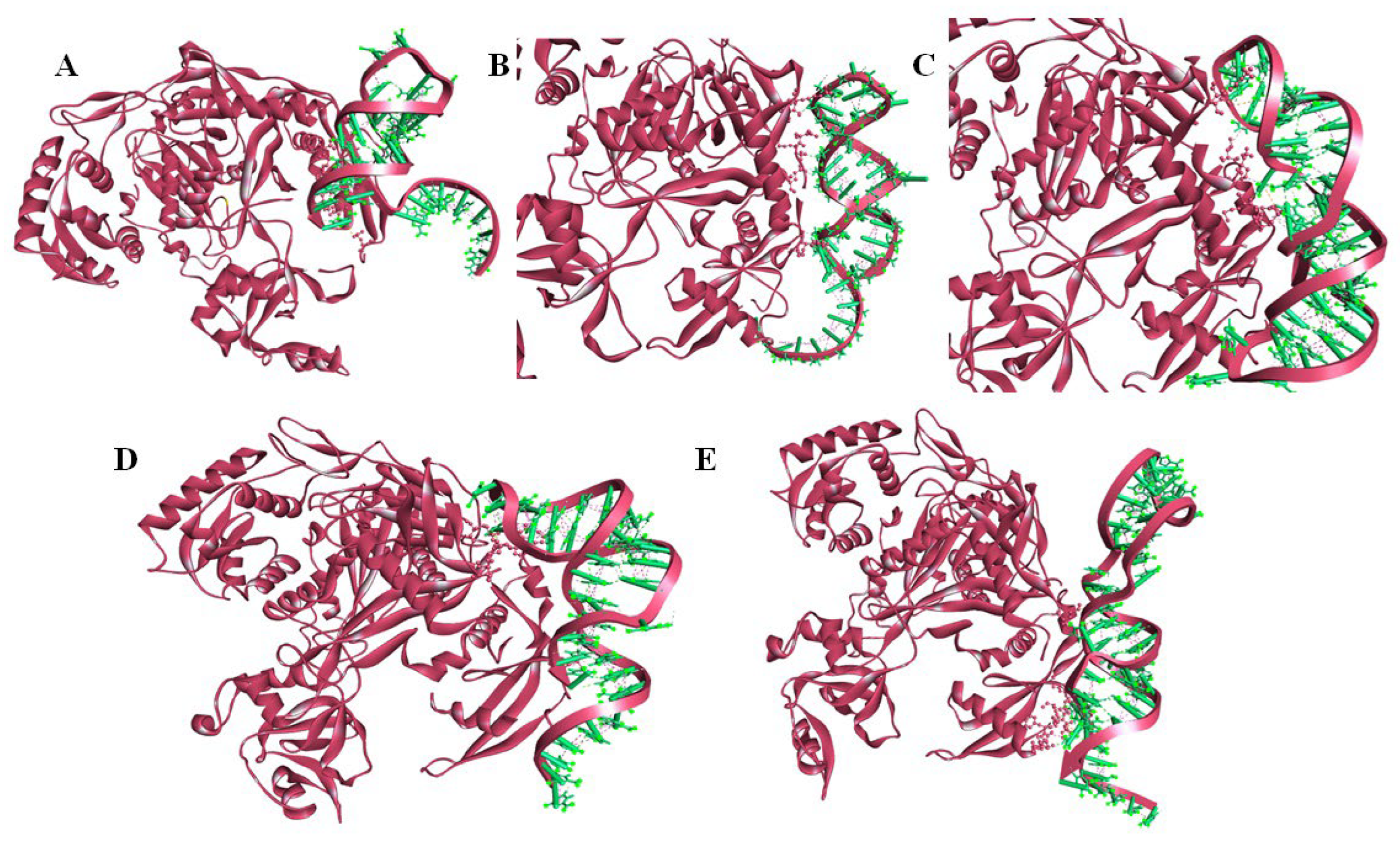
2.5. Molecular Docking of miRNA–mRNA–AGO Protein Complex
3. Discussion
4. Materials and Methods
4.1. Prediction of miRNA Interactors
4.2. Functional Classification
4.3. Network Clustering, Nodes, and Sub-Nodes Identification
4.4. Enrichment Analysis of Transcription Factors
4.5. Analysis of Significant miR-122 Interactors
4.6. Binding Site Analysis
4.7. Structural Prediction of miR122–mRNA Duplexes
4.8. Structural Extraction of Gene Regulator Protein
4.9. Molecular Docking of miRNA–mRNA Duplex and AGO Protein
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taft, R.J.; Pang, K.C.; Mercer, T.R.; Dinger, M.; Mattick, J.S. Non-coding RNAs: Regulators of disease. J. Pathol. 2010, 220, 126–139. [Google Scholar] [PubMed]
- MacFarlane, L.A.; Murphy, P.R. MicroRNA: Biogenesis, function and role in cancer. Curr. Genom. 2010, 11, 537–561. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Hall, B.; Shi, X. A network view of microRNA and gene interactions in different pathological stages of colon cancer. BMC. Med. Genom. 2019, 12, 158. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Wang, Q.; Zhang, X.; Wang, D.; Tang, H.C.; Meng, X.; Ding, X. Identification of an lncRNA-miRNA-mRNA interaction mechanism in breast cancer based on bioinformatic analysis. Mol. Med. Rep. 2017, 16, 5113–5120. [Google Scholar] [CrossRef] [PubMed]
- Usuba, W.; Urabe, F.; Yamamoto, Y.; Matsuzaki, J.; Sasaki, H.; Ichikawa, M.; Takizawa, S.; Aoki, Y.; Niida, S.; Kato, K.; et al. Circulating miRNA panels for specific and early detection in bladder cancer. Cancer Sci. 2019, 110, 408–419. [Google Scholar] [CrossRef]
- Schetter, A.J.; Okayama, H.; Harris, C.C. The role of microRNAs in colorectal cancer. Cancer J. 2012, 18, 244. [Google Scholar] [PubMed]
- Ng, E.K.; Chong, W.W.; Jin, H.; Lam, E.K.; Shin, V.Y.; Yu, J.; Poon, T.C.; Ng, S.S.; Sung, J.J. Differential expression of microRNAs in plasma of patients with colorectal cancer: A potential marker for colorectal cancer screening. Gut 2009, 58, 1375–1381. [Google Scholar] [CrossRef]
- Qu, H.; Xu, W.; Huang, Y.; Yang, S. Circulating miRNAs: Promising biomarkers of human cancer. Asian Pac. J. Cancer Prev. 2011, 12, 1117–1125. [Google Scholar]
- Wang, X.; Zhang, H.; Yang, H.; Bai, M.; Ning, T.; Deng, T.; Liu, R.; Fan, Q.; Zhu, K.; Li, J.; et al. Exosome-delivered circRNA promotes glycolysis to induce chemoresistance through the miR-122-PKM2 axis in colorectal cancer. Mol. Oncol. 2020, 14, 539–555. [Google Scholar] [CrossRef]
- Sun, L.; Liu, X.; Pan, B.; Hu, X.; Zhu, Y.; Su, Y.; Guo, Z.; Zhang, G.; Xu, M.; Xu, X.; et al. Serum exosomal miR-122 as a potential diagnostic and prognostic biomarker of colorectal cancer with liver metastasis. J. Cancer 2020, 11, 630. [Google Scholar] [CrossRef]
- Asadi, M.; Talesh, S.T.; Gjerstorff, M.F.; Shanehbandi, D.; Baradaran, B.; Hashemzadeh, S.; Zafari, V. Identification of miRNAs correlating with stage and progression of colorectal cancer. Color. Cancer 2019, 8, CRC06. [Google Scholar] [CrossRef]
- Yin, W.; Xu, J.; Li, C.; Dai, X.; Wu, T.; Wen, J. Circular RNA circ_0007142 facilitates colorectal cancer progression by modulating CDC25A expression via miR-122-5p. OncoTargets Ther. 2020, 13, 3689. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Zhu, Y.; Zhang, J.; Zhu, Z.; Ning, Z.; Chen, H.; Liu, L.; Chen, Z.; Meng, Z. miR-122 targets X-linked inhibitor of apoptosis protein to sensitize oxaliplatin-resistant colorectal cancer cells to oxaliplatin-mediated cytotoxicity. Cell. Physiol. Biochem. 2018, 51, 2148–2159. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, X.; Jin, Z.; Yin, T.; Duan, C.; Sun, J.; Xiong, R.; Li, Z. MiR-122 promotes the development of colon cancer by targeting ALDOA in vitro. Technol. Can. Res. Treat. 2019, 18, 1533033819871300. [Google Scholar] [CrossRef]
- Malayaperumal, S.; Sriramulu, S.; Banerjee, A.; Pathak, S. Over-Expression of MicroRNA-122 Inhibits Proliferation and Induces Apoptosis in Colon Cancer Cells. MicroRNA 2020, 9, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Maierthaler, M.; Benner, A.; Hoffmeister, M.; Surowy, H.; Jansen, L.; Knebel, P.; Chang-Claude, J.; Brenner, H.; Burwinkel, B. Plasma miR-122 and miR-200 family are prognostic markers in colorectal cancer. Int. J. Cancer 2017, 140, 176–187. [Google Scholar] [CrossRef]
- Höck, J.; Meister, G. The Argonaute protein family. Genome Biol. 2008, 9, 210. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Yu, H. A distributed infomap algorithm for scalable and high-quality community detection. In Proceedings of the 47th International Conference on Parallel Processing, Eugene, OR, USA, 13–16 August 2018; pp. 1–11. [Google Scholar]
- Song, H.; Ding, N.; Li, S.; Liao, J.; Xie, A.; Yu, Y.; Zhang, C.; Ni, C. Identification of hub genes associated with hepatocellular carcinoma using robust rank aggregation combined with weighted gene co-expression network analysis. Front. Genet. 2020, 11, 895. [Google Scholar] [CrossRef] [PubMed]
- Dweep, H.; Gretz, N. miRWalk2.0: A comprehensive atlas of microRNA-target interactions. Nat. Methods 2015, 12, 697. [Google Scholar] [CrossRef]
- Kou, Y.; Chen, E.Y.; Clark, N.R.; Duan, Q.; Tan, C.M.; Ma‘ayan, A. ChEA2: Gene-set libraries from ChIP-X experiments to decode the transcription regulome. In Proceedings of the International Conference on Availability, Reliability, and Security, Regensburg, Germany, 2–6 September 2013; Springer: Berlin/Heidelberg, Germany, 2013; pp. 416–430. [Google Scholar]
- Gosline, S.J. Beyond Argonaute: Understanding microRNA dysregulation in cancer and its effects on protein interaction and transcriptional regulatory networks. In Proceedings of the Intergrative RNA Biology Special Interest Group Meeting, Boston, MA, USA, 14 July 2014; p. 18. [Google Scholar]
- Amaro, A.; Croce, M.; Ferrini, S.; Barisione, G.; Gualco, M.; Perri, P.; Pfeffer, U.; Jager, M.J.; Coupland, S.E.; Mosci, C.; et al. Potential onco-suppressive role of miR122 and miR144 in uveal melanoma through ADAM10 and C-Met inhibition. Cancers 2020, 12, 1468. [Google Scholar] [CrossRef]
- Cervena, K.; Novosadova, V.; Pardini, B.; Naccarati, A.; Opattova, A.; Horak, J.; Vodenkova, S.; Buchler, T.; Skrobanek, P.; Levy, M.; et al. Analysis of microRNA expression changes during the course of therapy in rectal cancer patients. Front. Oncol. 2021, 11, 702258. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.M.; Xu, Z.; Shek, F.H.; Wong, K.F.; Lee, N.P.; Poon, R.T.; Chen, J.; Luk, J.M. miR-122 targets pyruvate kinase M2 and affects metabolism of hepatocellular carcinoma. PLoS ONE 2014, 9, e86872. [Google Scholar] [CrossRef] [PubMed]
- Béres, N.J.; Szabó, D.; Kocsis, D.; Szűcs, D.; Kiss, Z.; Müller, K.E.; Lendvai, G.; Kiss, A.; Arató, A.; Sziksz, E.; et al. Role of altered expression of miR-146a, miR-155, and miR-122 in pediatric patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2016, 22, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, Y.; Zheng, Y. MiR-122/Wnt/β-catenin regulatory circuitry sustains glioma progression. Tumor Biol. 2014, 35, 8565–8572. [Google Scholar] [CrossRef]
- Kim, J.G.; Islam, R.; Cho, J.Y.; Jeong, H.; Cap, K.C.; Park, Y.; Hossain, A.J.; Park, J.B. Regulation of RhoA GTPase and various transcription factors in the RhoA pathway. J. Cell. Physiol. 2018, 233, 6381–6392. [Google Scholar] [CrossRef]
- Hridita, R.T. The Role of SIRT6 in down Regulation of Colorectal Cancer. Ph.D. Thesis, Brac University, Dhaka, Bangladesh, 2019. [Google Scholar]
- Kong, L.; Wu, Q.; Zhao, L.; Ye, J.; Li, N.; Yang, H. Upregulated lncRNA-UCA1 contributes to metastasis of bile duct carcinoma through regulation of miR-122/CLIC1 and activation of the ERK/MAPK signaling pathway. Cell Cycle 2019, 18, 1212–1228. [Google Scholar] [CrossRef]
- Chen, C.; Wang, X.; Jiang, N.; Zong, D.; Yao, C.; Zhao, L.; Yin, L.; Huang, S.; Ji, H.; Xia, H. MiR-122 exerts anti-proliferative and apoptotic effects on nasopharyngeal carcinoma cells via the PI3K/AKT signaling pathway. Cell. Mol. Biol. 2018, 64, 21–25. [Google Scholar] [CrossRef]
- Alizadeh, E.; Akbarzadeh, A.; Eslaminejad, M.B.; Barzegar, A.; Hashemzadeh, S.; Nejati-Koshki, K.; Zarghami, N. Up regulation of liver-enriched transcription factors HNF 4a and HNF 6 and liver-specific microRNA (miR-122) by inhibition of Let-7b in mesenchymal stem cells. Chem. Biol. Drug Des. 2015, 85, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Gulubova, M.; Manolova, I.; Ananiev, J.; Julianov, A.; Yovchev, Y.; Peeva, K. Role of TGF-β1, its receptor TGFβRII, and Smad proteins in the progression of colorectal cancer. Int. J. Color. Dis. 2010, 25, 591–599. [Google Scholar] [CrossRef]
- Tanaka, A.; Zhou, Y.; Ogawa, M.; Shia, J.; Klimstra, D.S.; Wang, J.Y.; Roehrl, M.H. STAT1 as a potential prognosis marker for poor outcomes of early-stage colorectal cancer with microsatellite instability. PloS ONE 2020, 15, e0229252. [Google Scholar] [CrossRef] [PubMed]
- Shao, K.; Pu, W.; Zhang, J.; Guo, S.; Qian, F.; Glurich, I.; Jin, Q.; Ma, Y.; Ju, S.; Zhang, Z.; et al. DNA hypermethylation contributes to colorectal cancer metastasis by regulating the binding of CEBPB and TFCP2 to the CPEB1 promoter. Clin. Epigenet. 2021, 13, 89. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, X.; Yan, B.; Yu, W.; Shan, L. CircAGFG1 drives metastasis and stemness in colorectal cancer by modulating YY1/CTNNB1. Cell Death Dis. 2020, 11, 542. [Google Scholar] [CrossRef] [PubMed]
- Al-Sohaily, S.; Biankin, A.; Leong, R.; Kohonen-Corish, M.; Warusavitarne, J. Molecular pathways in colorectal cancer. J. Gastroenterol. Hepatol. 2012, 27, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.C. The genetic basis of colorectal cancer: Insights into critical pathways of tumorigenesis. Gastroenterology 2000, 119, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Luque-García, J.L.; Martínez-Torrecuadrada, J.L.; Epifano, C.; Cañamero, M.; Babel, I.; Casal, J.I. Differential protein expression on the cell surface of colorectal cancer cells associated to tumor metastasis. Proteomics 2010, 10, 940–952. [Google Scholar] [CrossRef]
- Anzai, T.; Matsumura, Y. Topological analysis of TMEM180, a newly identified membrane protein that is highly expressed in colorectal cancer cells. Biochem. Biophys. Res. Commun. 2019, 520, 566–572. [Google Scholar] [CrossRef]
- Choi, D.S.; Yang, J.S.; Choi, E.J.; Jang, S.C.; Park, S.; Kim, O.Y.; Hwang, D.; Kim, K.P.; Kim, Y.K.; Kim, S.; et al. The protein interaction network of extracellular vesicles derived from human colorectal cancer cells. J. Proteome Res. 2012, 11, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Sun, X.; Wei, Y.; Liang, H.; Yuan, M.; Jin, F.; Chen, X.; Liu, Y.; Zhang, C.Y.; Li, L.; et al. Nuclear miR-122 directly regulates the biogenesis of cell survival oncomiR miR-21 at the posttranscriptional level. Nucleic Acids. Res. 2018, 46, 2012–2029. [Google Scholar] [CrossRef]
- Qi, Y.; Ding, L.; Zhang, S.; Yao, S.; Ong, J.; Li, Y.; Wu, H.; Du, P. A plant immune protein enables broad antitumor response by rescuing microRNA deficiency. Cell 2022, 185, 1888–1904. [Google Scholar] [CrossRef]
- Rath, S.N.; Das, D.; Konkimalla, V.B.; Pradhan, S.K. In silico study of miRNA based gene regulation, involved in solid cancer, by the assistance of argonaute protein. Genom. Inform. 2016, 14, 112–124. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Z.; Ma, L.; Pei, C.; Qin, L.; Gao, N.; Li, J.; Yin, Y. MiRNAs regulate oxidative stress related genes via binding to the 3′ UTR and TATA-box regions: A new hypothesis for cataract pathogenesis. BMC Ophthalmol. 2017, 17, 142. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Zhou, G.; Soufan, O.; Xia, J. miRNet 2.0: Network-based visual analytics for miRNA functional analysis and systems biology. Nucleic Acids Res. 2020, 48, W244–W251. [Google Scholar] [CrossRef]
- Mering, C.V.; Huynen, M.; Jaeggi, D.; Schmidt, S.; Bork, P.; Snel, B. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 2003, 31, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32 (Suppl. 1), D115–D119. [Google Scholar] [CrossRef]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.P.; Mushayamaha, T.; Thomas, P.D. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef] [PubMed]
- Sriramulu, S.; Nandy, S.K.; Ganesan, H.; Banerjee, A.; Pathak, S. In silico analysis and prediction of transcription factors of the proteins interacting with astrocyte elevated gene 1. Comput. Biol. Chem. 2021, 92, 107478. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Shishkin, A.A.; Giannoukos, G.; Kucukural, A.; Ciulla, D.; Busby, M.; Surka, C.; Chen, J.; Bhattacharyya, R.P.; Rudy, R.F.; Patel, M.M.; et al. Simultaneous generation of many RNA-seq libraries in a single reaction. Nat. Methods 2015, 12, 323–325. [Google Scholar] [CrossRef]
- Karnovsky, A.; Weymouth, T.; Hull, T.; Tarcea, V.G.; Scardoni, G.; Laudanna, C.; Sartor, M.A.; Stringer, K.A.; Jagadish, H.V.; Burant, C.; et al. Metscape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data. Bioinformatics 2012, 28, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Hu, F.; Xiong, W.; Wei, Q.; Liu, F.F. Network-based transcriptomic analysis reveals novel melatonin-sensitive genes in cardiovascular system. Endocrine 2019, 64, 414–419. [Google Scholar] [CrossRef]
- Pathan, M.; Keerthikumar, S.; Ang, C.S.; Gangoda, L.; Quek, C.Y.; Williamson, N.A.; Mouradov, D.; Sieber, O.M.; Simpson, R.J.; Salim, A.; et al. FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics 2015, 15, 2597–2601. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38 (Suppl. 2), W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Rehnström, M.; Frederiksen, S.D.; Ansar, S.; Edvinsson, L. Transcriptome profiling revealed early vascular smooth muscle cell gene activation following focal ischemic stroke in female rats–comparisons with males. BMC Genom. 2020, 21, 883. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.H.; Apeltsin, L.; Newman, A.M.; Baumbach, J.; Wittkop, T.; Su, G.; Bader, G.D.; Ferrin, T.E. clusterMaker: A multi-algorithm clustering plugin for Cytoscape. BMC Bioinform. 2011, 12, 436. [Google Scholar]
- Saito, R.; Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Lotia, S.; Pico, A.R.; Bader, G.D.; Ideker, T. A travel guide to Cytoscape plugins. Nat. Methods 2012, 9, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Lou, W.; Liu, J.; Ding, B.; Chen, D.; Xu, L.; Ding, J.; Jiang, D.; Zhou, L.; Zheng, S.; Fan, W. Identification of potential miRNA–mRNA regulatory network contributing to pathogenesis of HBV-related HCC. J. Transl. Med. 2019, 17, 7. [Google Scholar] [CrossRef]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed]
- Hofacker, I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003, 31, 3429–3431. [Google Scholar] [CrossRef] [PubMed]
- Antczak, M.; Popenda, M.; Zok, T.; Sarzynska, J.; Ratajczak, T.; Tomczyk, K.; Adamiak, R.W.; Szachniuk, M. New functionality of RNAComposer: Application to shape the axis of miR160 precursor structure. Acta Biochim. Pol. 2016, 63, 737–744. [Google Scholar] [CrossRef]
- Wang, Y.; Juranek, S.; Li, H.; Sheng, G.; Tuschl, T.; Patel, D.J. Structure of an argonaute silencing complex with a seed-containing guide DNA and target RNA duplex. Nature 2008, 456, 921–926. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; Van Dijk, M.; Bonvin, A.M. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Uniprot ID | Tissue Specificity (Overexpressed Tissues) | Subcellular Location |
|---|---|---|---|
| AKT1 | Q96B36 | Liver, colon, and heart; high levels in cancer cell lines (A-549 and HeLa) | Nucleus, cytosol, and cytoplasm |
| AKT3 | Q9Y243 | Brain, lung, and kidney | Nucleus, cytosol, and cytoplasm |
| GSK3B | P49841 | Testis, thymus, prostate, ovary, lungs, and intestine | Nucleus, plasma membrane, cytosol, and cytoplasm |
| MAPK1 | P28482 | Putamen, brain | Cytoplasm, cytosol, nucleus, and cytoskeleton |
| MAPK11 | Q15759 | Brain | Cytosol and nucleus |
| MTDH (AEG-1) | Q86UE4 | Skeletal muscle, heart, small intestine, and in endocrine glands; Over expressed in various cancers including breast, brain, and colon cancer | Nucleus and endoplasmic reticulum |
| MYC | P01106 | Thoracic mammary gland | Nucleus |
| NCOR2 | Q9Y618 | Colon, lung, spleen, and brain | Nucleus |
| PIK3CG | P48736 | Pancreas, skeletal muscle, liver, and heart | Plasma membrane, cytoplasm, and cytosol |
| PIK3R2 | O00459 | Temporal lobe | Cytosol and nucleus |
| PTPN1 | P18031 | Keratinocytes | Endoplasmic reticulum |
| RHOA | P61586 | Metanephric glomerulus | Cytoskeleton and plasma membrane |
| SMAD4 | Q13485 | Kidney | Nucleus, cytoplasm, and cytosol |
| TGFBR1 | P36897 | Abundant in most types of tissues; Also expressed in a variety of cancer cell lines | Plasma membrane |
| Mirna ID | Refseq ID | Gene Symbol | Start | End | No. of Pairings | Binding Region Length | Binding Score | Binding Energy kcal/mol | Longest Consecutive Pairings | Position |
|---|---|---|---|---|---|---|---|---|---|---|
| hsa-miR-122-5p | NM_001282426 | PIK3CG | 5528 | 5549 | 19 | 21 | 1 | −23 | 12 | 3UTR |
| hsa-miR-122-3p | NM_178812 | MTDH | 4741 | 4776 | 15 | 18 | 1 | −21.9 | 13 | 3UTR |
| hsa-miR-122-5p | NM_001313943 | RHOA | 1063 | 1111 | 15 | 48 | 1 | −21.3 | 7 | 3UTR |
| hsa-miR-122-5p | NM_178812 | MTDH | 3008 | 3026 | 15 | 18 | 1 | −19.2 | 8 | 3UTR |
| hsa-miR-122-3p | NM_002093 | GSK3B | 4464 | 4501 | 12 | 14 | 1 | −18.1 | 12 | 3UTR |
| hsa-miR-122-5p | NM_001278618 | PTPN1 | 2530 | 2553 | 17 | 23 | 0.923 | −20.6 | 7 | 3UTR |
| hsa-miR-122-5p | NM_001282426 | PIK3CG | 6288 | 6306 | 15 | 18 | 0.923 | −19.5 | 9 | 3UTR |
| hsa-miR-122-5p | NM_178812 | MTDH | 4855 | 4872 | 14 | 17 | 0.923 | −19.4 | 8 | 3UTR |
| hsa-miR-122-5p | NM_001278618 | PTPN1 | 3077 | 3091 | 13 | 14 | 0.923 | −18.7 | 13 | 3UTR |
| hsa-miR-122-3p | NM_001278618 | PTPN1 | 2915 | 2930 | 13 | 15 | 0.923 | −17.9 | 7 | 3UTR |
| hsa-miR-122-5p | NM_002751 | MAPK11 | 1262 | 1281 | 15 | 19 | 0.846 | −22 | 8 | 3UTR |
| hsa-miR-122-5p | NM_001282426 | PIK3CG | 5221 | 5244 | 19 | 23 | 0.846 | −20.6 | 8 | 3UTR |
| hsa-miR-122-5p | NM_001282426 | PIK3CG | 3899 | 3916 | 14 | 17 | 0.846 | −20 | 6 | 3UTR |
| hsa-miR-122-5p | NM_001278618 | PTPN1 | 2459 | 2474 | 14 | 15 | 0.846 | −19.7 | 14 | 3UTR |
| hsa-miR-122-3p | NM_178812 | MTDH | 4338 | 4352 | 13 | 14 | 0.846 | −19.4 | 13 | 3UTR |
| hsa-miR-122-5p | NM_005359 | SMAD4 | 3600 | 3653 | 19 | 33 | 0.846 | −19.4 | 10 | 3UTR |
| hsa-miR-122-5p | NM_002467 | MYC | 2646 | 2666 | 17 | 20 | 0.846 | −18.9 | 13 | 3UTR |
| hsa-miR-122-3p | NM_001282426 | PIK3CG | 3940 | 3967 | 14 | 17 | 0.846 | −18.7 | 9 | 3UTR |
| hsa-miR-122b-3p | NM_001077261 | NCOR2 | 2961 | 2976 | 13 | 15 | 0.961 | −20.2 | 9 | CDS |
| hsa-miR-122-5p | NM_181690 | AKT3 | 990 | 1043 | 20 | 34 | 0.923 | −24.5 | 8 | CDS |
| hsa-miR-122-5p | NM_002093 | GSK3B | 1441 | 1474 | 20 | 33 | 0.923 | -23.2 | 10 | CDS |
| hsa-miR-122-5p | NM_004612 | TGFBR1 | 1177 | 1201 | 19 | 24 | 0.846 | −23.6 | 12 | CDS |
| hsa-miR-122-5p | NM_001313943 | RHOA | 683 | 706 | 16 | 23 | 0.846 | −19.8 | 8 | CDS |
| hsa-miR-122-5p | NM_005359 | SMAD4 | 1042 | 1061 | 17 | 19 | 0.846 | −19.4 | 14 | CDS |
| hsa-miR-122-5p | NM_001206654 | NCOR2 | 596 | 642 | 16 | 21 | 0.846 | −19.1 | 10 | CDS |
| hsa-miR-122-5p | NM_001306210 | TGFBR1 | 1189 | 1213 | 19 | 24 | 0.846 | −18.6 | 12 | CDS |
| hsa-miR-122b-3p | NM_005163 | AKT1 | 1536 | 1553 | 15 | 17 | 0.846 | −17.5 | 12 | CDS |
| hsa-miR-122-5p | NM_138957 | MAPK1 | 806 | 829 | 16 | 23 | 0.846 | −17.2 | 6 | CDS |
| hsa-miR-122b-5p | NM_005163 | AKT1 | 1298 | 1323 | 15 | 20 | 0.846 | −17 | 8 | CDS |
| hsa-miR-122-5p | NM_001278618 | PTPN1 | 677 | 701 | 19 | 24 | 0.846 | −16.7 | 12 | CDS |
| hsa-miR-122-5p | NM_001077261 | NCOR2 | 622 | 642 | 16 | 20 | 0.846 | −16.5 | 10 | CDS |
| hsa-miR-122-3p | NM_002093 | GSK3B | 1779 | 1802 | 13 | 17 | 0.846 | −16.3 | 11 | CDS |
| hsa-miR-122-5p | NM_001313943 | RHOA | 382 | 402 | 18 | 20 | 0.846 | −15.1 | 9 | CDS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganesan, H.; Nandy, S.K.; Banerjee, A.; Pathak, S.; Zhang, H.; Sun, X.-F. RNA-Interference-Mediated miR-122-Based Gene Regulation in Colon Cancer, a Structural In Silico Analysis. Int. J. Mol. Sci. 2022, 23, 15257. https://doi.org/10.3390/ijms232315257
Ganesan H, Nandy SK, Banerjee A, Pathak S, Zhang H, Sun X-F. RNA-Interference-Mediated miR-122-Based Gene Regulation in Colon Cancer, a Structural In Silico Analysis. International Journal of Molecular Sciences. 2022; 23(23):15257. https://doi.org/10.3390/ijms232315257
Chicago/Turabian StyleGanesan, Harsha, Suman K. Nandy, Antara Banerjee, Surajit Pathak, Hong Zhang, and Xiao-Feng Sun. 2022. "RNA-Interference-Mediated miR-122-Based Gene Regulation in Colon Cancer, a Structural In Silico Analysis" International Journal of Molecular Sciences 23, no. 23: 15257. https://doi.org/10.3390/ijms232315257
APA StyleGanesan, H., Nandy, S. K., Banerjee, A., Pathak, S., Zhang, H., & Sun, X.-F. (2022). RNA-Interference-Mediated miR-122-Based Gene Regulation in Colon Cancer, a Structural In Silico Analysis. International Journal of Molecular Sciences, 23(23), 15257. https://doi.org/10.3390/ijms232315257