
Phosphorylation of CaMK and CREB-Mediated Cardiac Aldosterone Synthesis Induced by Arginine Vasopressin in Rats with Myocardial Infarction


Abstract
1. Introduction
2. Results
2.1. Left Ventricular Mass Index and Cardiac Function in Rats with MI
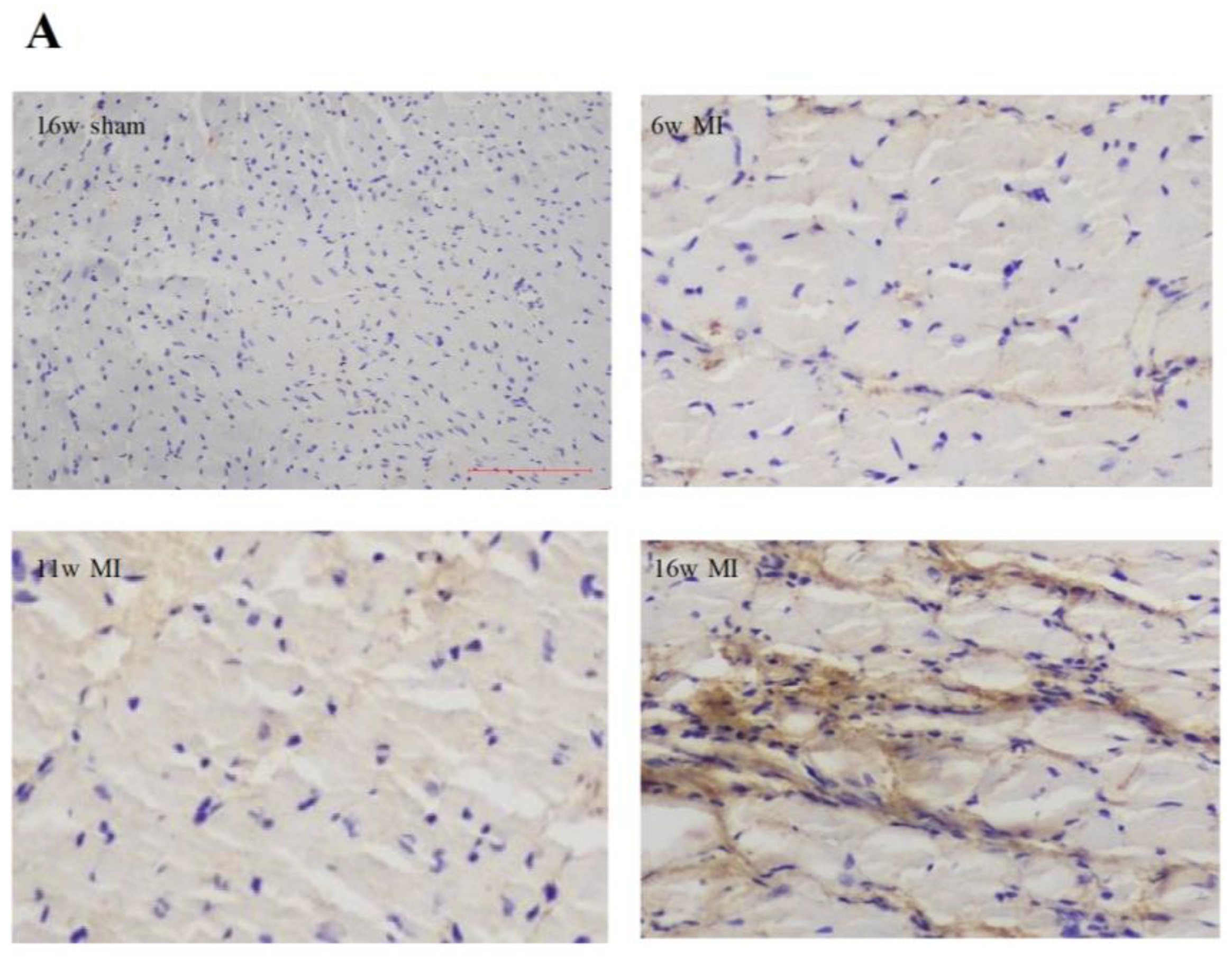
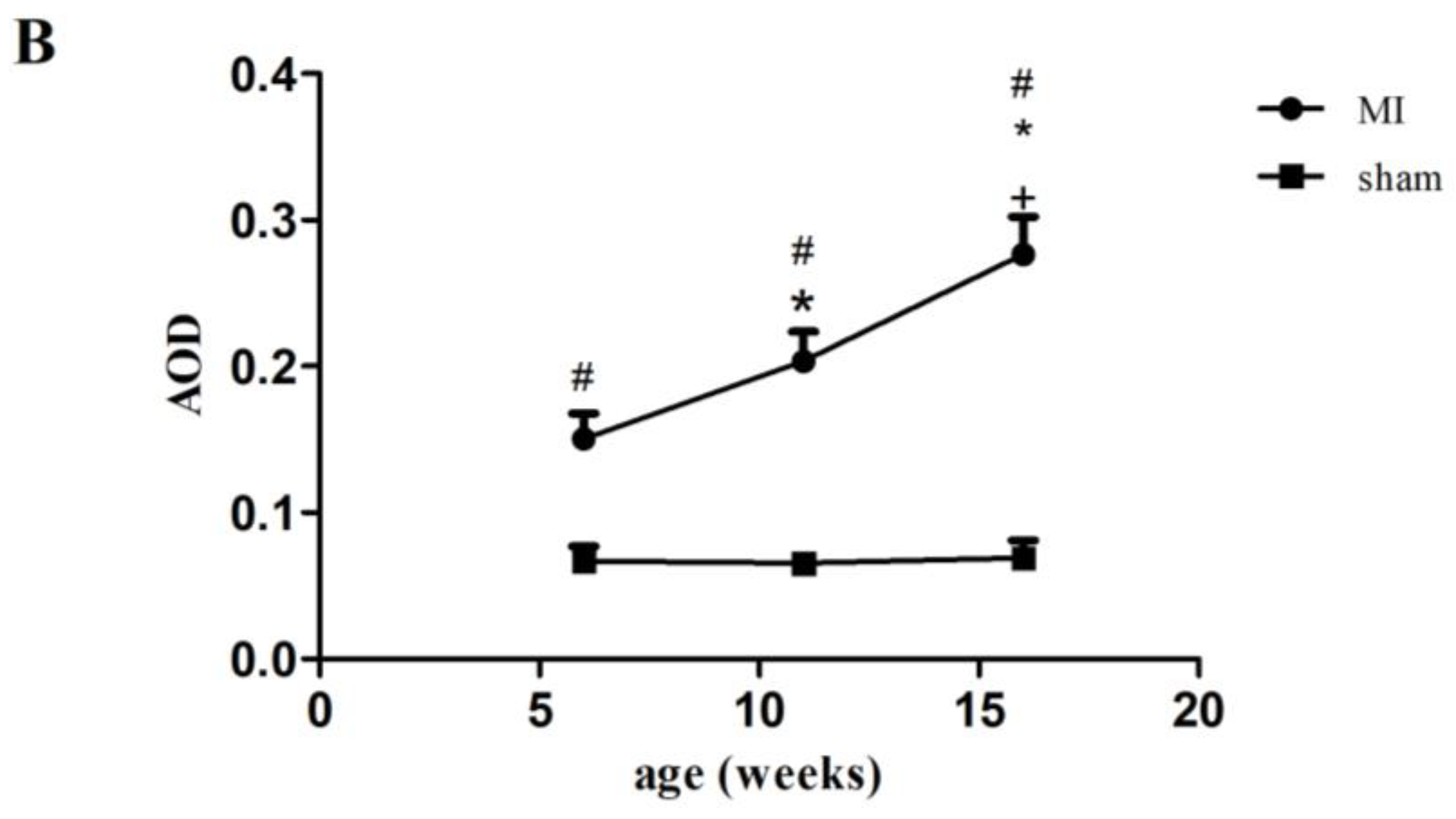
2.2. Type I Collagen Expression in Rats with MI
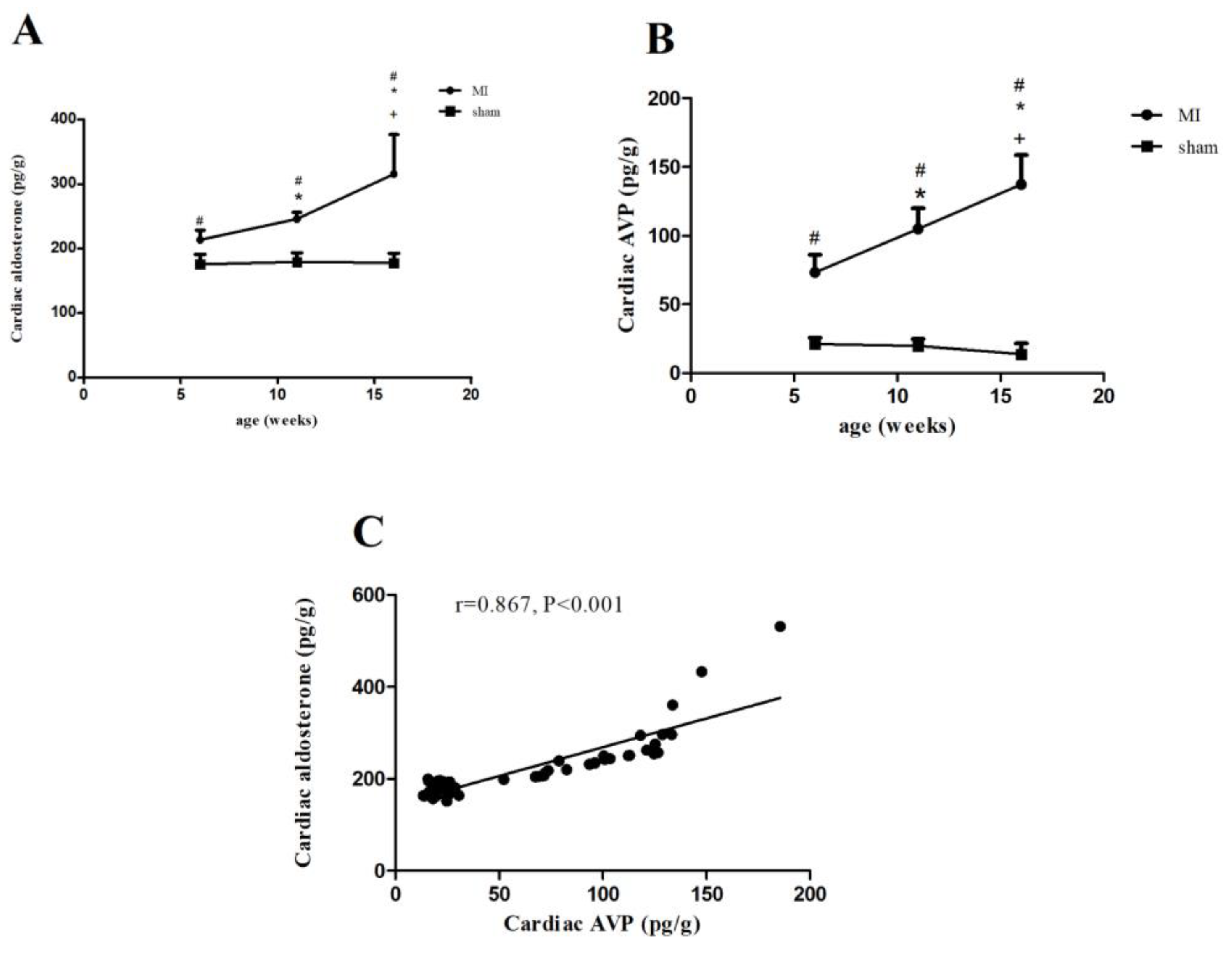
2.3. Aldosterone and AVP Concentrations in Rats with MI
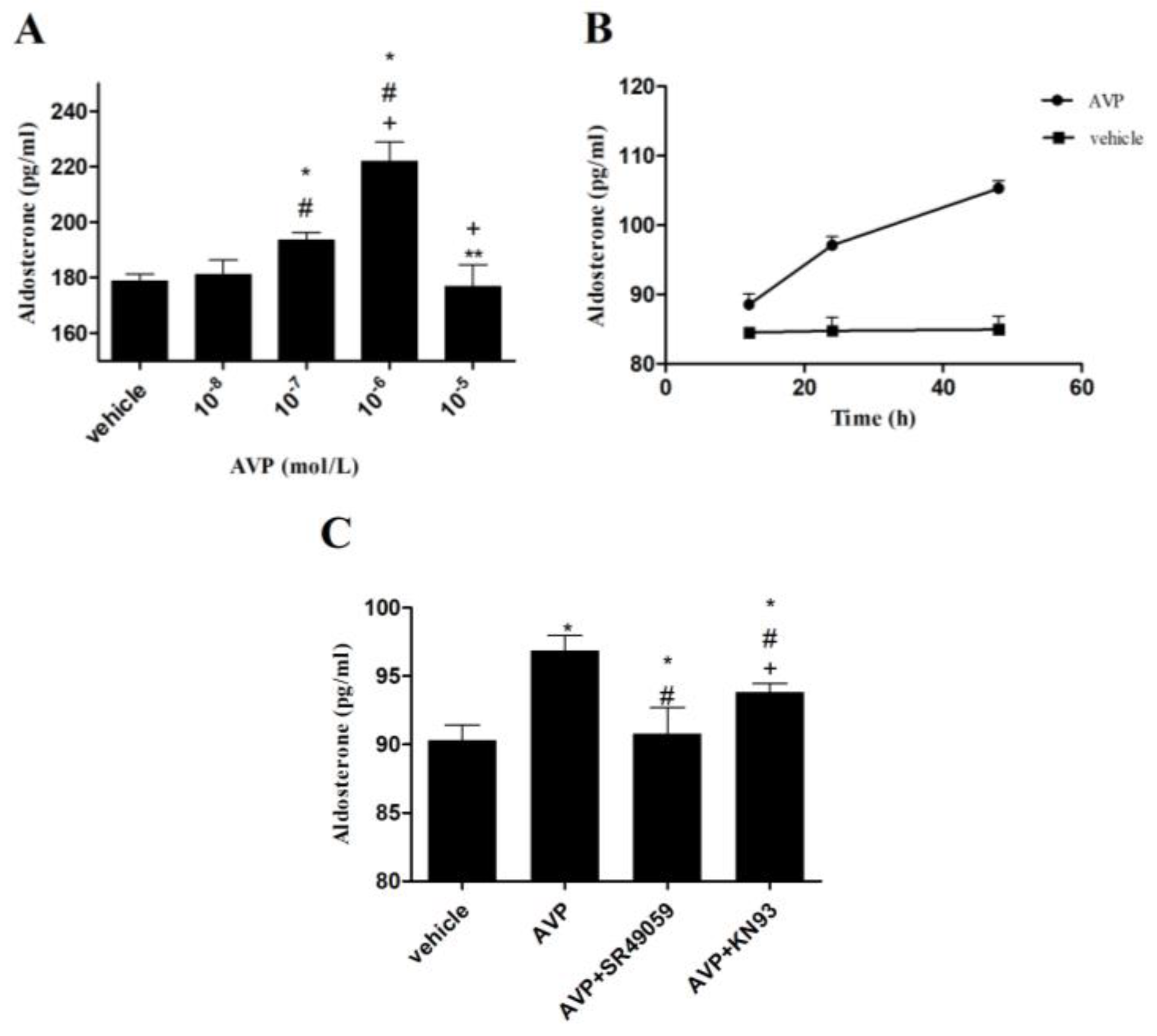
2.4. Effects of AVP on Aldosterone Secretion in CMECs
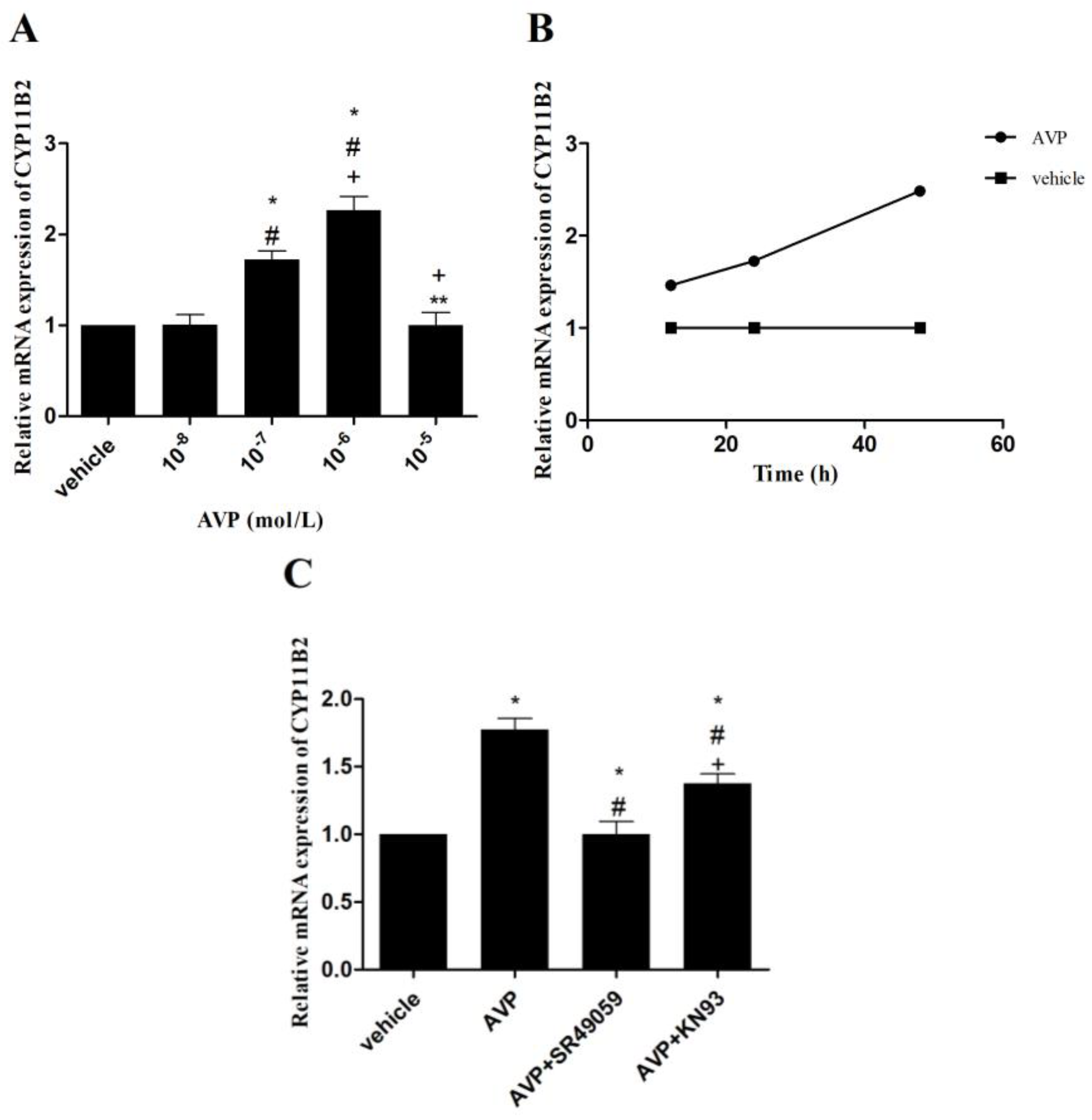
2.5. Effects of AVP on CYP11B2 mRNA Expression in CMECs
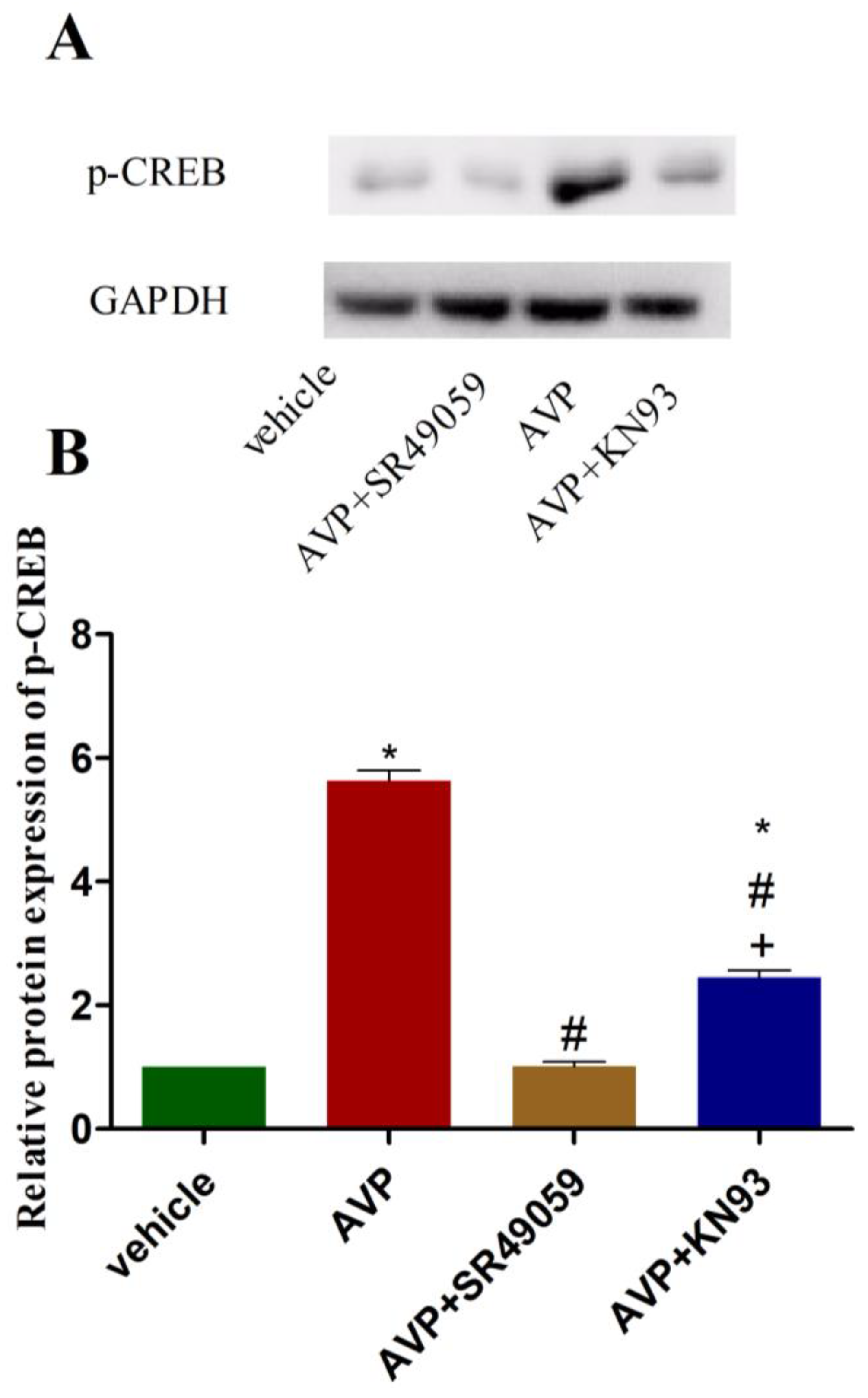
2.6. Effects of AVP on Protein Expression of Phosphorylated CREB in CMECs
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Protocol
4.3. Cardiac Function Assessment via Echocardiography
4.4. Immunohistochemical Analysis of Type I Collagen
4.5. Cardiac Microvascular Endothelial Cell Culture In Vitro
4.6. Enzyme-Linked Immunosorbent Assay
4.7. Real-Time Polymerase Chain Reaction
4.8. Western Blotting
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AVP | arginine vasopressin |
| MI | myocardial infarction |
| CMECs | cardiac microvascular endothelial cells |
| AMI | acute myocardial infarction |
| HF | heart failure |
| ECM | extracellular matrix |
| CaMK | calmodulin kinase |
| cAMP | cyclic adenosine monophosphate |
| CREB | cAMP-response element-binding |
| VSMCs | vascular smooth muscle cells |
| V1R | V1 receptor |
| MR | mineralocorticoid receptor |
| MRA | mineralocorticoid receptor antagonist |
| LVMI | left ventricular mass index |
| LVESD | left ventricular end-systolic diameter |
| LVEDD | left ventricular end-diastolic diameter |
| LVEF | left ventricular ejection fraction |
| p-CREB | phosphorylated CREB |
| αSMA | α-smooth muscle actin |
| SF-1 | steroidogenic factor- |
| COUP-TF | chicken ovalbumin upstream promoter transcription factor |
| iNOS | inducible nitric oxide synthase |
| NO | nitric oxide |
| PKC | protein kinase C |
| MAPK | mitogen-activated protein kinase |
| AOD | average optical density |
References
- Sulo, G.; Igland, J.; Vollset, S.E.; Nygard, O.; Ebbing, M.; Sulo, E.; Egeland, G.M.; Tell, G.S. Heart Failure Complicating Acute Myocardial Infarction; Burden and Timing of Occurrence: A Nation-wide Analysis Including 86 771 Patients From the Cardiovascular Disease in Norway (CVDNOR) Project. J. Am. Heart Assoc. 2016, 5, e002667. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef] [PubMed]
- Azibani, F.; Benard, L.; Schlossarek, S.; Merval, R.; Tournoux, F.; Fazal, L.; Polidano, E.; Launay, J.M.; Carrier, L.; Chatziantoniou, C.; et al. Aldosterone inhibits antifibrotic factors in mouse hypertensive heart. Hypertension 2012, 59, 1179–1187. [Google Scholar] [CrossRef]
- Silvestre, J.S.; Heymes, C.; Oubenaissa, A.; Robert, V.; Aupetit-Faisant, B.; Carayon, A.; Swynghedauw, B.; Delcayre, C. Activation of cardiac aldosterone production in rat myocardial infarction: Effect of angiotensin II receptor blockade and role in cardiac fibrosis. Circulation 1999, 99, 2694–2701. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Callera, G.E. A new look at the eye: Aldosterone and mineralocorticoid receptors as novel targets in retinal vasculopathy. Circ Res. 2009, 104, 9–11. [Google Scholar] [CrossRef][Green Version]
- Silvestre, J.S.; Robert, V.; Heymes, C.; Aupetit-Faisant, B.; Mouas, C.; Moalic, J.M.; Swynghedauw, B.; Delcayre, C. Myocardial production of aldosterone and corticosterone in the rat. Physiological regulation. J. Biol. Chem. 1998, 273, 4883–4891. [Google Scholar] [CrossRef]
- Bassett, M.H.; Suzuki, T.; Sasano, H.; White, P.C.; Rainey, W.E. The orphan nuclear receptors NURR1 and NGFIB regulate adrenal aldosterone production. Mol. Endocrinol. 2004, 18, 279–290. [Google Scholar] [CrossRef]
- Goldsmith, S.R.; Gheorghiade, M. Vasopressin antagonism in heart failure. J. Am. Coll. Cardiol. 2005, 46, 1785–1791. [Google Scholar] [CrossRef]
- Hupf, H.; Grimm, D.; Riegger, G.A.; Schunkert, H. Evidence for a vasopressin system in the rat heart. Circ. Res. 1999, 84, 365–370. [Google Scholar] [CrossRef]
- Gilotra, N.A.; Russell, S.D. Arginine vasopressin as a target in the treatment of acute heart failure. World J. Cardiol. 2014, 6, 1252–1261. [Google Scholar] [CrossRef]
- Chen, X.; Lu, G.; Tang, K.; Li, Q.; Gao, X. The secretion patterns and roles of cardiac and circulating arginine vasopressin during the development of heart failure. Neuropeptides 2015, 51, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Tesch, G.H.; Young, M.J. Mineralocorticoid Receptor Signaling as a Therapeutic Target for Renal and Cardiac Fibrosis. Front. Pharmacol. 2017, 8, 313. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M.; et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef]
- Cannavo, A.; Bencivenga, L.; Liccardo, D.; Elia, A.; Marzano, F.; Gambino, G.; D’Amico, M.L.; Perna, C.; Ferrara, N.; Rengo, G.; et al. Aldosterone and Mineralocorticoid Receptor System in Cardiovascular Physiology and Pathophysiology. Oxidative Med. Cell Longev. 2018, 2018, 1204598. [Google Scholar] [CrossRef]
- Mazzocchi, G.; Malendowicz, L.K.; Rocco, S.; Musajo, F.; Nussdorfer, G.G. Arginine-vasopressin release mediates the aldosterone secretagogue effect of neurotensin in rats. Neuropeptides 1993, 24, 105–108. [Google Scholar] [CrossRef]
- Bashey, R.I.; Donnelly, M.; Insinga, F.; Jimenez, S.A. Growth properties and biochemical characterization of collagens synthesized by adult rat heart fibroblasts in culture. J. Mol. Cell. Cardiol. 1992, 24, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Yoshimura, M.; Nakamura, S.; Nakayama, M.; Shimasaki, Y.; Harada, E.; Mizuno, Y.; Yamamuro, M.; Harada, M.; Saito, Y.; et al. Inhibitory effect of natriuretic peptides on aldosterone synthase gene expression in cultured neonatal rat cardiocytes. Circulation 2003, 107, 807–810. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hatakeyama, H.; Miyamori, I.; Fujita, T.; Takeda, Y.; Takeda, R.; Yamamoto, H. Vascular aldosterone. Biosynthesis and a link to angiotensin II-induced hypertrophy of vascular smooth muscle cells. J. Biol. Chem. 1994, 269, 24316–24320. [Google Scholar] [CrossRef]
- Brown, N.J. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat. Rev. Nephrol. 2013, 9, 459–469. [Google Scholar] [CrossRef]
- AlQudah, M.; Hale, T.M.; Czubryt, M.P. Targeting the renin-angiotensin-aldosterone system in fibrosis. Matrix Biol. 2020, 91–92, 92–108. [Google Scholar] [CrossRef] [PubMed]
- Calvier, L.; Miana, M.; Reboul, P.; Cachofeiro, V.; Martinez-Martinez, E.; de Boer, R.A.; Poirier, F.; Lacolley, P.; Zannad, F.; Rossignol, P.; et al. Galectin-3 mediates aldosterone-induced vascular fibrosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 67–75. [Google Scholar] [CrossRef]
- Chang, Y.; Ben, Y.; Li, H.; Xiong, Y.; Chen, G.; Hao, J.; Ma, X.; Gao, X.; Qiang, P.; Shimosawa, T.; et al. Eplerenone Prevents Cardiac Fibrosis by Inhibiting Angiogenesis in Unilateral Urinary Obstruction Rats. J. Renin Angiotensin Aldosterone Syst. 2022, 2022, 1283729. [Google Scholar] [CrossRef] [PubMed]
- Lother, A.; Deng, L.; Huck, M.; Furst, D.; Kowalski, J.; Esser, J.S.; Moser, M.; Bode, C.; Hein, L. Endothelial cell mineralocorticoid receptors oppose VEGF-induced gene expression and angiogenesis. J. Endocrinol. 2019, 240, 15–26. [Google Scholar] [CrossRef]
- Staessen, J.; Lijnen, P.; Fagard, R.; Verschueren, L.J.; Amery, A. Rise in plasma concentration of aldosterone during long-term angiotensin II suppression. J. Endocrinol. 1981, 91, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Dzau, V.J.; Hirsch, A.T. Emerging role of the tissue renin-angiotensin systems in congestive heart failure. Eur. Heart J. 1990, 11 (Suppl. B), 65–71. [Google Scholar] [CrossRef]
- Curnow, K.M.; Tusie-Luna, M.T.; Pascoe, L.; Natarajan, R.; Gu, J.L.; Nadler, J.L.; White, P.C. The product of the CYP11B2 gene is required for aldosterone biosynthesis in the human adrenal cortex. Mol. Endocrinol. 1991, 5, 1513–1522. [Google Scholar] [CrossRef]
- MacKenzie, S.M.; van Kralingen, J.C.; Davies, E. Regulation of Aldosterone Secretion. Vitam. Horm. 2019, 109, 241–263. [Google Scholar]
- Clyne, C.D.; Zhang, Y.; Slutsker, L.; Mathis, J.M.; White, P.C.; Rainey, W.E. Angiotensin II and potassium regulate human CYP11B2 transcription through common cis-elements. Mol. Endocrinol. 1997, 11, 638–649. [Google Scholar] [CrossRef]
- Sun, P.; Lou, L.; Maurer, R.A. Regulation of activating transcription factor-1 and the cAMP response element-binding protein by Ca2+/calmodulin-dependent protein kinases type I, II, and IV. J. Biol. Chem. 1996, 271, 3066–3073. [Google Scholar] [CrossRef]
- Sun, P.; Enslen, H.; Myung, P.S.; Maurer, R.A. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994, 8, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.H.; Zhao, L.Y.; Zheng, Q.S.; Dong, H.; Wang, H.C.; Yang, X.D. Arginine vasopressin increases iNOS-NO system activity in cardiac fibroblasts through NF-kappaB activation and its relation with myocardial fibrosis. Life Sci. 2007, 81, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Hiroyama, M.; Wang, S.; Aoyagi, T.; Oikawa, R.; Sanbe, A.; Takeo, S.; Tanoue, A. Vasopressin promotes cardiomyocyte hypertrophy via the vasopressin V1A receptor in neonatal mice. Eur. J. Pharmacol. 2007, 559, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.D.; Zhao, L.Y.; Zheng, Q.S.; Li, X. Effects of arginine vasopressin on growth of rat cardiac fibroblasts: Role of V1 receptor. J. Cardiovasc. Pharmacol. 2003, 42, 132–135. [Google Scholar] [CrossRef]
- Thibonnier, M.; Conarty, D.M.; Plesnicher, C.L. Mediators of the mitogenic action of human V(1) vascular vasopressin receptors. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2529–H2539. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Gao, X.; Wu, Q.; Peng, L.; Lin, J.; Zuo, Z. Fluvastatin decreases cardiac fibrosis possibly through regulation of TGF-beta(1)/Smad 7 expression in the spontaneously hypertensive rats. Eur. J. Pharmacol. 2008, 587, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Ito, J.; Minemura, T.; Walchli, S.; Niimi, T.; Fujihara, Y.; Kuroda, S.; Takimoto, K.; Maturana, A.D. Id2 Represses Aldosterone-Stimulated Cardiac T-Type Calcium Channels Expression. Int. J. Mol. Sci. 2021, 22, 3561. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | 6w Sham | 11w Sham | 16w Sham | 6w MI | 11w MI | 16w MI |
|---|---|---|---|---|---|---|
| n = 10 | n = 10 | n = 10 | n = 9 | n = 9 | n = 8 | |
| BW (g) | 414 ± 17 | 504 ± 28 | 514 ± 24 | 406 ± 16 | 497 ± 43 b | 480 ± 16 ab |
| LVM (mg) | 773 ± 45 | 950 ± 86 | 968 ± 82 | 833 ± 67 a | 1101 ± 119 ab | 1212 ± 74 abc |
| LVMI (mg/g) | 1.87 ± 0.1 | 1.88 ± 0.1 | 1.9 ± 0.1 | 2.1 ± 0.1 a | 2.2 ± 0.1 ab | 2.5 ± 0.1 abc |
| LVESD (mm) | 5.42 ± 1.33 | 5.54 ± 0.45 | 5.15 ± 1.08 | 6.97 ± 0.88 a | 7.92 ± 0.67 ab | 10.05 ± 1.08 abc |
| LVEDD (mm) | 7.72 ± 1.17 | 7.76 ± 0.54 | 7.45 ± 1.06 | 8.73 ± 0.92 a | 9.34 ± 75.97 a | 11.18 ± 1.19 abc |
| LVEF (%) | 55.6 ± 12.0 | 53.2 ± 5.63 | 57.6 ± 8.3 | 39.7 ± 5.2 a | 30.7 ± 4.0 ab | 20.8 ± 2.5 abc |
| Genes | Forward Sequence | Reverse Sequence |
|---|---|---|
| Actin | 5′-CGTTGACATCCGTAAAGACCTC-3′ | 5′-TAGGAGCCAGGGCAGTAATCT-3′ |
| CYP11B2 | 5′-ACCATGGATGTCCAGCAA-3’ | 5′-GAGAGCTGCCGAGTCTGA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhai, Y.-S.; Li, J.; Peng, L.; Lu, G.; Gao, X. Phosphorylation of CaMK and CREB-Mediated Cardiac Aldosterone Synthesis Induced by Arginine Vasopressin in Rats with Myocardial Infarction. Int. J. Mol. Sci. 2022, 23, 15061. https://doi.org/10.3390/ijms232315061
Zhai Y-S, Li J, Peng L, Lu G, Gao X. Phosphorylation of CaMK and CREB-Mediated Cardiac Aldosterone Synthesis Induced by Arginine Vasopressin in Rats with Myocardial Infarction. International Journal of Molecular Sciences. 2022; 23(23):15061. https://doi.org/10.3390/ijms232315061
Chicago/Turabian StyleZhai, Yuan-Sheng, Jie Li, Longyun Peng, Guihua Lu, and Xiuren Gao. 2022. "Phosphorylation of CaMK and CREB-Mediated Cardiac Aldosterone Synthesis Induced by Arginine Vasopressin in Rats with Myocardial Infarction" International Journal of Molecular Sciences 23, no. 23: 15061. https://doi.org/10.3390/ijms232315061
APA StyleZhai, Y.-S., Li, J., Peng, L., Lu, G., & Gao, X. (2022). Phosphorylation of CaMK and CREB-Mediated Cardiac Aldosterone Synthesis Induced by Arginine Vasopressin in Rats with Myocardial Infarction. International Journal of Molecular Sciences, 23(23), 15061. https://doi.org/10.3390/ijms232315061