

Pathological Roles of Pulmonary Cells in Acute Lung Injury: Lessons from Clinical Practice

Abstract
1. Introduction
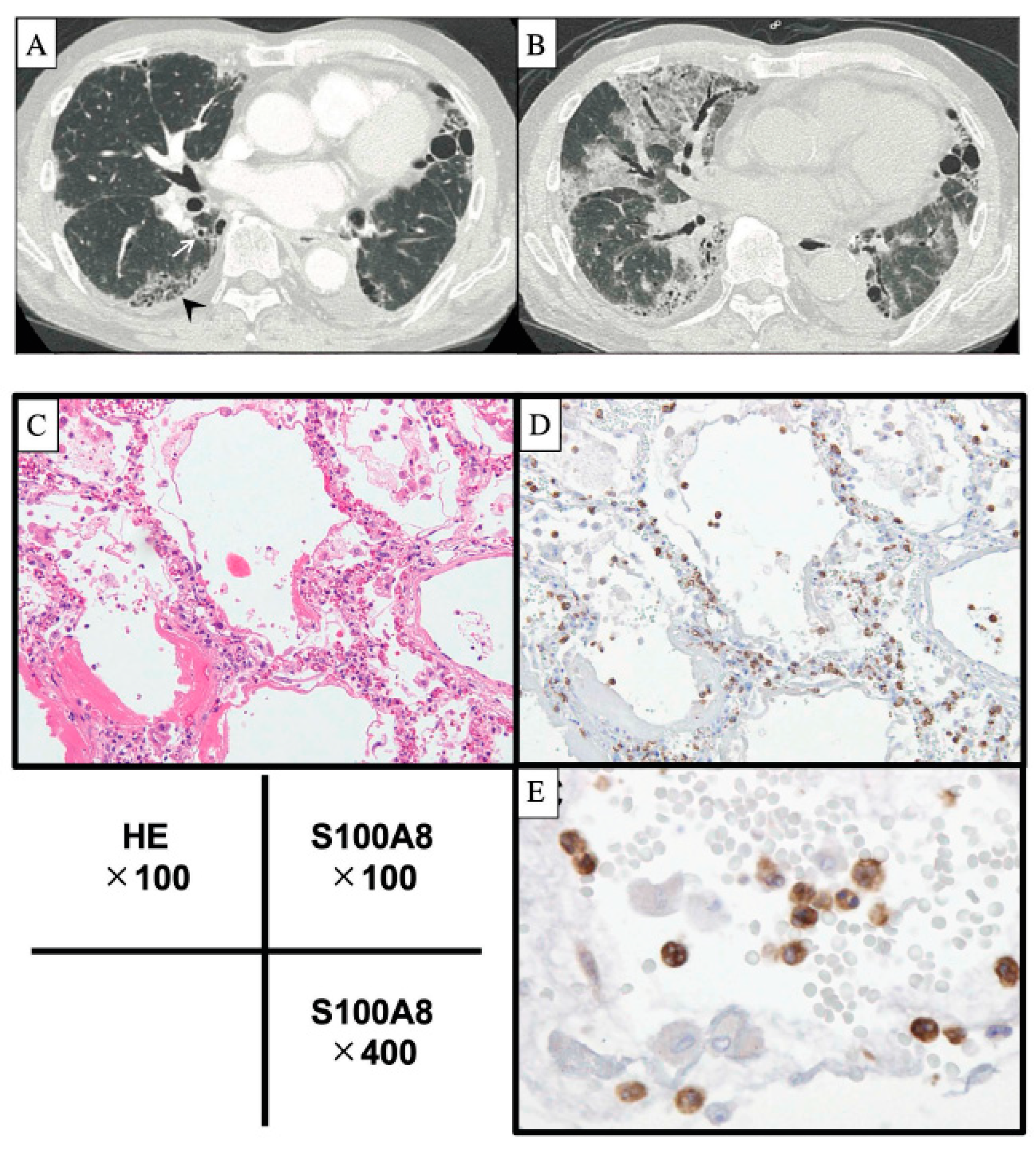
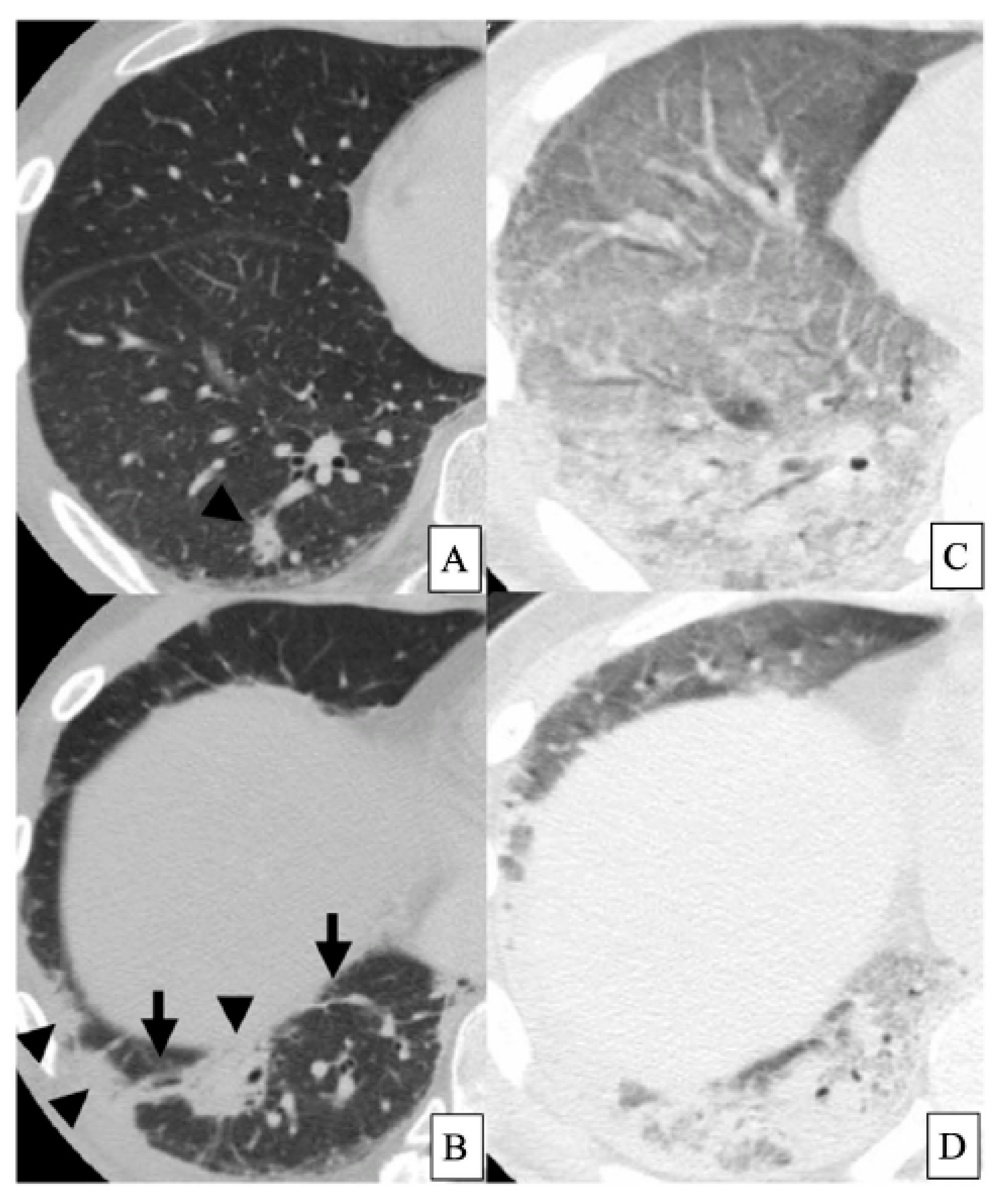
2. Acute Exacerbation of IPF
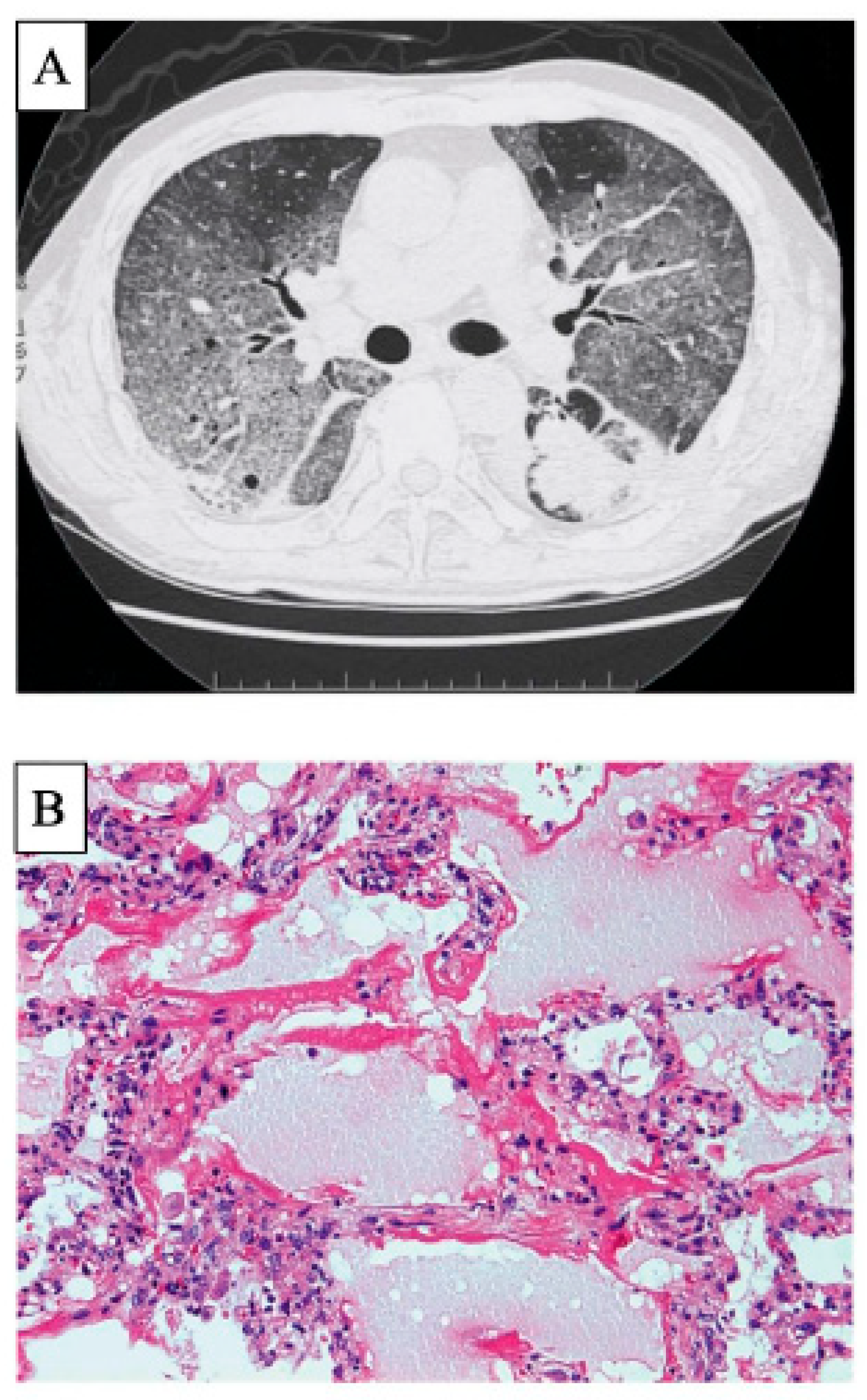
3. Clinically Amyopathic Dermatomyositis-Related ILD
4. EGFR-TKI-Induced Lung Injury
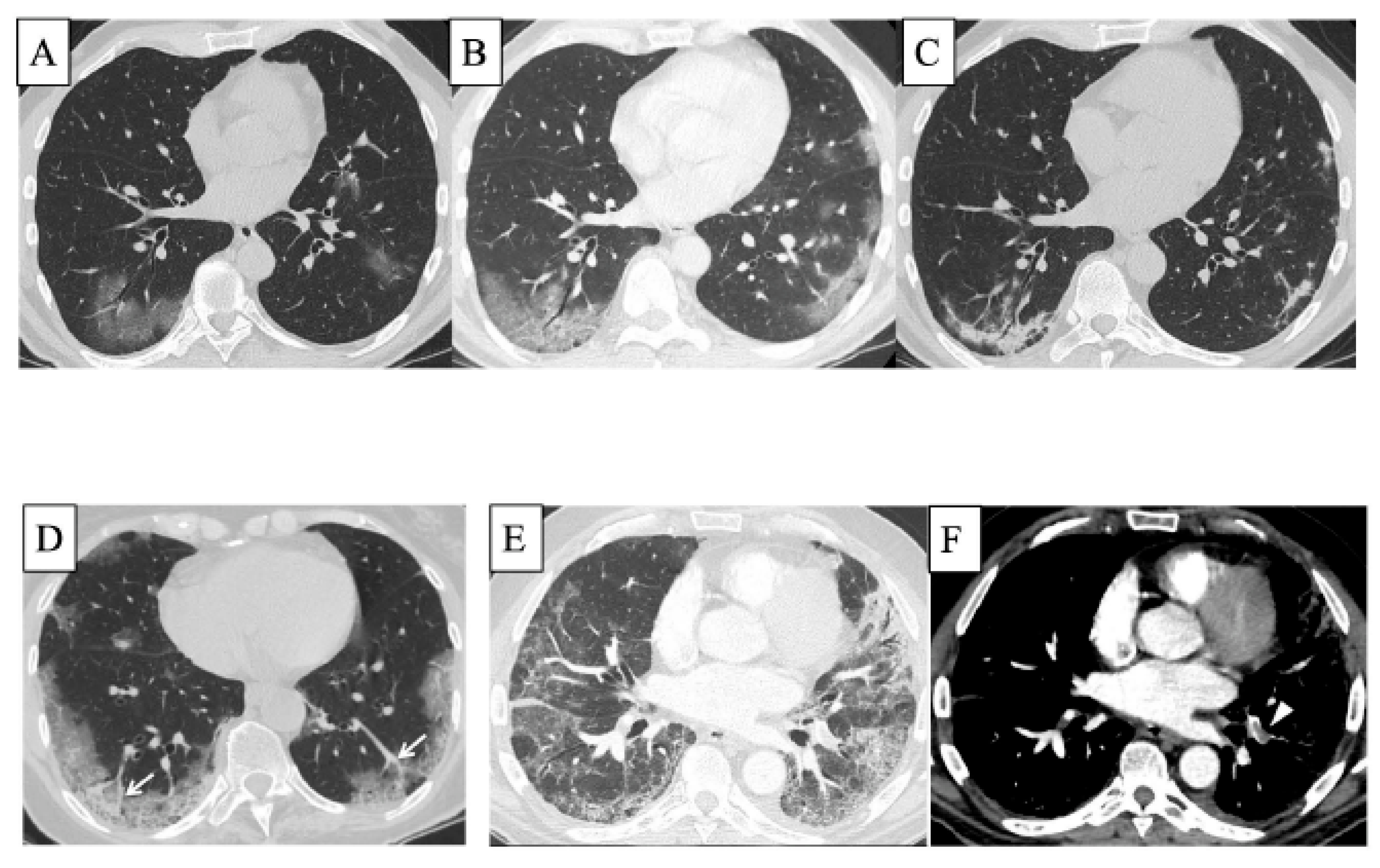
5. COVID-19 and ILD
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oda, K.; Ishimoto, H.; Yamada, S.; Kushima, H.; Ishii, H.; Imanaga, T.; Harada, T.; Ishimatsu, Y.; Matsumoto, N.; Naito, K.; et al. Autopsy analyses in acute exacerbation of idiopathic pulmonary fibrosis. Respir. Res. 2014, 15, 109. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Enomoto, N.; Hozumi, H.; Isayama, T.; Naoi, H.; Aono, Y.; Katsumata, M.; Yasui, H.; Karayama, M.; Suzuki, Y.; et al. Serum S100A8 and S100A9 as prognostic biomarkers in acute exacerbation of idiopathic pulmonary fibrosis. Respir. Investig. 2021, 59, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Matsuki, Y.; Yamashita, H.; Takahashi, Y.; Kano, T.; Shimizu, A.; Itoh, K.; Kaneko, H.; Mimori, A. Diffuse alveolar damage in patients with dermatomyositis: A six-case series. Mod. Rheumatol. 2012, 22, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Saijo, Y.; Maemondo, M.; Gomi, K.; Tokue, Y.; Kimura, Y.; Ebina, M.; Kikuchi, T.; Moriya, T.; Nukiwa, T. Severe acute interstitial pneumonia and gefitinib. Lancet 2003, 361, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Moore, B.B.; Flaherty, K.R.; Brown, K.K.; Kaner, R.J.; King, T.E., Jr.; Lasky, J.A.; Loyd, J.E.; Noth, I.; Olman, M.A.; et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2007, 176, 636–643. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- Collard, H.R.; Ryerson, C.J.; Corte, T.J.; Jenkins, G.; Kondoh, Y.; Lederer, D.J.; Lee, J.S.; Maher, T.M.; Wells, A.U.; Antoniou, K.M.; et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report. Am. J. Respir. Crit. Care Med. 2016, 194, 265–275. [Google Scholar] [CrossRef]
- Kondoh, Y.; Taniguchi, H.; Kawabata, Y.; Yokoi, T.; Suzuki, K.; Takagi, K. Acute exacerbation in idiopathic pulmonary fibrosis. Analysis of clinical and pathologic findings in three cases. Chest 1993, 103, 1808–1812. [Google Scholar] [CrossRef]
- Azuma, A.; Hagiwara, K.; Kudoh, S. Basis of acute exacerbation of idiopathic pulmonary fibrosis in Japanese patients. Am. J. Respir. Crit. Care Med. 2008, 177, 1397–1398. [Google Scholar] [CrossRef]
- Kakugawa, T.; Sakamoto, N.; Sato, S.; Yura, H.; Harada, T.; Nakashima, S.; Hara, A.; Oda, K.; Ishimoto, H.; Yatera, K.; et al. Risk factors for an acute exacerbation of idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 79. [Google Scholar] [CrossRef]
- Enomoto, N.; Oyama, Y.; Enomoto, Y.; Yasui, H.; Karayama, M.; Kono, M.; Hozumi, H.; Suzuki, Y.; Furuhashi, K.; Fujisawa, T.; et al. Differences in clinical features of acute exacerbation between connective tissue disease-associated interstitial pneumonia and idiopathic pulmonary fibrosis. Chron. Respir. Dis. 2019, 16, 1479972318809476. [Google Scholar] [CrossRef] [PubMed]
- Akira, M.; Hamada, H.; Sakatani, M.; Kobayashi, C.; Nishioka, M.; Yamamoto, S. CT findings during phase of accelerated deterioration in patients with idiopathic pulmonary fibrosis. AJR Am. J. Roentgenol. 1997, 168, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Akira, M.; Kozuka, T.; Yamamoto, S.; Sakatani, M. Computed tomography findings in acute exacerbation of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S. Acute exacerbations in patients with idiopathic pulmonary fibrosis. Respir. Res. 2013, 14, 86. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, Y.; Taniguchi, H.; Katsuta, T.; Kataoka, K.; Kimura, T.; Nishiyama, O.; Sakamoto, K.; Johkoh, T.; Nishimura, M.; Ono, K.; et al. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc. Diffuse Lung Dis. 2010, 27, 103–110. [Google Scholar]
- Song, J.W.; Hong, S.B.; Lim, C.M.; Koh, Y.; Kim, D.S. Acute exacerbation of idiopathic pulmonary fibrosis: Incidence, risk factors and outcome. Eur. Respir. J. 2011, 37, 356–363. [Google Scholar] [CrossRef]
- Collard, H.R.; Richeldi, L.; Kim, D.S.; Taniguchi, H.; Tschoepe, I.; Luisetti, M.; Roman, J.; Tino, G.; Schlenker-Herceg, R.; Hallmann, C.; et al. Acute exacerbations in the INPULSIS trials of nintedanib in idiopathic pulmonary fibrosis. Eur. Respir. J. 2017, 49, 1601339. [Google Scholar] [CrossRef]
- Ley, B.; Collard, H.R.; King, T.E., Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 431–440. [Google Scholar] [CrossRef]
- Natsuizaka, M.; Chiba, H.; Kuronuma, K.; Otsuka, M.; Kudo, K.; Mori, M.; Bando, M.; Sugiyama, Y.; Takahashi, H. Epidemiologic survey of Japanese patients with idiopathic pulmonary fibrosis and investigation of ethnic differences. Am. J. Respir. Crit. Care Med. 2014, 190, 773–779. [Google Scholar] [CrossRef]
- Kim, D.S.; Park, J.H.; Park, B.K.; Lee, J.S.; Nicholson, A.G.; Colby, T. Acute exacerbation of idiopathic pulmonary fibrosis: Frequency and clinical features. Eur. Respir. J. 2006, 27, 143–150. [Google Scholar] [CrossRef]
- Enomoto, N.; Naoi, H.; Aono, Y.; Katsumata, M.; Horiike, Y.; Yasui, H.; Karayama, M.; Hozumi, H.; Suzuki, Y.; Furuhashi, K.; et al. Acute exacerbation of unclassifiable idiopathic interstitial pneumonia: Comparison with idiopathic pulmonary fibrosis. Ther. Adv. Respir. Dis. 2020, 14, 1753466620935774. [Google Scholar] [CrossRef] [PubMed]
- Park, I.N.; Kim, D.S.; Shim, T.S.; Lim, C.M.; Lee, S.D.; Koh, Y.; Kim, W.S.; Kim, W.D.; Jang, S.J.; Colby, T.V. Acute exacerbation of interstitial pneumonia other than idiopathic pulmonary fibrosis. Chest 2007, 132, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Kaida, Y.; Nakamura, Y.; Enomoto, N.; Fujisawa, T.; Imokawa, S.; Hashizume, H.; Naito, T.; Hashimoto, D.; Takehara, Y.; et al. Acute exacerbation of interstitial pneumonia associated with collagen vascular diseases. Respir. Med. 2009, 103, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, N.; Naoi, H.; Mochizuka, Y.; Isayama, T.; Tanaka, Y.; Fukada, A.; Aono, Y.; Katsumata, M.; Yasui, H.; Mori, K.; et al. Frequency, proportion of PF-ILD, and prognostic factors in patients with acute exacerbation of ILD related to systemic autoimmune diseases. BMC Pulm. Med. 2022, 22, 387. [Google Scholar] [CrossRef]
- Hozumi, H.; Kono, M.; Hasegawa, H.; Kato, S.; Inoue, Y.; Suzuki, Y.; Karayama, M.; Furuhashi, K.; Enomoto, N.; Fujisawa, T.; et al. Acute exacerbation of rheumatoid arthritis-associated interstitial lung disease: Mortality and its prediction model. Respir. Res. 2022, 23, 57. [Google Scholar] [CrossRef]
- Simon-Blancal, V.; Freynet, O.; Nunes, H.; Bouvry, D.; Naggara, N.; Brillet, P.Y.; Denis, D.; Cohen, Y.; Vincent, F.; Valeyre, D.; et al. Acute exacerbation of idiopathic pulmonary fibrosis: Outcome and prognostic factors. Respiration 2012, 83, 28–35. [Google Scholar] [CrossRef]
- Collard, H.R.; Yow, E.; Richeldi, L.; Anstrom, K.J.; Glazer, C. Suspected acute exacerbation of idiopathic pulmonary fibrosis as an outcome measure in clinical trials. Respir. Res. 2013, 14, 73. [Google Scholar] [CrossRef]
- Kondoh, Y.; Kataoka, K.; Ando, M.; Awaya, Y.; Ichikado, K.; Kataoka, M.; Komase, Y.; Mineshita, M.; Ohno, Y.; Okamoto, H.; et al. COVID-19 and acute exacerbation of interstitial lung disease. Respir. Investig. 2021, 59, 675–678. [Google Scholar] [CrossRef]
- Sgalla, G.; Magri, T.; Lerede, M.; Comes, A.; Richeldi, L. COVID-19 Vaccine in Patients with Exacerbation of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2022, 206, 219–221. [Google Scholar] [CrossRef]
- Sato, T.; Teramukai, S.; Kondo, H.; Watanabe, A.; Ebina, M.; Kishi, K.; Fujii, Y.; Mitsudomi, T.; Yoshimura, M.; Maniwa, T.; et al. Impact and predictors of acute exacerbation of interstitial lung diseases after pulmonary resection for lung cancer. J. Thorac. Cardiovasc. Surg. 2014, 147, 1604–1611.e3. [Google Scholar] [CrossRef]
- Kondoh, Y.; Taniguchi, H.; Kitaichi, M.; Yokoi, T.; Johkoh, T.; Oishi, T.; Kimura, T.; Nishiyama, O.; Kato, K.; du Bois, R.M. Acute exacerbation of interstitial pneumonia following surgical lung biopsy. Respir. Med. 2006, 100, 1753–1759. [Google Scholar] [CrossRef]
- Kaarteenaho, R. The current position of surgical lung biopsy in the diagnosis of idiopathic pulmonary fibrosis. Respir. Res. 2013, 14, 43. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Taniguchi, H.; Kondoh, Y.; Wakai, K.; Kimura, T.; Kataoka, K.; Hashimoto, N.; Nishiyama, O.; Hasegawa, Y. Acute exacerbation of IPF following diagnostic bronchoalveolar lavage procedures. Respir. Med. 2012, 106, 436–442. [Google Scholar] [CrossRef]
- Sakamoto, S.; Shimizu, H.; Isshiki, T.; Nakamura, Y.; Usui, Y.; Kurosaki, A.; Isobe, K.; Takai, Y.; Homma, S. New risk scoring system for predicting 3-month mortality after acute exacerbation of idiopathic pulmonary fibrosis. Sci. Rep. 2022, 12, 1134. [Google Scholar] [CrossRef]
- Konishi, K.; Gibson, K.F.; Lindell, K.O.; Richards, T.J.; Zhang, Y.; Dhir, R.; Bisceglia, M.; Gilbert, S.; Yousem, S.A.; Song, J.W.; et al. Gene expression profiles of acute exacerbations of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2009, 180, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Izuhara, K.; Conway, S.J.; Moore, B.B.; Matsumoto, H.; Holweg, C.T.; Matthews, J.G.; Arron, J.R. Roles of Periostin in Respiratory Disorders. Am. J. Respir. Crit. Care Med. 2016, 193, 949–956. [Google Scholar] [CrossRef]
- Shimizu, H.; Sakamoto, S.; Okamoto, M.; Isshiki, T.; Ono, J.; Shimizu, S.; Hoshino, T.; Izuhara, K.; Homma, S. Association of serum monomeric periostin level with outcomes of acute exacerbation of idiopathic pulmonary fibrosis and fibrosing nonspecific interstitial pneumonia. Ann. Transl. Med. 2021, 9, 739. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.G.; Choi, S.M.; Lee, J.H.; Yoon, J.K.; Song, J.W. Changes in blood Krebs von den Lungen-6 predict the mortality of patients with acute exacerbation of interstitial lung disease. Sci. Rep. 2022, 12, 4916. [Google Scholar] [CrossRef]
- Abe, S.; Seo, Y.; Hayashi, H.; Matsuda, K.; Usuki, J.; Azuma, A.; Kudoh, S.; Gemma, A. Neutrophil adsorption by polymyxin B-immobilized fiber column for acute exacerbation in patients with interstitial pneumonia: A pilot study. Blood Purif. 2010, 29, 321–326. [Google Scholar] [CrossRef]
- Kono, M.; Miyashita, K.; Hirama, R.; Oshima, Y.; Takeda, K.; Mochizuka, Y.; Tsutsumi, A.; Miwa, H.; Miki, Y.; Hashimoto, D.; et al. Prognostic significance of bronchoalveolar lavage cellular analysis in patients with acute exacerbation of interstitial lung disease. Respir. Med. 2021, 186, 106534. [Google Scholar] [CrossRef]
- Schupp, J.C.; Binder, H.; Jager, B.; Cillis, G.; Zissel, G.; Muller-Quernheim, J.; Prasse, A. Macrophage activation in acute exacerbation of idiopathic pulmonary fibrosis. PLoS ONE 2015, 10, e0116775. [Google Scholar] [CrossRef]
- Prasse, A.; Probst, C.; Bargagli, E.; Zissel, G.; Toews, G.B.; Flaherty, K.R.; Olschewski, M.; Rottoli, P.; Muller-Quernheim, J. Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Kagawa, T.; Sasaki, Y.; Sugawara, R.; Sugimoto, C.; Tachibana, K.; Fujita, Y.; Hayashi, S.; Inoue, Y. Hemosiderin-Laden Macrophages in Bronchoalveolar Lavage: Predictive Role for Acute Exacerbation of Idiopathic Interstitial Pneumonias. Can. Respir. J. 2021, 2021, 4595019. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, N.; Oyama, Y.; Enomoto, Y.; Mikamo, M.; Karayama, M.; Hozumi, H.; Suzuki, Y.; Kono, M.; Furuhashi, K.; Fujisawa, T.; et al. Prognostic evaluation of serum ferritin in acute exacerbation of idiopathic pulmonary fibrosis. Clin. Respir. J. 2018, 12, 2378–2389. [Google Scholar] [CrossRef] [PubMed]
- Serezani, A.P.M.; Pascoalino, B.D.; Bazzano, J.M.R.; Vowell, K.N.; Tanjore, H.; Taylor, C.J.; Calvi, C.L.; McCall, A.S.; Bacchetta, M.D.; Shaver, C.M.; et al. Multiplatform Single-Cell Analysis Identifies Immune Cell Types Enhanced in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2022, 67, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Shenderov, K.; Collins, S.L.; Powell, J.D.; Horton, M.R. Immune dysregulation as a driver of idiopathic pulmonary fibrosis. J. Clin. Investig. 2021, 131, e143226. [Google Scholar] [CrossRef]
- Lee, J.S.; Song, J.W.; Wolters, P.J.; Elicker, B.M.; King, T.E., Jr.; Kim, D.S.; Collard, H.R. Bronchoalveolar lavage pepsin in acute exacerbation of idiopathic pulmonary fibrosis. Eur. Respir. J. 2012, 39, 352–358. [Google Scholar] [CrossRef]
- D’Alessandro-Gabazza, C.N.; Yasuma, T.; Kobayashi, T.; Toda, M.; Abdel-Hamid, A.M.; Fujimoto, H.; Hataji, O.; Nakahara, H.; Takeshita, A.; Nishihama, K.; et al. Inhibition of lung microbiota-derived proapoptotic peptides ameliorates acute exacerbation of pulmonary fibrosis. Nat. Commun. 2022, 13, 1558. [Google Scholar] [CrossRef]
- D’Alessandro-Gabazza, C.N.; Kobayashi, T.; Yasuma, T.; Toda, M.; Kim, H.; Fujimoto, H.; Hataji, O.; Takeshita, A.; Nishihama, K.; Okano, T.; et al. A Staphylococcus pro-apoptotic peptide induces acute exacerbation of pulmonary fibrosis. Nat. Commun. 2020, 11, 1539. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Shan, P.; Hunt, C.R.; Pandita, T.K.; Lee, P.J. A protective Hsp70-TLR4 pathway in lethal oxidant lung injury. J. Immunol. 2013, 191, 1393–1403. [Google Scholar] [CrossRef]
- Kahloon, R.A.; Xue, J.; Bhargava, A.; Csizmadia, E.; Otterbein, L.; Kass, D.J.; Bon, J.; Soejima, M.; Levesque, M.C.; Lindell, K.O.; et al. Patients with idiopathic pulmonary fibrosis with antibodies to heat shock protein 70 have poor prognoses. Am. J. Respir. Crit. Care Med. 2013, 187, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Ebina, M.; Taniguchi, H.; Miyasho, T.; Yamada, S.; Shibata, N.; Ohta, H.; Hisata, S.; Ohkouchi, S.; Tamada, T.; Nishimura, H.; et al. Gradual increase of high mobility group protein b1 in the lungs after the onset of acute exacerbation of idiopathic pulmonary fibrosis. Pulm. Med. 2011, 2011, 916486. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Calfee, C.S.; Wolters, P.J.; Song, J.W.; Hong, S.B.; Brady, S.; Ishizaka, A.; Jones, K.D.; King, T.E., Jr.; Matthay, M.A.; et al. Plasma biomarker profiles in acute exacerbation of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 299, L3–L7. [Google Scholar] [CrossRef] [PubMed]
- Abe, S.; Hayashi, H.; Seo, Y.; Matsuda, K.; Kamio, K.; Saito, Y.; Usuki, J.; Azuma, A.; Kudo, S.; Gemma, A. Reduction in serum high mobility group box-1 level by polymyxin B-immobilized fiber column in patients with idiopathic pulmonary fibrosis with acute exacerbation. Blood Purif. 2011, 32, 310–316. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Iwamoto, H.; Sakamoto, S.; Horimasu, Y.; Masuda, T.; Miyamoto, S.; Nakashima, T.; Ohshimo, S.; Fujitaka, K.; Hamada, H.; et al. Serum high-mobility group box 1 is associated with the onset and severity of acute exacerbation of idiopathic pulmonary fibrosis. Respirology 2020, 25, 275–280. [Google Scholar] [CrossRef]
- Enomoto, N.; Oyama, Y.; Yasui, H.; Karayama, M.; Hozumi, H.; Suzuki, Y.; Kono, M.; Furuhashi, K.; Fujisawa, T.; Inui, N.; et al. Analysis of serum adiponectin and leptin in patients with acute exacerbation of idiopathic pulmonary fibrosis. Sci. Rep. 2019, 9, 10484. [Google Scholar] [CrossRef]
- Kondoh, Y.; Taniguchi, H.; Ebina, M.; Azuma, A.; Ogura, T.; Taguchi, Y.; Suga, M.; Takahashi, H.; Nakata, K.; Sugiyama, Y.; et al. Risk factors for acute exacerbation of idiopathic pulmonary fibrosis--Extended analysis of pirfenidone trial in Japan. Respir. Investig. 2015, 53, 271–278. [Google Scholar] [CrossRef]
- Enomoto, N.; Mikamo, M.; Oyama, Y.; Kono, M.; Hashimoto, D.; Fujisawa, T.; Inui, N.; Nakamura, Y.; Yasuda, H.; Kato, A.; et al. Treatment of acute exacerbation of idiopathic pulmonary fibrosis with direct hemoperfusion using a polymyxin B-immobilized fiber column improves survival. BMC Pulm. Med. 2015, 15, 15. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47. [Google Scholar] [CrossRef]
- Idiopathic Pulmonary Fibrosis Clinical Research, N.; Raghu, G.; Anstrom, K.J.; King, T.E., Jr.; Lasky, J.A.; Martinez, F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar] [CrossRef]
- Naccache, J.M.; Jouneau, S.; Didier, M.; Borie, R.; Cachanado, M.; Bourdin, A.; Reynaud-Gaubert, M.; Bonniaud, P.; Israel-Biet, D.; Prevot, G.; et al. Cyclophosphamide added to glucocorticoids in acute exacerbation of idiopathic pulmonary fibrosis (EXAFIP): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir. Med. 2022, 10, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, Y.; Azuma, A.; Inoue, Y.; Ogura, T.; Sakamoto, S.; Tsushima, K.; Johkoh, T.; Fujimoto, K.; Ichikado, K.; Matsuzawa, Y.; et al. Thrombomodulin Alfa for Acute Exacerbation of Idiopathic Pulmonary Fibrosis. A Randomized, Double-Blind Placebo-controlled Trial. Am. J. Respir. Crit. Care Med. 2020, 201, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Han, M.; Song, J.W. Antifibrotic treatment improves clinical outcomes in patients with idiopathic pulmonary fibrosis: A propensity score matching analysis. Sci. Rep. 2020, 10, 15620. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Cottin, V.; du Bois, R.M.; Selman, M.; Kimura, T.; Bailes, Z.; Schlenker-Herceg, R.; Stowasser, S.; Brown, K.K. Nintedanib in patients with idiopathic pulmonary fibrosis: Combined evidence from the TOMORROW and INPULSIS((R)) trials. Respir. Med. 2016, 113, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Abe, S.; Azuma, A.; Mukae, H.; Ogura, T.; Taniguchi, H.; Bando, M.; Sugiyama, Y. Polymyxin B-immobilized fiber column (PMX) treatment for idiopathic pulmonary fibrosis with acute exacerbation: A multicenter retrospective analysis. Intern. Med. 2012, 51, 1487–1491. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, N.; Suda, T.; Uto, T.; Kato, M.; Kaida, Y.; Ozawa, Y.; Miyazaki, H.; Kuroishi, S.; Hashimoto, D.; Naito, T.; et al. Possible therapeutic effect of direct haemoperfusion with a polymyxin B immobilized fibre column (PMX-DHP) on pulmonary oxygenation in acute exacerbations of interstitial pneumonia. Respirology 2008, 13, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Suda, T.; Enomoto, N.; Nakamura, Y.; Kaida, Y.; Hashimoto, D.; Inui, N.; Mizuguchi, T.; Kato, A.; Shirai, T.; et al. Evaluation of different perfusion durations in direct hemoperfusion with polymyxin B-immobilized fiber column therapy for acute exacerbation of interstitial pneumonias. Blood Purif. 2011, 32, 75–81. [Google Scholar] [CrossRef]
- Oishi, K.; Aoe, K.; Mimura, Y.; Murata, Y.; Sakamoto, K.; Koutoku, W.; Matsumoto, T.; Ueoka, H.; Yano, M. Survival from an Acute Exacerbation of Idiopathic Pulmonary Fibrosis with or without Direct Hemoperfusion with a Polymyxin B-immobilized Fiber Column: A Retrospective Analysis. Intern. Med. 2016, 55, 3551–3559. [Google Scholar] [CrossRef]
- Furusawa, H.; Sugiura, M.; Mitaka, C.; Inase, N. Direct hemoperfusion with polymyxin B-immobilized fibre treatment for acute exacerbation of interstitial pneumonia. Respirology 2017, 22, 1357–1362. [Google Scholar] [CrossRef]
- Fischer, A.; du Bois, R. Interstitial lung disease in connective tissue disorders. Lancet 2012, 380, 689–698. [Google Scholar] [CrossRef]
- Tyndall, A.J.; Bannert, B.; Vonk, M.; Airo, P.; Cozzi, F.; Carreira, P.E.; Bancel, D.F.; Allanore, Y.; Muller-Ladner, U.; Distler, O.; et al. Causes and risk factors for death in systemic sclerosis: A study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann. Rheum. Dis. 2010, 69, 1809–1815. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann-Vold, A.M.; Fretheim, H.; Halse, A.K.; Seip, M.; Bitter, H.; Wallenius, M.; Garen, T.; Salberg, A.; Brunborg, C.; Midtvedt, O.; et al. Tracking Impact of Interstitial Lung Disease in Systemic Sclerosis in a Complete Nationwide Cohort. Am. J. Respir. Crit. Care Med. 2019, 200, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, Y.; Yamada, H.; Ohkubo, M.; Yamasaki, M.; Azuma, K.; Ogawa, H.; Mizushima, M.; Ozaki, S. Longterm survival and associated risk factors in patients with adult-onset idiopathic inflammatory myopathies and amyopathic dermatomyositis: Experience in a single institute in Japan. J. Rheumatol. 2011, 38, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, D.S.; Park, I.N.; Jang, S.J.; Kitaichi, M.; Nicholson, A.G.; Colby, T.V. Prognosis of fibrotic interstitial pneumonia: Idiopathic versus collagen vascular disease-related subtypes. Am. J. Respir. Crit. Care Med. 2007, 175, 705–711. [Google Scholar] [CrossRef]
- Nakajima, A.; Inoue, E.; Tanaka, E.; Singh, G.; Sato, E.; Hoshi, D.; Shidara, K.; Hara, M.; Momohara, S.; Taniguchi, A.; et al. Mortality and cause of death in Japanese patients with rheumatoid arthritis based on a large observational cohort, IORRA. Scand. J. Rheumatol. 2010, 39, 360–367. [Google Scholar] [CrossRef]
- Suda, T.; Fujisawa, T.; Enomoto, N.; Nakamura, Y.; Inui, N.; Naito, T.; Hashimoto, D.; Sato, J.; Toyoshima, M.; Hashizume, H.; et al. Interstitial lung diseases associated with amyopathic dermatomyositis. Eur. Respir. J. 2006, 28, 1005–1012. [Google Scholar] [CrossRef]
- Fujisawa, T.; Hozumi, H.; Kono, M.; Enomoto, N.; Hashimoto, D.; Nakamura, Y.; Inui, N.; Yokomura, K.; Koshimizu, N.; Toyoshima, M.; et al. Prognostic factors for myositis-associated interstitial lung disease. PLoS ONE 2014, 9, e98824. [Google Scholar] [CrossRef]
- Distler, O.; Highland, K.B.; Gahlemann, M.; Azuma, A.; Fischer, A.; Mayes, M.D.; Raghu, G.; Sauter, W.; Girard, M.; Alves, M.; et al. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. N. Engl. J. Med. 2019, 380, 2518–2528. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Suda, T.; Nakamura, Y.; Enomoto, N.; Ide, K.; Toyoshima, M.; Uchiyama, H.; Tamura, R.; Ida, M.; Yagi, T.; et al. Differences in clinical features and prognosis of interstitial lung diseases between polymyositis and dermatomyositis. J. Rheumatol. 2005, 32, 58–64. [Google Scholar]
- Hozumi, H.; Fujisawa, T.; Nakashima, R.; Johkoh, T.; Sumikawa, H.; Murakami, A.; Enomoto, N.; Inui, N.; Nakamura, Y.; Hosono, Y.; et al. Comprehensive assessment of myositis-specific autoantibodies in polymyositis/dermatomyositis-associated interstitial lung disease. Respir. Med. 2016, 121, 91–99. [Google Scholar] [CrossRef]
- Sontheimer, R.D. Would a new name hasten the acceptance of amyopathic dermatomyositis (dermatomyositis sine myositis) as a distinctive subset within the idiopathic inflammatory dermatomyopathies spectrum of clinical illness? J. Am. Acad. Dermatol. 2002, 46, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Ghirardello, A.; Borella, E.; Beggio, M.; Franceschini, F.; Fredi, M.; Doria, A. Myositis autoantibodies and clinical phenotypes. Autoimmun. Highlights 2014, 5, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Hozumi, H.; Enomoto, N.; Kono, M.; Fujisawa, T.; Inui, N.; Nakamura, Y.; Sumikawa, H.; Johkoh, T.; Nakashima, R.; Imura, Y.; et al. Prognostic significance of anti-aminoacyl-tRNA synthetase antibodies in polymyositis/dermatomyositis-associated interstitial lung disease: A retrospective case control study. PLoS ONE 2015, 10, e0120313. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Li, S.; Wang, T.; Shi, J.; Wang, G. Clinical Heterogeneity of Interstitial Lung Disease in Polymyositis and Dermatomyositis Patients With or Without Specific Autoantibodies. Am. J. Med. Sci. 2018, 355, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, R.; Imura, Y.; Kobayashi, S.; Yukawa, N.; Yoshifuji, H.; Nojima, T.; Kawabata, D.; Ohmura, K.; Usui, T.; Fujii, T.; et al. The RIG-I-like receptor IFIH1/MDA5 is a dermatomyositis-specific autoantigen identified by the anti-CADM-140 antibody. Rheumatology 2010, 49, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Tanizawa, K.; Handa, T.; Nakashima, R.; Kubo, T.; Hosono, Y.; Aihara, K.; Ikezoe, K.; Watanabe, K.; Taguchi, Y.; Hatta, K.; et al. The prognostic value of HRCT in myositis-associated interstitial lung disease. Respir. Med. 2013, 107, 745–752. [Google Scholar] [CrossRef]
- Labrador-Horrillo, M.; Martinez, M.A.; Selva-O’Callaghan, A.; Trallero-Araguas, E.; Balada, E.; Vilardell-Tarres, M.; Juarez, C. Anti-MDA5 antibodies in a large Mediterranean population of adults with dermatomyositis. J. Immunol. Res. 2014, 2014, 290797. [Google Scholar] [CrossRef]
- Koga, T.; Fujikawa, K.; Horai, Y.; Okada, A.; Kawashiri, S.Y.; Iwamoto, N.; Suzuki, T.; Nakashima, Y.; Tamai, M.; Arima, K.; et al. The diagnostic utility of anti-melanoma differentiation-associated gene 5 antibody testing for predicting the prognosis of Japanese patients with DM. Rheumatology 2012, 51, 1278–1284. [Google Scholar] [CrossRef]
- Hoshino, K.; Muro, Y.; Sugiura, K.; Tomita, Y.; Nakashima, R.; Mimori, T. Anti-MDA5 and anti-TIF1-gamma antibodies have clinical significance for patients with dermatomyositis. Rheumatology 2010, 49, 1726–1733. [Google Scholar] [CrossRef]
- Nishina, N.; Sato, S.; Masui, K.; Gono, T.; Kuwana, M. Seasonal and residential clustering at disease onset of anti-MDA5-associated interstitial lung disease. RMD Open 2020, 6, e001202. [Google Scholar] [CrossRef]
- Muro, Y.; Sugiura, K.; Hoshino, K.; Akiyama, M.; Tamakoshi, K. Epidemiologic study of clinically amyopathic dermatomyositis and anti-melanoma differentiation-associated gene 5 antibodies in central Japan. Arthritis Res. Ther. 2011, 13, R214. [Google Scholar] [CrossRef]
- Tanizawa, K.; Handa, T.; Nakashima, R.; Kubo, T.; Hosono, Y.; Watanabe, K.; Aihara, K.; Oga, T.; Chin, K.; Nagai, S.; et al. HRCT features of interstitial lung disease in dermatomyositis with anti-CADM-140 antibody. Respir. Med. 2011, 105, 1380–1387. [Google Scholar] [CrossRef] [PubMed]
- Chino, H.; Sekine, A.; Baba, T.; Iwasawa, T.; Okudela, K.; Takemura, T.; Itoh, H.; Sato, S.; Suzuki, Y.; Ogura, T. Radiological and Pathological Correlation in Anti-MDA5 Antibody-positive Interstitial Lung Disease: Rapidly Progressive Perilobular Opacities and Diffuse Alveolar Damage. Intern. Med. 2016, 55, 2241–2246. [Google Scholar] [CrossRef] [PubMed]
- Gono, T.; Miyake, K.; Kawaguchi, Y.; Kaneko, H.; Shinozaki, M.; Yamanaka, H. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology 2012, 51, 1336–1338. [Google Scholar] [CrossRef] [PubMed]
- Rosario, C.; Zandman-Goddard, G.; Meyron-Holtz, E.G.; D’Cruz, D.P.; Shoenfeld, Y. The hyperferritinemic syndrome: Macrophage activation syndrome, Still’s disease, septic shock and catastrophic antiphospholipid syndrome. BMC Med. 2013, 11, 185. [Google Scholar] [CrossRef]
- Gono, T.; Kawaguchi, Y.; Satoh, T.; Kuwana, M.; Katsumata, Y.; Takagi, K.; Masuda, I.; Tochimoto, A.; Baba, S.; Okamoto, Y.; et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology 2010, 49, 1713–1719. [Google Scholar] [CrossRef]
- Horiike, Y.; Suzuki, Y.; Fujisawa, T.; Yasui, H.; Karayama, M.; Hozumi, H.; Furuhashi, K.; Enomoto, N.; Nakamura, Y.; Inui, N.; et al. Successful classification of macrophage-mannose receptor CD206 in severity of anti-MDA5 antibody positive dermatomyositis associated ILD. Rheumatology 2019, 58, 2143–2152. [Google Scholar] [CrossRef]
- Zhao, S.; Ma, X.; Zhang, X.; Jin, Z.; Hu, W.; Hua, B.; Wang, H.; Feng, X.; Sun, L.; Chen, Z. Clinical significance of HScore and MS score comparison in the prognostic evaluation of anti-MDA5-positive patients with dermatomyositis and interstitial lung disease. Mod. Rheumatol. 2022, 32, 373–379. [Google Scholar] [CrossRef]
- Aggarwal, N.R.; King, L.S.; D’Alessio, F.R. Diverse macrophage populations mediate acute lung inflammation and resolution. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L709–L725. [Google Scholar] [CrossRef]
- Colafrancesco, S.; Priori, R.; Alessandri, C.; Astorri, E.; Perricone, C.; Blank, M.; Agmon-Levin, N.; Shoenfeld, Y.; Valesini, G. The hyperferritinemic syndromes and CD163: A marker of macrophage activation. Isr. Med. Assoc. J. 2014, 16, 662–663. [Google Scholar]
- Peng, Q.L.; Zhang, Y.L.; Shu, X.M.; Yang, H.B.; Zhang, L.; Chen, F.; Lu, X.; Wang, G.C. Elevated Serum Levels of Soluble CD163 in Polymyositis and Dermatomyositis: Associated with Macrophage Infiltration in Muscle Tissue. J. Rheumatol. 2015, 42, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Hozumi, H.; Yasui, H.; Suzuki, Y.; Karayama, M.; Furuhashi, K.; Enomoto, N.; Nakamura, Y.; Inui, N.; Suda, T. Clinical Significance of Serum Chitotriosidase Level in Anti-MDA5 Antibody-positive Dermatomyositis-associated Interstitial Lung Disease. J. Rheumatol. 2019, 46, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Hozumi, H.; Fujisawa, T.; Enomoto, N.; Nakashima, R.; Enomoto, Y.; Suzuki, Y.; Kono, M.; Karayama, M.; Furuhashi, K.; Murakami, A.; et al. Clinical Utility of YKL-40 in Polymyositis/dermatomyositis-associated Interstitial Lung Disease. J. Rheumatol. 2017, 44, 1394–1401. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, Y.; Suzuki, Y.; Hozumi, H.; Mori, K.; Kono, M.; Karayama, M.; Furuhashi, K.; Fujisawa, T.; Enomoto, N.; Nakamura, Y.; et al. Clinical significance of soluble CD163 in polymyositis-related or dermatomyositis-related interstitial lung disease. Arthritis Res. Ther. 2017, 19, 9. [Google Scholar] [CrossRef]
- Gono, T.; Okazaki, Y.; Kuwana, M. Antiviral proinflammatory phenotype of monocytes in anti-MDA5 antibody-associated interstitial lung disease. Rheumatology 2022, 61, 806–814. [Google Scholar] [CrossRef]
- Oda, K.; Kotani, T.; Takeuchi, T.; Ishida, T.; Shoda, T.; Isoda, K.; Yoshida, S.; Nishimura, Y.; Makino, S. Chemokine profiles of interstitial pneumonia in patients with dermatomyositis: A case control study. Sci. Rep. 2017, 7, 1635. [Google Scholar] [CrossRef]
- Lv, X.; Jin, Y.; Zhang, D.; Li, Y.; Fu, Y.; Wang, S.; Ye, Y.; Wu, W.; Ye, S.; Yan, B.; et al. Low Circulating Monocytes Is in Parallel with Lymphopenia Which Predicts Poor Outcome in Anti-melanoma Differentiation-Associated Gene 5 Antibody-Positive Dermatomyositis-Associated Interstitial Lung Disease. Front. Med. 2021, 8, 808875. [Google Scholar] [CrossRef]
- Chen, T.T.; Li, L.; Chung, D.H.; Allen, C.D.; Torti, S.V.; Torti, F.M.; Cyster, J.G.; Chen, C.Y.; Brodsky, F.M.; Niemi, E.C.; et al. TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis. J. Exp. Med. 2005, 202, 955–965. [Google Scholar] [CrossRef]
- Shirakashi, M.; Nakashima, R.; Tsuji, H.; Tanizawa, K.; Handa, T.; Hosono, Y.; Akizuki, S.; Murakami, K.; Hashimoto, M.; Yoshifuji, H.; et al. Efficacy of plasma exchange in anti-MDA5-positive dermatomyositis with interstitial lung disease under combined immunosuppressive treatment. Rheumatology 2020, 59, 3284–3292. [Google Scholar] [CrossRef]
- Gono, T.; Kaneko, H.; Kawaguchi, Y.; Hanaoka, M.; Kataoka, S.; Kuwana, M.; Takagi, K.; Ichida, H.; Katsumata, Y.; Ota, Y.; et al. Cytokine profiles in polymyositis and dermatomyositis complicated by rapidly progressive or chronic interstitial lung disease. Rheumatology 2014, 53, 2196–2203. [Google Scholar] [CrossRef]
- Takada, K.; Katada, Y.; Ito, S.; Hayashi, T.; Kishi, J.; Itoh, K.; Yamashita, H.; Hirakata, M.; Kawahata, K.; Kawakami, A.; et al. Impact of adding tacrolimus to initial treatment of interstitial pneumonitis in polymyositis/dermatomyositis: A single-arm clinical trial. Rheumatology 2020, 59, 1084–1093. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Hozumi, H.; Kamiya, Y.; Kaida, Y.; Akamatsu, T.; Kusagaya, H.; Satake, Y.; Mori, K.; Mikamo, M.; Matsuda, H.; et al. Prednisolone and tacrolimus versus prednisolone and cyclosporin A to treat polymyositis/dermatomyositis-associated ILD: A randomized, open-label trial. Respirology 2021, 26, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T. Management of Myositis-Associated Interstitial Lung Disease. Medicina 2021, 57, 347. [Google Scholar] [CrossRef]
- Kondoh, Y.; Makino, S.; Ogura, T.; Suda, T.; Tomioka, H.; Amano, H.; Anraku, M.; Enomoto, N.; Fujii, T.; Fujisawa, T.; et al. 2020 guide for the diagnosis and treatment of interstitial lung disease associated with connective tissue disease. Respir. Investig. 2021, 59, 709–740. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, H.; Nakashima, R.; Hosono, Y.; Imura, Y.; Yagita, M.; Yoshifuji, H.; Hirata, S.; Nojima, T.; Sugiyama, E.; Hatta, K.; et al. Multicenter Prospective Study of the Efficacy and Safety of Combined Immunosuppressive Therapy with High-Dose Glucocorticoid, Tacrolimus, and Cyclophosphamide in Interstitial Lung Diseases Accompanied by Anti-Melanoma Differentiation-Associated Gene 5-Positive Dermatomyositis. Arthritis Rheumatol. 2020, 72, 488–498. [Google Scholar] [PubMed]
- Sugiyama, Y.; Yoshimi, R.; Tamura, M.; Takeno, M.; Kunishita, Y.; Kishimoto, D.; Yoshioka, Y.; Kobayashi, K.; Takase-Minegishi, K.; Watanabe, T.; et al. The predictive prognostic factors for polymyositis/dermatomyositis-associated interstitial lung disease. Arthritis Res. Ther. 2018, 20, 7. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Hayakawa, H.; Miwa, S.; Shirai, M.; Fujii, M.; Gemma, H.; Suda, T.; Chida, K. Intravenous immunoglobulin therapy for refractory interstitial lung disease associated with polymyositis/dermatomyositis. Lung 2009, 187, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.M.; Yang, Q.H.; Zhang, L.; Liu, S.Y.; Zhang, P.P.; Zhang, X.; Liu, X.J.; Han, L.S.; Li, T.F. Intravenous immunoglobulin for interstitial lung diseases of anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Rheumatology 2022, 61, 3704–3710. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.F.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Kubo, K.; Azuma, A.; Kanazawa, M.; Kameda, H.; Kusumoto, M.; Genma, A.; Saijo, Y.; Sakai, F.; Sugiyama, Y.; Tatsumi, K.; et al. Consensus statement for the diagnosis and treatment of drug-induced lung injuries. Respir. Investig. 2013, 51, 260–277. [Google Scholar] [CrossRef]
- Gemma, A.; Kudoh, S.; Ando, M.; Ohe, Y.; Nakagawa, K.; Johkoh, T.; Yamazaki, N.; Arakawa, H.; Inoue, Y.; Ebina, M.; et al. Final safety and efficacy of erlotinib in the phase 4 POLARSTAR surveillance study of 10 708 Japanese patients with non-small-cell lung cancer. Cancer Sci. 2014, 105, 1584–1590. [Google Scholar] [CrossRef] [PubMed]
- Sawada, T.; Inokuma, S.; Sato, T.; Otsuka, T.; Saeki, Y.; Takeuchi, T.; Matsuda, T.; Takemura, T.; Sagawa, A.; Study Committee for Leflunomide-induced Lung Injury, Japan College of Rheumatology. Leflunomide-induced interstitial lung disease: Prevalence and risk factors in Japanese patients with rheumatoid arthritis. Rheumatology 2009, 48, 1069–1072. [Google Scholar] [CrossRef] [PubMed]
- Kudoh, S.; Kato, H.; Nishiwaki, Y.; Fukuoka, M.; Nakata, K.; Ichinose, Y.; Tsuboi, M.; Yokota, S.; Nakagawa, K.; Suga, M.; et al. Interstitial lung disease in Japanese patients with lung cancer: A cohort and nested case-control study. Am. J. Respir. Crit. Care Med. 2008, 177, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Suh, C.H.; Park, H.S.; Kim, K.W.; Pyo, J.; Hatabu, H.; Nishino, M. Pneumonitis in advanced non-small-cell lung cancer patients treated with EGFR tyrosine kinase inhibitor: Meta-analysis of 153 cohorts with 15,713 patients: Meta-analysis of incidence and risk factors of EGFR-TKI pneumonitis in NSCLC. Lung Cancer 2018, 123, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Okamoto, I.; Nakagawa, K. Pooled safety analysis of EGFR-TKI treatment for EGFR mutation-positive non-small cell lung cancer. Lung Cancer 2015, 88, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Takatani, K.; Miyazaki, E.; Nureki, S.; Ando, M.; Ueno, T.; Okubo, T.; Takenaka, R.; Hiroshige, S.; Kumamoto, T. High-resolution computed tomography patterns and immunopathogenetic findings in drug-induced pneumonitis. Respir. Med. 2008, 102, 892–898. [Google Scholar] [CrossRef]
- Ohnishi, H.; Yokoyama, A.; Yasuhara, Y.; Watanabe, A.; Naka, T.; Hamada, H.; Abe, M.; Nishimura, K.; Higaki, J.; Ikezoe, J.; et al. Circulating KL-6 levels in patients with drug induced pneumonitis. Thorax 2003, 58, 872–875. [Google Scholar] [CrossRef]
- Kameda, H.; Tokuda, H.; Sakai, F.; Johkoh, T.; Mori, S.; Yoshida, Y.; Takayanagi, N.; Taki, H.; Hasegawa, Y.; Hatta, K.; et al. Clinical and radiological features of acute-onset diffuse interstitial lung diseases in patients with rheumatoid arthritis receiving treatment with biological agents: Importance of Pneumocystis pneumonia in Japan revealed by a multicenter study. Intern. Med. 2011, 50, 305–313. [Google Scholar] [CrossRef]
- Noguchi, T.; Sekiguchi, Y.; Kudoh, Y.; Naganuma, R.; Kagi, T.; Nishidate, A.; Maeda, K.; Ishii, C.; Toyama, T.; Hirata, Y.; et al. Gefitinib initiates sterile inflammation by promoting IL-1beta and HMGB1 release via two distinct mechanisms. Cell Death Dis. 2021, 12, 49. [Google Scholar] [CrossRef]
- Kagi, T.; Noguchi, T.; Matsuzawa, A. Mechanisms of gefitinib-induced interstitial pneumonitis: Why and how the TKI perturbs innate immune systems? Oncotarget 2021, 12, 1321–1322. [Google Scholar] [CrossRef]
- Namba, T.; Tanaka, K.; Hoshino, T.; Azuma, A.; Mizushima, T. Suppression of expression of heat shock protein 70 by gefitinib and its contribution to pulmonary fibrosis. PLoS ONE 2011, 6, e27296. [Google Scholar] [CrossRef] [PubMed]
- Fujibayashi, T.; Hashimoto, N.; Jijiwa, M.; Hasegawa, Y.; Kojima, T.; Ishiguro, N. Protective effect of geranylgeranylacetone, an inducer of heat shock protein 70, against drug-induced lung injury/fibrosis in an animal model. BMC Pulm. Med. 2009, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- COVID-19 Excess Mortality Collaborators. Estimating excess mortality due to the COVID-19 pandemic: A systematic analysis of COVID-19-related mortality, 2020–2021. Lancet 2022, 399, 1513–1536. [Google Scholar]
- Adjei, S.; Hong, K.; Molinari, N.M.; Bull-Otterson, L.; Ajani, U.A.; Gundlapalli, A.V.; Harris, A.M.; Hsu, J.; Kadri, S.S.; Starnes, J.; et al. Mortality Risk Among Patients Hospitalized Primarily for COVID-19 during the Omicron and Delta Variant Pandemic Periods-United States, April 2020–June 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 1182–1189. [Google Scholar] [CrossRef]
- Simonson, T.S.; Baker, T.L.; Banzett, R.B.; Bishop, T.; Dempsey, J.A.; Feldman, J.L.; Guyenet, P.G.; Hodson, E.J.; Mitchell, G.S.; Moya, E.A.; et al. Silent hypoxaemia in COVID-19 patients. J. Physiol. 2021, 599, 1057–1065. [Google Scholar] [CrossRef]
- Terada, M.; Ohtsu, H.; Saito, S.; Hayakawa, K.; Tsuzuki, S.; Asai, Y.; Matsunaga, N.; Kutsuna, S.; Sugiura, W.; Ohmagari, N. Risk factors for severity on admission and the disease progression during hospitalisation in a large cohort of patients with COVID-19 in Japan. BMJ Open 2021, 11, e047007. [Google Scholar] [CrossRef]
- Swenson, K.E.; Swenson, E.R. Pathophysiology of Acute Respiratory Distress Syndrome and COVID-19 Lung Injury. Crit. Care Clin. 2021, 37, 749–776. [Google Scholar] [CrossRef]
- Zhao, J.; Metra, B.; George, G.; Roman, J.; Mallon, J.; Sundaram, B.; Li, M.; Summer, R. Mortality among Patients with COVID-19 and Different Interstitial Lung Disease Subtypes: A Multicenter Cohort Study. Ann. Am. Thorac. Soc. 2022, 19, 1435–1437. [Google Scholar] [CrossRef]
- Bain, W.; Yang, H.; Shah, F.A.; Suber, T.; Drohan, C.; Al-Yousif, N.; DeSensi, R.S.; Bensen, N.; Schaefer, C.; Rosborough, B.R.; et al. COVID-19 versus Non-COVID-19 Acute Respiratory Distress Syndrome: Comparison of Demographics, Physiologic Parameters, Inflammatory Biomarkers, and Clinical Outcomes. Ann. Am. Thorac. Soc. 2021, 18, 1202–1210. [Google Scholar] [CrossRef]
- Nishimoto, Y.; Yachi, S.; Takeyama, M.; Tsujino, I.; Nakamura, J.; Yamamoto, N.; Nakata, H.; Ikeda, S.; Umetsu, M.; Aikawa, S.; et al. The current status of thrombosis and anticoagulation therapy in patients with COVID-19 in Japan: From the CLOT-COVID study. J. Cardiol. 2022, 80, 285–291. [Google Scholar] [CrossRef]
- Su, H.; Yang, M.; Wan, C.; Yi, L.X.; Tang, F.; Zhu, H.Y.; Yi, F.; Yang, H.C.; Fogo, A.B.; Nie, X.; et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020, 98, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Larici, A.R.; Cicchetti, G.; Marano, R.; Bonomo, L.; Storto, M.L. COVID-19 pneumonia: Current evidence of chest imaging features, evolution and prognosis. Chin. J. Acad. Radiol. 2021, 4, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Obermayer, A.; Jakob, L.M.; Haslbauer, J.D.; Matter, M.S.; Tzankov, A.; Stoiber, W. Neutrophil Extracellular Traps in Fatal COVID-19-Associated Lung Injury. Dis. Markers 2021, 2021, 5566826. [Google Scholar] [CrossRef] [PubMed]
- Iwasawa, T.; Sato, M.; Yamaya, T.; Sato, Y.; Uchida, Y.; Kitamura, H.; Hagiwara, E.; Komatsu, S.; Utsunomiya, D.; Ogura, T. Ultra-high-resolution computed tomography can demonstrate alveolar collapse in novel coronavirus (COVID-19) pneumonia. Jpn. J. Radiol. 2020, 38, 394–398. [Google Scholar] [CrossRef]
- Lang, M.; Som, A.; Carey, D.; Reid, N.; Mendoza, D.P.; Flores, E.J.; Li, M.D.; Shepard, J.O.; Little, B.P. Pulmonary Vascular Manifestations of COVID-19 Pneumonia. Radiol. Cardiothorac. Imaging 2020, 2, e200277. [Google Scholar] [CrossRef]
- Grimmer, B.; Kuebler, W.M. The endothelium in hypoxic pulmonary vasoconstriction. J. Appl. Physiol. 2017, 123, 1635–1646. [Google Scholar] [CrossRef]
- D’Agnillo, F.; Walters, K.A.; Xiao, Y.; Sheng, Z.M.; Scherler, K.; Park, J.; Gygli, S.; Rosas, L.A.; Sadtler, K.; Kalish, H.; et al. Lung epithelial and endothelial damage, loss of tissue repair, inhibition of fibrinolysis, and cellular senescence in fatal COVID-19. Sci. Transl. Med. 2021, 13, eabj7790. [Google Scholar] [CrossRef]
- Batah, S.S.; Fabro, A.T. Pulmonary pathology of ARDS in COVID-19: A pathological review for clinicians. Respir. Med. 2021, 176, 106239. [Google Scholar] [CrossRef]
- Torres Acosta, M.A.; Singer, B.D. Pathogenesis of COVID-19-induced ARDS: Implications for an ageing population. Eur. Respir. J. 2020, 56, 2002049. [Google Scholar] [CrossRef]
- Helal, M.A.; Shouman, S.; Abdelwaly, A.; Elmehrath, A.O.; Essawy, M.; Sayed, S.M.; Saleh, A.H.; El-Badri, N. Molecular basis of the potential interaction of SARS-CoV-2 spike protein to CD147 in COVID-19 associated-lymphopenia. J. Biomol. Struct. Dyn. 2022, 40, 1109–1119. [Google Scholar] [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e15. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.A.; Cascino, K.; Ordonez, A.A.; Zhou, W.; Vaghasia, A.; Hamacher-Brady, A.; Brady, N.R.; Sun, I.H.; Wang, R.; Rosenberg, A.Z.; et al. Metabolic programs define dysfunctional immune responses in severe COVID-19 patients. Cell Rep. 2021, 34, 108863. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes. Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef]
- Paolini, A.; Borella, R.; De Biasi, S.; Neroni, A.; Mattioli, M.; Lo Tartaro, D.; Simonini, C.; Franceschini, L.; Cicco, G.; Piparo, A.M.; et al. Cell Death in Coronavirus Infections: Uncovering Its Role during COVID-19. Cells 2021, 10, 1585. [Google Scholar] [CrossRef]
- Gibellini, L.; De Biasi, S.; Paolini, A.; Borella, R.; Boraldi, F.; Mattioli, M.; Lo Tartaro, D.; Fidanza, L.; Caro-Maldonado, A.; Meschiari, M.; et al. Altered bioenergetics and mitochondrial dysfunction of monocytes in patients with COVID-19 pneumonia. EMBO Mol. Med. 2020, 12, e13001. [Google Scholar] [CrossRef]
- Silvin, A.; Chapuis, N.; Dunsmore, G.; Goubet, A.G.; Dubuisson, A.; Derosa, L.; Almire, C.; Henon, C.; Kosmider, O.; Droin, N.; et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell 2020, 182, 1401–1418.e18. [Google Scholar] [CrossRef]
- Severe Covid, G.G.; Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernandez, J.; Prati, D.; Baselli, G.; et al. Genomewide Association Study of Severe COVID-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar]
- Laing, A.G.; Lorenc, A.; Del Molino Del Barrio, I.; Das, A.; Fish, M.; Monin, L.; Munoz-Ruiz, M.; McKenzie, D.R.; Hayday, T.S.; Francos-Quijorna, I.; et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020, 26, 1623–1635. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients with Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. [Google Scholar] [CrossRef]
- Gangaev, A.; Ketelaars, S.L.C.; Isaeva, O.I.; Patiwael, S.; Dopler, A.; Hoefakker, K.; De Biasi, S.; Gibellini, L.; Mussini, C.; Guaraldi, G.; et al. Identification and characterization of a SARS-CoV-2 specific CD8(+) T cell response with immunodominant features. Nat. Commun. 2021, 12, 2593. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Martins, A.J.; Lau, W.W.; Rachmaninoff, N.; Chen, J.; Imberti, L.; Mostaghimi, D.; Fink, D.L.; Burbelo, P.D.; Dobbs, K.; et al. Time-resolved systems immunology reveals a late juncture linked to fatal COVID-19. Cell 2021, 184, 1836–1857.e22. [Google Scholar] [CrossRef] [PubMed]
- Peruzzi, B.; Bencini, S.; Capone, M.; Mazzoni, A.; Maggi, L.; Salvati, L.; Vanni, A.; Orazzini, C.; Nozzoli, C.; Morettini, A.; et al. Quantitative and qualitative alterations of circulating myeloid cells and plasmacytoid DC in SARS-CoV-2 infection. Immunology 2020, 161, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Ehrchen, J.M.; Sunderkotter, C.; Foell, D.; Vogl, T.; Roth, J. The endogenous Toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J. Leukoc. Biol. 2009, 86, 557–566. [Google Scholar] [CrossRef]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.A.; Phoon, M.C.; van Rooijen, N.; Chow, V.T. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e17. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.L.; Zhou, P.; et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct. Target. Ther. 2020, 5, 235. [Google Scholar] [CrossRef]
- Nakamura, H.; Kinjo, T.; Arakaki, W.; Miyagi, K.; Tateyama, M.; Fujita, J. Serum levels of receptor-interacting protein kinase-3 in patients with COVID-19. Crit. Care 2020, 24, 484. [Google Scholar] [CrossRef]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.Y.; Zhou, W.; Qiu, Y.; et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol. Immunol. 2020, 17, 881–883. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.J.; Oster, Y.; Moses, A.E.; Spitzer, A.; Benenson, S.; Israeli-Hospitals 4th Vaccine Working Group. Association of Receiving a Fourth Dose of the BNT162b Vaccine with SARS-CoV-2 Infection among Health Care Workers in Israel. JAMA Netw Open 2022, 5, e2224657. [Google Scholar] [CrossRef] [PubMed]
- Barthwal, M.S.; Dole, S.; Sahasrabudhe, T. Management of COVID-19: A comprehensive and practical approach. Med. J. Armed Forces India 2022. [Google Scholar] [CrossRef]
- Hammond, J.; Leister-Tebbe, H.; Gardner, A.; Abreu, P.; Bao, W.; Wisemandle, W.; Baniecki, M.; Hendrick, V.M.; Damle, B.; Simon-Campos, A.; et al. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with COVID-19. N. Engl. J. Med. 2022, 386, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Jayk Bernal, A.; Gomes da Silva, M.M.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; Delos Reyes, V.; Martin-Quiros, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for Oral Treatment of COVID-19 in Nonhospitalized Patients. N. Engl. J. Med. 2022, 386, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Marconi, V.C.; Ramanan, A.V.; de Bono, S.; Kartman, C.E.; Krishnan, V.; Liao, R.; Piruzeli, M.L.B.; Goldman, J.D.; Alatorre-Alexander, J.; de Cassia Pellegrini, R.; et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): A randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir. Med. 2021, 9, 1407–1418. [Google Scholar] [CrossRef]
- Kalil, A.C.; Patterson, T.F.; Mehta, A.K.; Tomashek, K.M.; Wolfe, C.R.; Ghazaryan, V.; Marconi, V.C.; Ruiz-Palacios, G.M.; Hsieh, L.; Kline, S.; et al. Baricitinib plus Remdesivir for Hospitalized Adults with COVID-19. N. Engl. J. Med. 2021, 384, 795–807. [Google Scholar] [CrossRef]
- WHO Solidarity Trial Consortium; Pan, H.; Peto, R.; Henao-Restrepo, A.M.; Preziosi, M.P.; Sathiyamoorthy, V.; Abdool Karim, Q.; Alejandria, M.M.; Hernandez Garcia, C.; Kieny, M.P.; et al. Repurposed Antiviral Drugs for COVID-19-Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2021, 384, 497–511. [Google Scholar] [CrossRef]
- Group, R.C.; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar]
- Investigators, R.-C.; Gordon, A.C.; Mouncey, P.R.; Al-Beidh, F.; Rowan, K.M.; Nichol, A.D.; Arabi, Y.M.; Annane, D.; Beane, A.; van Bentum-Puijk, W.; et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 384, 1491–1502. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alveolar Epithelial Cell Injury | Lung Endothelial Cell Injury | Activated Alveolar Macrophage | DC | Lymphocyte | Monocyte | Activated Neutrophil | Other Cells | Histopathological Patterns and Other Findings | |
|---|---|---|---|---|---|---|---|---|---|
| AE-IPF | KL-6↑, SP-D↑ α-defensin↑ Periostin↑ Corisin↑ (staphylococcus) Pepsin↑, HMGB1↑ Apoptosis↑ | HSP70↓ Thrombomodulin↓ | CCL18↑ Ferritin↑ Hemosiderin score↑ HMGB1↑ | DC↑ | Memory T cells↑ | S100A8↑ S100A9↑ MMP9↑ IL-8↑ | Adiponectin↑ Leptin↑ | DAD Pulmonary haemorrhage Thromboembolism | |
| CADM-ILD | YKL-40↑ | Anti-MDA5 ab↑ Ferritin↑, YKL-40↑ Chitotriosidase↑ CD163↑, CD206↑ CCL2↑ Interferon β↑ Hemophagocytic score↑ | Lymphocyte↓ (H-Ferritin↑, TIM2) | Monocytes↓ (H-Ferritin↑, TIM2) CCL2↑ IFIT3↑ | Chitotriosidase↑ IL-8↑ | DAD | |||
| EGFR-TKI-induced lung injury | HSP70↓ Apoptosis↑ | Mitochondrial ROS↑ NLRP3↑, IL-1β↑ HMGB1↑ Pyroptosis↑ | Neutrophils↑ | DAD | |||||
| COVID-19 and ILD | Direct invasion of virus PANoptosis↑ | Direct invasion of virus PANoptosis↑ Thrombosis↑ | Plasmacytoid DC↓ Myeloid DC↓ | Direct invasion of virus Lymphocytes↓ Mitochondrial ROS↑, NLRP3↑ PD-1↑, PDL-1↑ CXCR6↓, TNFα↑ IFNγ↑ PANoptosis↑ | Monocytes↓ Mitochondrial damage↑ PD-1↑, PDL-1↑ | S100A8↑ S100A9↑ NETs↑ | DAD Thrombosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enomoto, N. Pathological Roles of Pulmonary Cells in Acute Lung Injury: Lessons from Clinical Practice. Int. J. Mol. Sci. 2022, 23, 15027. https://doi.org/10.3390/ijms232315027
Enomoto N. Pathological Roles of Pulmonary Cells in Acute Lung Injury: Lessons from Clinical Practice. International Journal of Molecular Sciences. 2022; 23(23):15027. https://doi.org/10.3390/ijms232315027
Chicago/Turabian StyleEnomoto, Noriyuki. 2022. "Pathological Roles of Pulmonary Cells in Acute Lung Injury: Lessons from Clinical Practice" International Journal of Molecular Sciences 23, no. 23: 15027. https://doi.org/10.3390/ijms232315027
APA StyleEnomoto, N. (2022). Pathological Roles of Pulmonary Cells in Acute Lung Injury: Lessons from Clinical Practice. International Journal of Molecular Sciences, 23(23), 15027. https://doi.org/10.3390/ijms232315027