

Golgi Complex form and Function: A Potential Hub Role Also in Skeletal Muscle Pathologies?


1
Laboratory of Muscle Biophysics, Department of Biomedical Sciences, University of Padova, 35131 Padova, Italy
2
Department of Medicine, Surgery and Health Sciences, University of Trieste, 34149 Trieste, Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(23), 14989; https://doi.org/10.3390/ijms232314989
Submission received: 28 October 2022
/
Revised: 22 November 2022
/
Accepted: 28 November 2022
/
Published: 30 November 2022
(This article belongs to the Special Issue Genetic Basis and Epidemiology of Myopathies 2.0)
{kind=link}
Abstract
:A growing number of disorders has been associated with mutations in the components of the vesicular transport machinery. The early secretory pathway consists of Endoplasmic Reticulum, numerous vesicles, and the Golgi Complex (GC), which work together to modify and package proteins to deliver them to their destination. The GC is a hub organelle, crucial for organization of the other secretory pathway components. As a consequence, GC’s form and function are key players in the pathogenesis of several disorders. Skeletal muscle (SKM) damage can be caused by defective protein modifications and traffic, as observed in some Limb girdle muscular dystrophies. Interestingly, in turn, muscle damage in Duchenne dystrophic SKM cells also includes the alteration of GC morphology. Based on the correlation between GC’s form and function described in non-muscle diseases, we suggest a key role for this hub organelle also in the onset and progression of some SKM disorders. An altered GC could affect the secretory pathway via primary (e.g., mutation of a glycosylation enzyme), or secondary mechanisms (e.g., GC mis-localization in Duchenne muscles), which converge in SKM cell failure. This evidence induces considering the secretory pathway as a potential therapeutic target in the treatment of muscular dystrophies.
1. Introduction
In recent years, study of the correlation between the secretory pathway and inherited human diseases has become increasingly important, allowing for a link to a growing number of disorders and mutations in the components of the secretory pathway [1,2,3].
In the eucaryotic cell, the secretory pathway is constituted by several components with the fundamental tasks to receive, modify, and package proteins and lipids to drive them to their final destinations. In the conventional secretion pathway, proteins are synthetized in the rough endoplasmic reticulum (ER), they leave the ER at the level of the ER- exit sites (ERES) and are transported in COPII vesicles to the Golgi Complex (GC), where they undergo post-translational modifications (e.g., glycosylation) and are packed into post-GC vesicles to reach their target. Defective proteins return to the ER via COPI vesicles. The early secretory pathway, which consists of the ER, ERES, COPII and COPI vesicles, and the GC is the first secretory compartment that the proteins transit to reach their final destination. Since this part of the secretory pathway fulfill the task of selectively transporting native proteins to reach their final target, the ER to GC interface is a fundamental checkpoint for protein scrutinization [4].
In mature skeletal muscle (SKM) cells, the architectural disposition of the early secretory components is unique: the GC is formed by very small elements, localized near the nucleus and in the fiber in close association with ERES in a fiber-type dependent fashion [5,6,7]. This arrangement is the result of a reorganization process occurring during SKM differentiation, which leads to the formation of small GC elements localized near the ERES [5,6]. How this process takes place is not completely clear; however, our recent work [5], in agreement with data on non-muscle cells [8], suggests that during the SKM differentiation process, the GC plays a central role in the positioning and regulation of ERES and other early secretory pathway components.
Actually, these data agree with the evidence that, in other cell types, the GC is not only the central organelle for glycosylation, but it exerts a hub function in the regulation of the transport via the modulation of the assembly and localization of vesicles, as well as ERES distribution [9]. This provides for the GC a key role in the onset and progression of several diseases [10,11] and calls for the investigation of the correlation between SKM diseases and the form and function of the GC.
2. The Early Secretory Pathway and Skeletal Muscle Dystrophies
Congenital muscular dystrophies are a heterogeneous group of inherited disorders characterized by progressive muscle weakness and wasting [12,13]. Although a large number of mutations to the distinct components of the dystrophin associated protein complex (DAPC) have been shown as causative for several forms of dystrophy [12], there is growing evidence that defects in the biosynthetic pathway, such as protein modification, folding or intracellular traffic, have an important influence on SKM integrity [1,2].
Interestingly, despite the numerous proteins involved in the early secretory pathway, the very low level of mutations associated with a disease indicates its essential role in the cell physiology.
Accordingly, the literature reports several forms of muscular dystrophies and myopathies associated with mutations to genes involved in post-translational modifications (e.g., glycosylation enzymes [14,15,16]) in membrane traffic (e.g., Cav3 [17], TRAPPC11 [18,19]) and cell membrane remodeling (e.g., dysferlin [20]), while there are few examples of mutations to GC resident proteins or proteins involved in the ER to GC transport. Few exceptions are the description of the mutation to the GC matrix protein 130 (GM130) [21,22] and to the vesicle fusion protein BET1 [23].
In general, due to the ubiquitous expression of most of these proteins, the damage to the SKM tissue can be one of the features in a very complex clinical condition, as largely described for some alpha-dystroglycanopathies [13,14,15] and recently reported in patients with mutations to the GC matrix protein GM130 [21,22] and the vesicle fusion protein BET1 [23].
3. The Golgi Complex: A Hub Organelle in the Early Secretory Pathway
The GC is involved in protein quality control and is the fundamental organelle for the glycosylation of numerous proteins. To this aim, the GC is a highly dynamic organelle, organized in flattened polarized cisternae, organized in a cis region facing the ER and trans region directed toward the plasma membrane [8,24,25]. Transport between the ER and the GC is regulated by the COP II and COP I coat vesicles. The conventional protein secretion pathway entails the translocation of proteins directly from the ER or via COPII vesicles to the cis face of the GC (anterograde traffic), where they are modified and released via the trans face, to be then directed to the plasma membrane. Defective proteins are transported in COPI vesicles in the retrograde traffic pathway [26].
There is convincing evidence that the maintenance of the GC structure is important for its own function and that several diseases are correlated with modifications of the GC morphology and localization [10,11,25]. In fact, an alteration of its morphology potentially leads to damages in the organization of the sub-compartments and can introduce an impairment of the post-translation processing as a consequence. In turn, the accumulation of improperly modified proteins affects GC organization, vesicles assembly, and protein traffic itself [24,27]. Altogether, this evidence confers a central role to the GC [9].
A comparable role for the GC could be also advanced in the context of the SKM cell because, as suggested by Kanagawa in a description of alpha-dystroglycanopathies [14], proteins that also play a role in the structural maintenance and function of the GC may result in alterations to the glycosylation machinery, affecting the biosynthetic pathway of dystroglycans.
In support to the important role of the GC in the SKM cell, our recent work showed that in in vitro differentiating myoblasts, the depletion of GM130 interferes with GC morphology and ERES organization. It strongly inhibits the transport of M-cadherin, a membrane protein involved in myoblast fusion [28], to the plasma membrane, which remains confined at the ER level. As a final effect, the loss of M-cadherin at the plasma membranes significantly decreases the efficiency of cell fusion [5]. This suggests that the re-organization of ERES, modulated by the GC matrix protein GM130, plays a crucial role in the efficient transport from ER to GC of differentiation-specific cargos during SKM differentiation and provides the first example of specific traffic inhibition from the ER to GC in SKM cells.
Interestingly, it has been reported that mutations to the GC matrix protein GM130, in a pediatric patient from consanguineous parents, induce developmental delay, neuromuscular disorders and muscular damage [21]. Work from Shamseldin and collaborators showed that the mutation in the gene sequence produces a GM130 truncated protein, which is either not produced or very unstable, resembling a null mutation where GM130 functions are likely lost [21].
Although GM130 is a ubiquitous protein, and therefore the clinical picture is complicated with the malfunctioning of multiple organs and system (e.g., the central nervous system) [13,22,29], in vitro experiments, together with the characterization of the human mutated protein, suggest that GM130 plays an important role in SKM functioning. Moreover, these data suggest that the strict coordination among the distinct components of the early secretory pathway (e.g., ER to GC transport) is essential for a correct muscle physiology.
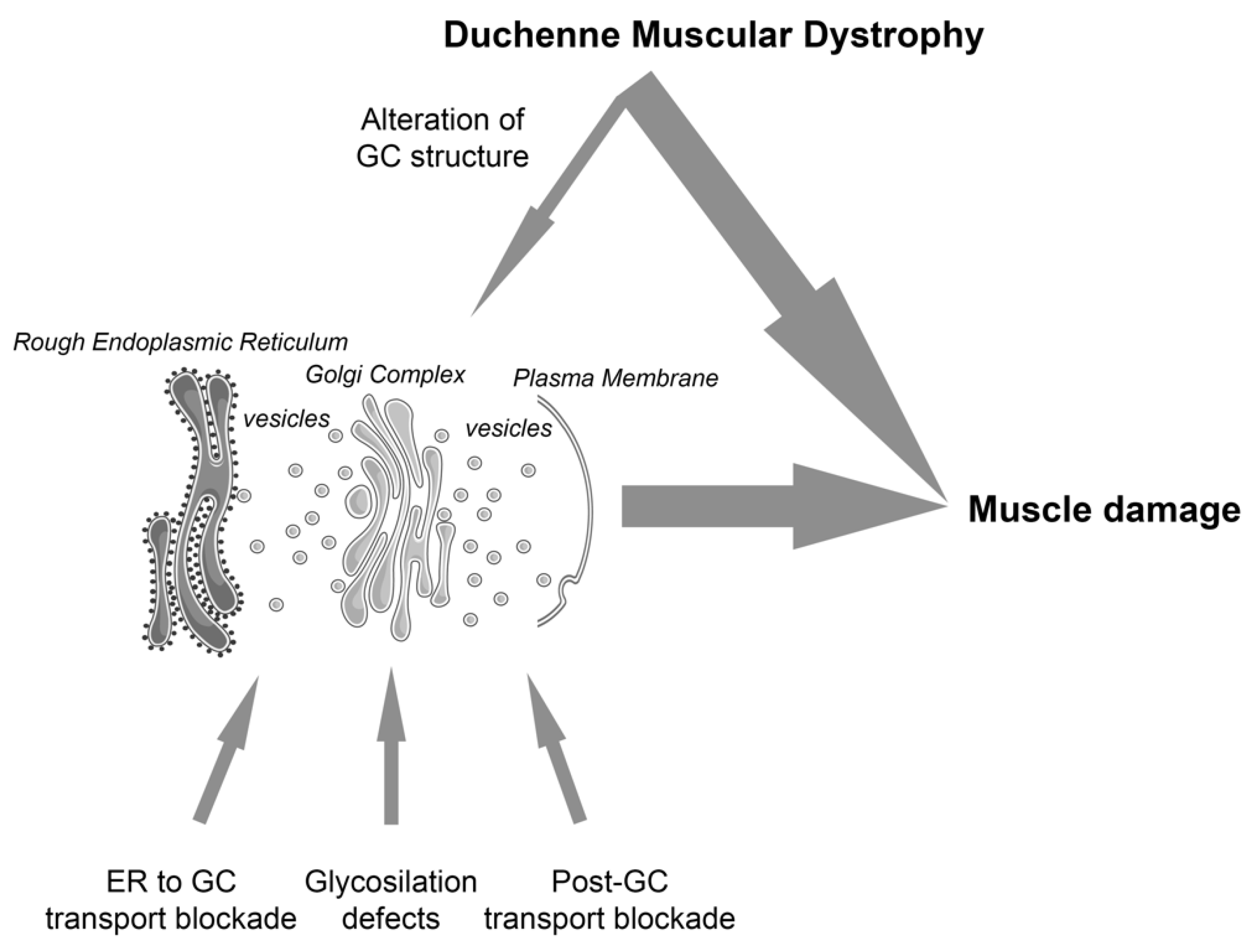
On the other hand, it has been shown that the alteration of muscle specific proteins, not directly involved in the early secretory pathway, could also have an impact on GC form and function. This is the case of the Duchenne muscular dystrophy (DMD), where the loss of dystrophin is accompanied by the alteration of the morphology and mis-localization of the GC, [30,31], with a parallel modification of the microtubule network [32]. Based on data showing the strict relation between form and function in non-muscle cells [10,11,25,27,33], we hypothesize that the alteration of GC morphology in DMD muscle cells introduces a secondary effect that, in a chain-reaction mode, may amplify the progression of the pathology (Figure 1).
In conclusion, in light of the above-mentioned correlation between GC form and function in non-muscle cells [10,11,27,33], according to our observations showing the role of GC form and function during muscle differentiation [5] and the loss of an adequate GC morphology in dystrophic muscle fibers [31,32], we suggest that the GC integrity and functionality can potentially contribute to the pathological mechanisms of muscular dystrophies in a dual way (Figure 1). Either it may contribute directly (e.g., aberrant glycosylation due to a mutation of a GC resident enzyme, such as in alpha-dystroglycanopathies), or/and can have a secondary role (e.g., GC mis-localization in Duchenne dystrophy). Both conditions potentially lead to an altered early secretory pathway. However, in the latter situation, the inefficiency of the protein traffic is an addition to the preexisting pathological mechanisms, which could lead to an amplification of the pathological response, possibly contributing to a worse progression of the SKM disorder.
4. The Components of the Early Secretory Pathway as Therapeutic Targets
Either for direct or secondary contributions, the resulting effect of a non-working early secretory pathway is an accumulation of proteins [4].
Similar to an airways traffic jam, where the passengers take alternative flights to reach their final destination, we can try to manipulate some of the components of the intracellular traffic by exploiting alternative routes and/or hasten the traffic, allowing proteins to reach their destination.
Potentially, different approaches can be applied to reduce the consequences of a defective secretory pathway [34]. These strategies could be summarized in the following categories: (1) agents that modulate the proteostasis, able to interact with the protein quality control, or to reduce ER and cellular stress; (2) a genetic approach to modify mutated proteins [31]; (3) exploitation of the unconventional protein secretion route [35].
Some compounds have already been identified (e.g., in the treatment of Cystic Fibrosis [34]); however, the application of drugs that target the secretory pathway requires further study, and, therefore, this therapeutic approach is still poorly pursued.
It can be suggested that the application of secretory pathway regulators could be of help in also treating muscular dystrophies. Evidently, the choice of the treatment depends on the type of disease and whether the secretory pathway defects result from a direct (e.g., Fukuyama myopathy) or secondary effects (e.g., Duchenne muscular dystrophy). Actually, in the latter situation, the therapeutic targeting of the secretory pathway is not sufficient to prevent or delete the principal effects of a defective protein (e.g., dystrophin), but could have an adjuvant effect by modulating the load of the above-mentioned secondary effects (Figure 1).
However, since the different components of the secretory pathway are highly connected, it is not excluded that a single strategy can have multiple effects and can be helpful for several muscular pathologies.
5. Conclusions
In this perspective, we briefly reported and discussed data on the relevance of the early secretory pathway and its hub organelle, the GC, in the context of muscular dystrophies. Despite the paucity of data on the secretory pathway in the SKM cell, observations from the literature lead us to suggest that the integrity of the GC and vesicular components is necessary for maintenance of the form and function of this tissue.
Based on observations in other cell types, where the alteration of the secretory pathway function results in unproper targeting and the accumulation of proteins, we hypothesize that the secretory pathway can be a potential therapeutic target in the treatment of SKM diseases.
A desirable outcome of future research is to gain further understanding of mechanisms regulating the secretory machinery in the SKM and to test the possibility of targeting the secretory pathway to ameliorate SKM function in pathologic conditions.
Author Contributions
Conceptualization, L.T. and E.G.; writing—original draft preparation, E.G.; writing—review and editing, L.T., G.S., N.F. and E.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dalkilic, I.; Kunkel, L.M. Muscular Dystrophies: Genes to Pathogenesis. Curr. Opin. Genet. Dev. 2003, 13, 231–238. [Google Scholar] [CrossRef]
- Gissen, P.; Maher, E.R. Cargos and Genes: Insights into Vesicular Transport from Inherited Human Disease. J. Med. Genet. 2007, 44, 545–555. [Google Scholar] [CrossRef] [Green Version]
- García-Cazorla, A.; Oyarzábal, A.; Saudubray, J.-M.; Martinelli, D.; Dionisi-Vici, C. Genetic Disorders of Cellular Trafficking. Trends Genet. 2022, 38, 724–751. [Google Scholar] [CrossRef]
- Anelli, T.; Sitia, R. Protein Quality Control in the Early Secretory Pathway. EMBO J. 2008, 27, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Giacomello, E.; Ronchi, P.; Pepperkok, R. GM130 and P115 Play a Key Role in the Organisation of the Early Secretory Pathway during Skeletal Muscle Differentiation. J. Cell Sci. 2019, 132, jcs222083. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Joseph, D.; Bugnard, E.; Zaal, K.J.; Ralston, E. Golgi Complex Reorganization during Muscle Differentiation: Visualization in Living Cells and Mechanism. Mol. Biol. Cell 2001, 12, 795–808. [Google Scholar] [CrossRef] [Green Version]
- Ralston, E.; Lu, Z.; Ploug, T. The Organization of the Golgi Complex and Microtubules in Skeletal Muscle Is Fiber Type-Dependent. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 10694–10705. [Google Scholar] [CrossRef] [Green Version]
- Ronchi, P.; Tischer, C.; Acehan, D.; Pepperkok, R. Positive Feedback between Golgi Membranes, Microtubules and ER Exit Sites Directs de Novo Biogenesis of the Golgi. J. Cell Sci. 2014, 127, 4620–4633. [Google Scholar] [CrossRef] [Green Version]
- Luini, A.; Parashuraman, S. Signaling at the Golgi: Sensing and Controlling the Membrane Fluxes. Curr. Opin. Cell Biol. 2016, 39, 37–42. [Google Scholar] [CrossRef]
- Bexiga, M.G.; Simpson, J.C. Human Diseases Associated with Form and Function of the Golgi Complex. Int. J. Mol. Sci. 2013, 14, 18670–18681. [Google Scholar] [CrossRef]
- Petrosyan, A. Unlocking Golgi: Why Does Morphology Matter? Biochem. Biokhimiia 2019, 84, 1490–1501. [Google Scholar] [CrossRef]
- Cohn, R.D.; Campbell, K.P. Molecular Basis of Muscular Dystrophies. Muscle Nerve 2000, 23, 1456–1471. [Google Scholar] [CrossRef]
- Falsaperla, R.; Praticò, A.D.; Ruggieri, M.; Parano, E.; Rizzo, R.; Corsello, G.; Vitaliti, G.; Pavone, P. Congenital Muscular Dystrophy: From Muscle to Brain. Ital. J. Pediatr. 2016, 42, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanagawa, M. Dystroglycanopathy: From Elucidation of Molecular and Pathological Mechanisms to Development of Treatment Methods. Int. J. Mol. Sci. 2021, 22, 13162. [Google Scholar] [CrossRef]
- Angelini Corrado, L.N. Heterogeneous Pathogenesis of LGMD2: Consequences for Therapy. Basic Appl. Myol. 2007, 17, 173–179. [Google Scholar]
- Brockington, M.; Yuva, Y.; Prandini, P.; Brown, S.C.; Torelli, S.; Benson, M.A.; Herrmann, R.; Anderson, L.V.; Bashir, R.; Burgunder, J.M.; et al. Mutations in the Fukutin-Related Protein Gene (FKRP) Identify Limb Girdle Muscular Dystrophy 2I as a Milder Allelic Variant of Congenital Muscular Dystrophy MDC1C. Hum. Mol. Genet. 2001, 10, 2851–2859. [Google Scholar] [CrossRef]
- Minetti, C.; Sotgia, F.; Bruno, C.; Scartezzini, P.; Broda, P.; Bado, M.; Masetti, E.; Mazzocco, M.; Egeo, A.; Donati, M.A.; et al. Mutations in the Caveolin-3 Gene Cause Autosomal Dominant Limb-Girdle Muscular Dystrophy. Nat. Genet. 1998, 18, 365–368. [Google Scholar] [CrossRef]
- Bogershausen, N.; Shahrzad, N.; Chong, J.X.; von Kleist-Retzow, J.C.; Stanga, D.; Li, Y.; Bernier, F.P.; Loucks, C.M.; Wirth, R.; Puffenberger, E.G.; et al. Recessive TRAPPC11 Mutations Cause a Disease Spectrum of Limb Girdle Muscular Dystrophy and Myopathy with Movement Disorder and Intellectual Disability. Am. J. Hum. Genet. 2013, 93, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Fee, D.B.; Harmelink, M.; Monrad, P.; Pyzik, E. Siblings with Mutations in TRAPPC11 Presenting with Limb-Girdle Muscular Dystrophy 2S. J. Clin. Neuromuscul. Dis. 2017, 19, 27–30. [Google Scholar] [CrossRef]
- Bashir, R.; Britton, S.; Strachan, T.; Keers, S.; Vafiadaki, E.; Lako, M.; Richard, I.; Marchand, S.; Bourg, N.; Argov, Z.; et al. A Gene Related to Caenorhabditis Elegans Spermatogenesis Factor Fer-1 Is Mutated in Limb-Girdle Muscular Dystrophy Type 2B. Nat. Genet. 1998, 20, 37–42. [Google Scholar] [CrossRef]
- Shamseldin, H.E.; Bennett, A.H.; Alfadhel, M.; Gupta, V.; Alkuraya, F.S. GOLGA2, Encoding a Master Regulator of Golgi Apparatus, Is Mutated in a Patient with a Neuromuscular Disorder. Hum. Genet. 2016, 135, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, U.; Mistri, M.; Shah, N.; Shah, P.S.; Gupta, V.A. Bi-Allelic Loss of Function Variants in GOLGA2 Are Associated with a Complex Neurological Phenotype: Report of a Second Family. Clin. Genet. 2021, 100, 748–751. [Google Scholar] [CrossRef] [PubMed]
- Donkervoort, S.; Krause, N.; Dergai, M.; Yun, P.; Koliwer, J.; Gorokhova, S.; Geist Hauserman, J.; Cummings, B.B.; Hu, Y.; Smith, R.; et al. BET1 Variants Establish Impaired Vesicular Transport as a Cause for Muscular Dystrophy with Epilepsy. EMBO Mol. Med. 2021, 13, e13787. [Google Scholar] [CrossRef] [PubMed]
- Linders, P.T.A.; Peters, E.; ter Beest, M.; Lefeber, D.J.; van den Bogaart, G. Sugary Logistics Gone Wrong: Membrane Trafficking and Congenital Disorders of Glycosylation. Int. J. Mol. Sci. 2020, 21, 4654. [Google Scholar] [CrossRef]
- Saraste, J.; Prydz, K. A New Look at the Functional Organization of the Golgi Ribbon. Front. Cell Dev. Biol. 2019, 7, 171. [Google Scholar] [CrossRef]
- Duden, R. ER-to-Golgi Transport: COP I and COP II Function (Review). Mol. Membr. Biol. 2003, 20, 197–207. [Google Scholar] [CrossRef]
- Hellicar, J.; Stevenson, N.L.; Stephens, D.J.; Lowe, M. Supply Chain Logistics—The Role of the Golgi Complex in Extracellular Matrix Production and Maintenance. J. Cell Sci. 2022, 135, jcs258879. [Google Scholar] [CrossRef]
- Donalies, M.; Cramer, M.; Ringwald, M.; Starzinski-Powitz, A. Expression of M-Cadherin, a Member of the Cadherin Multigene Family, Correlates with Differentiation of Skeletal Muscle Cells. Proc. Natl. Acad. Sci. USA 1991, 88, 8024–8028. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Mei, M.; Li, Q.; Roboti, P.; Pang, Q.; Ying, Z.; Gao, F.; Lowe, M.; Bao, S. Loss of the Golgin GM130 Causes Golgi Disruption, Purkinje Neuron Loss, and Ataxia in Mice. Proc. Natl. Acad. Sci. USA 2017, 114, 346–351. [Google Scholar] [CrossRef] [Green Version]
- Percival, J.M.; Froehner, S.C. Golgi Complex Organization in Skeletal Muscle: A Role for Golgi-Mediated Glycosylation in Muscular Dystrophies? Traffic 2007, 8, 184–194. [Google Scholar] [CrossRef]
- Percival, J.M.; Gregorevic, P.; Odom, G.L.; Banks, G.B.; Chamberlain, J.S.; Froehner, S.C. RAAV6-Microdystrophin Rescues Aberrant Golgi Complex Organization in Mdx Skeletal Muscles. Traffic 2007, 8, 1424–1439. [Google Scholar] [CrossRef] [PubMed]
- Oddoux, S.; Randazzo, D.; Kenea, A.; Alonso, B.; Zaal, K.J.M.; Ralston, E. Misplaced Golgi Elements Produce Randomly Oriented Microtubules and Aberrant Cortical Arrays of Microtubules in Dystrophic Skeletal Muscle Fibers. Front. Cell Dev. Biol. 2019, 7, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, M. The Physiological Functions of the Golgin Vesicle Tethering Proteins. Front. Cell Dev. Biol. 2019, 7, 94. [Google Scholar] [CrossRef]
- Sicari, D.; Igbaria, A.; Chevet, E. Control of Protein Homeostasis in the Early Secretory Pathway: Current Status and Challenges. Cells 2019, 8, 1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Gee, H.Y.; Lee, M.G. Unconventional Protein Secretion—New Insights into the Pathogenesis and Therapeutic Targets of Human Diseases. J. Cell Sci. 2018, 131, jcs213686. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The GC is a central organelle in the biosynthetic pathway. We suggest that GC integrity and functionality contributes to the pathological mechanisms of muscular dystrophies via direct (bottom arrows highlight three different conditions that affect protein traffic in the context of muscular dystrophies: ER to GC blockade [5,21,22]; glycosylation defects [14,16]; post GC transport blockade [18,19]) or secondary mechanisms (top arrow; [30,31,32]), contributing to muscle damage. The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
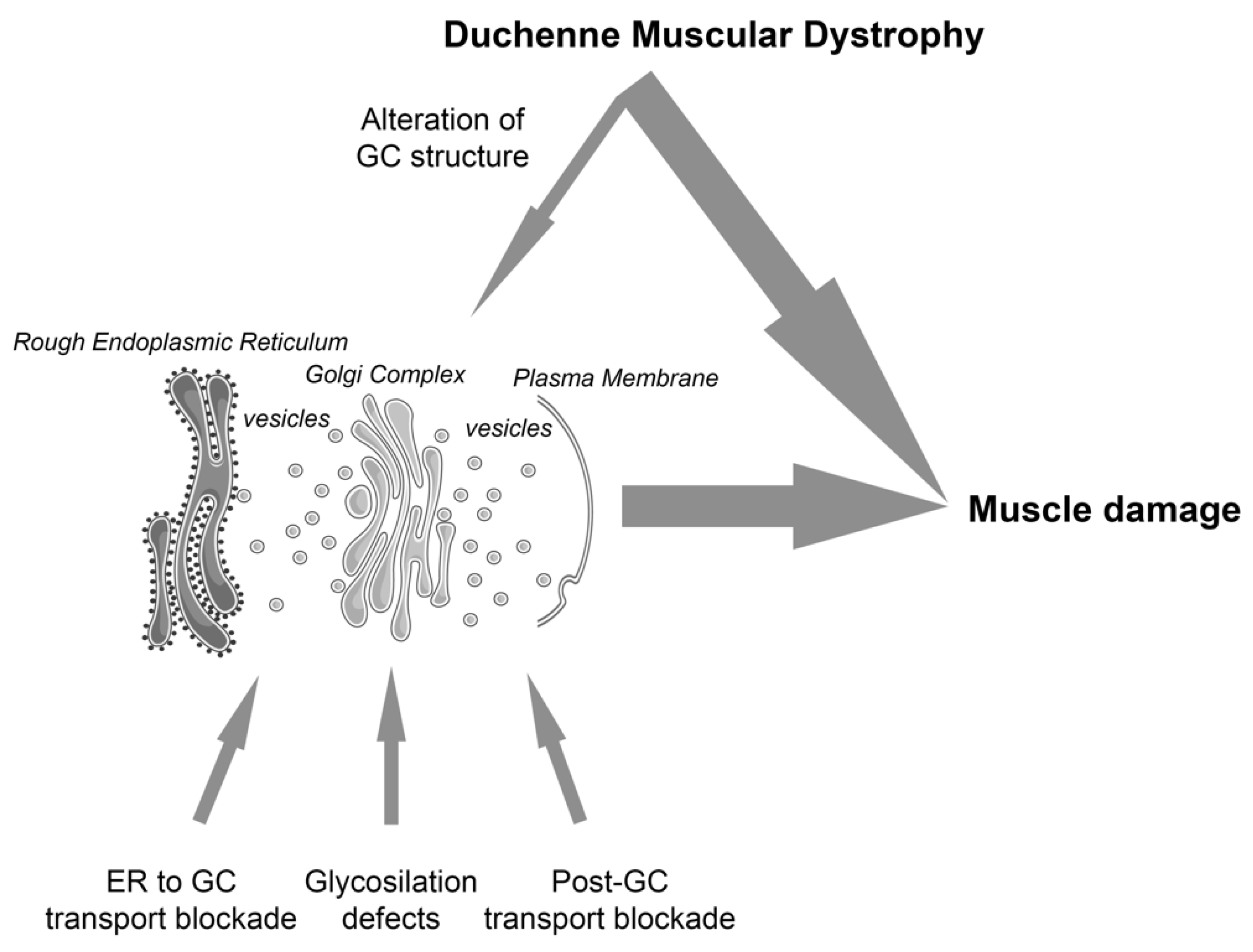
Figure 1.
The GC is a central organelle in the biosynthetic pathway. We suggest that GC integrity and functionality contributes to the pathological mechanisms of muscular dystrophies via direct (bottom arrows highlight three different conditions that affect protein traffic in the context of muscular dystrophies: ER to GC blockade [5,21,22]; glycosylation defects [14,16]; post GC transport blockade [18,19]) or secondary mechanisms (top arrow; [30,31,32]), contributing to muscle damage. The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Toniolo, L.; Sirago, G.; Fiotti, N.; Giacomello, E. Golgi Complex form and Function: A Potential Hub Role Also in Skeletal Muscle Pathologies? Int. J. Mol. Sci. 2022, 23, 14989. https://doi.org/10.3390/ijms232314989
AMA Style
Toniolo L, Sirago G, Fiotti N, Giacomello E. Golgi Complex form and Function: A Potential Hub Role Also in Skeletal Muscle Pathologies? International Journal of Molecular Sciences. 2022; 23(23):14989. https://doi.org/10.3390/ijms232314989
Chicago/Turabian StyleToniolo, Luana, Giuseppe Sirago, Nicola Fiotti, and Emiliana Giacomello. 2022. "Golgi Complex form and Function: A Potential Hub Role Also in Skeletal Muscle Pathologies?" International Journal of Molecular Sciences 23, no. 23: 14989. https://doi.org/10.3390/ijms232314989
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.