
Synthetic Melanin Acts as Efficient Peptide Carrier in Cancer Vaccine Strategy
 , ,
, ,  and
and 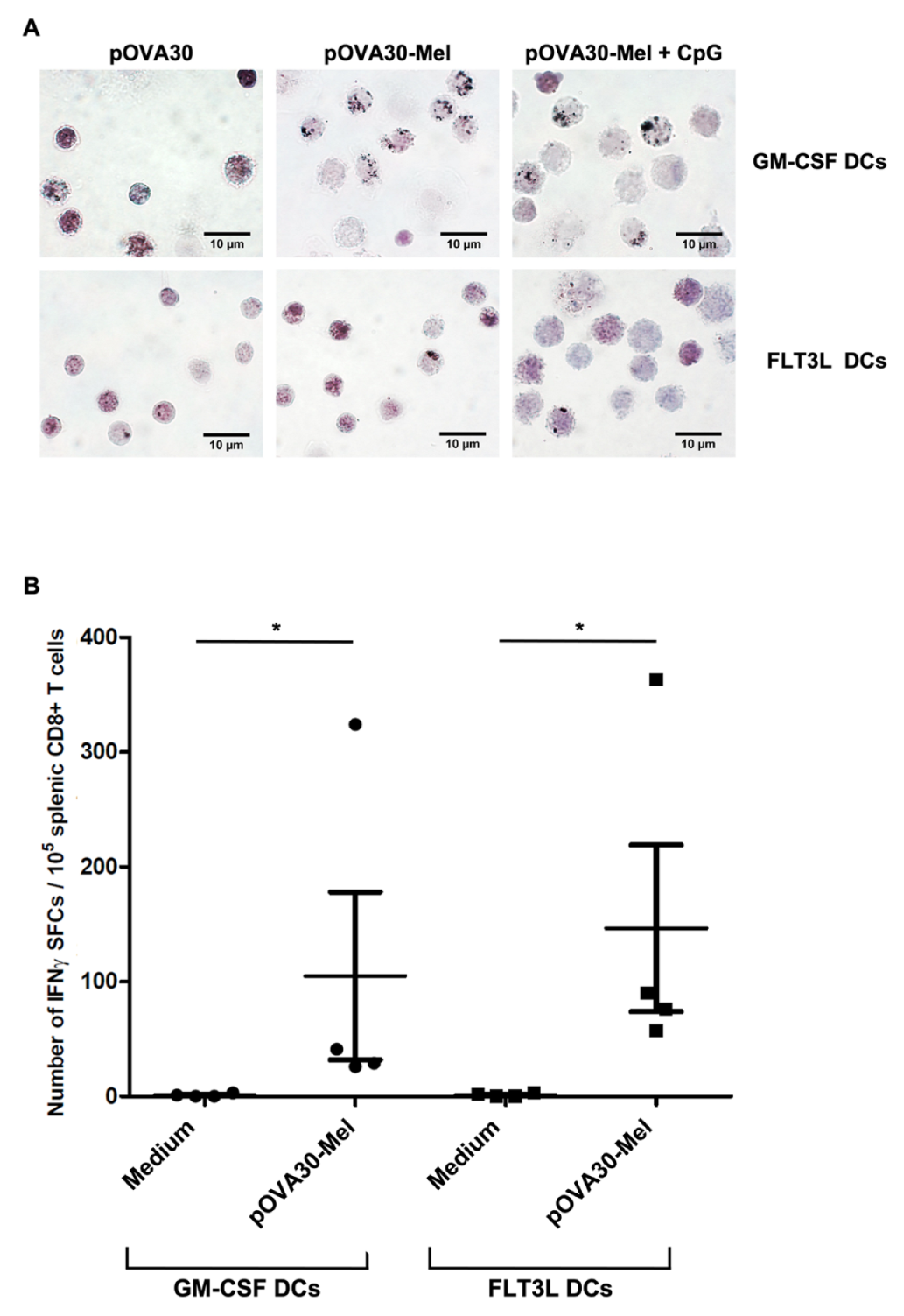{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
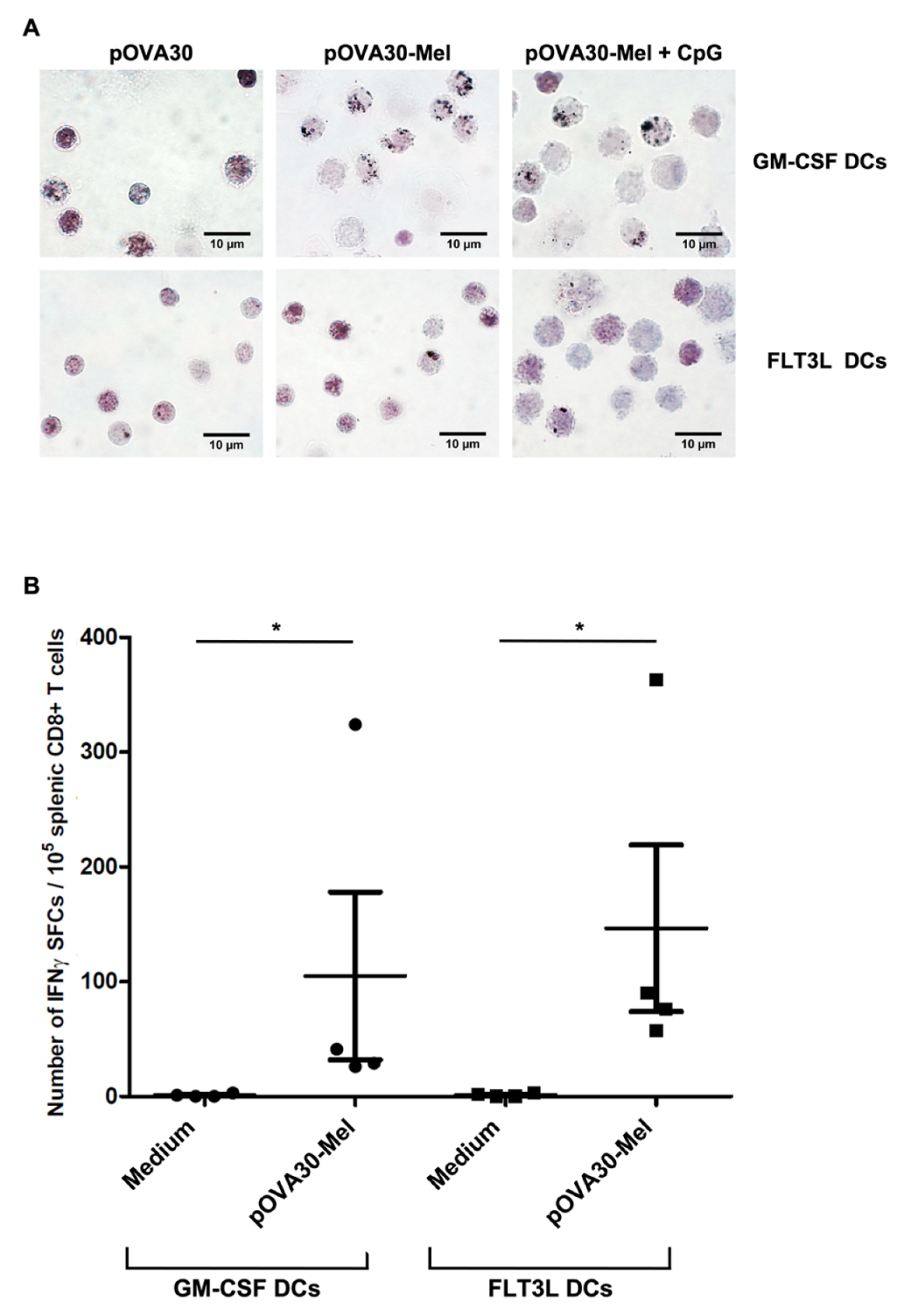
2.1. Dendritic Cells Efficiently Phagocytize Melanin–Peptide Nanoaggregates and Cross-Present the Antigen to T Lymphocyte
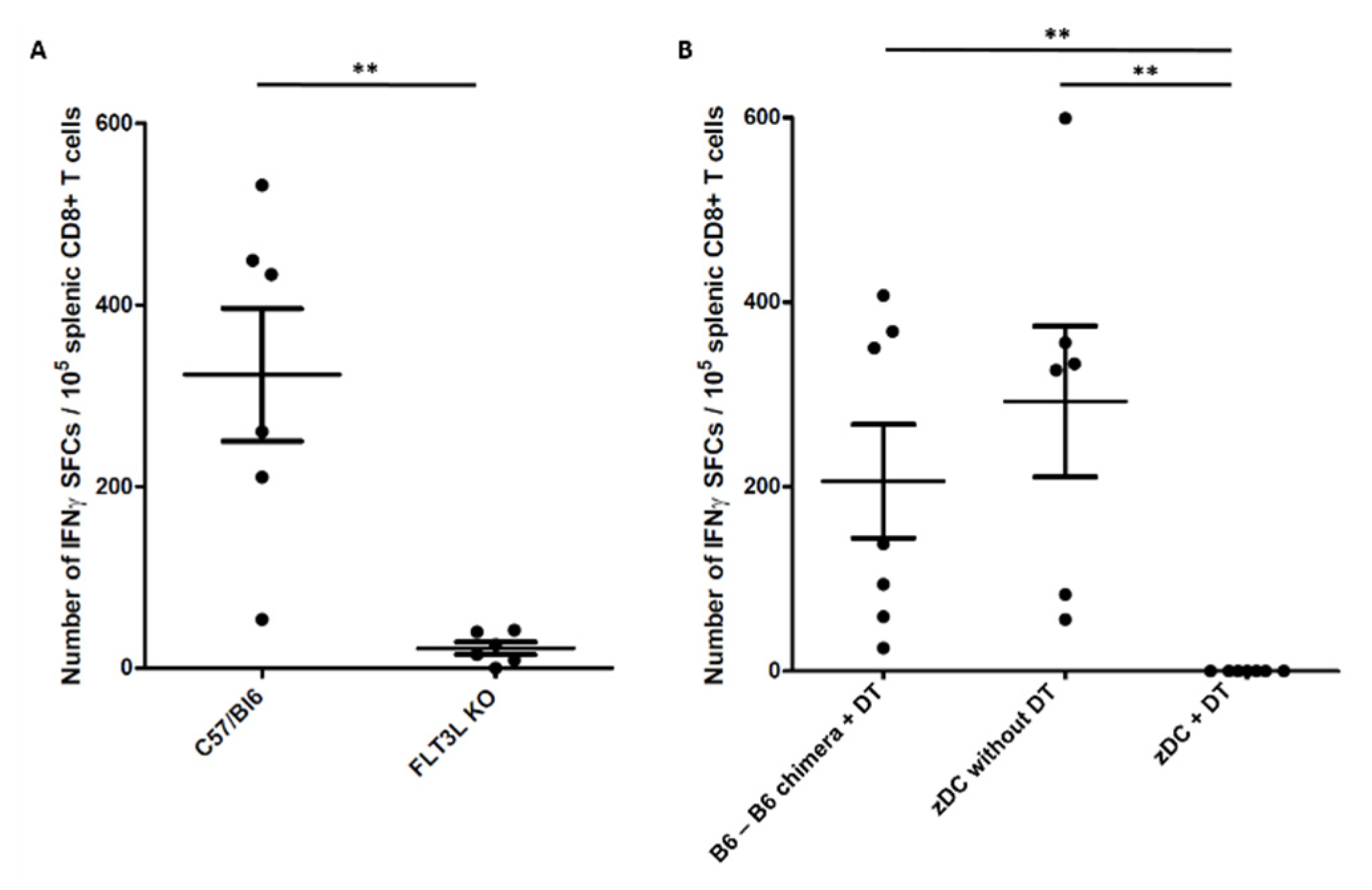
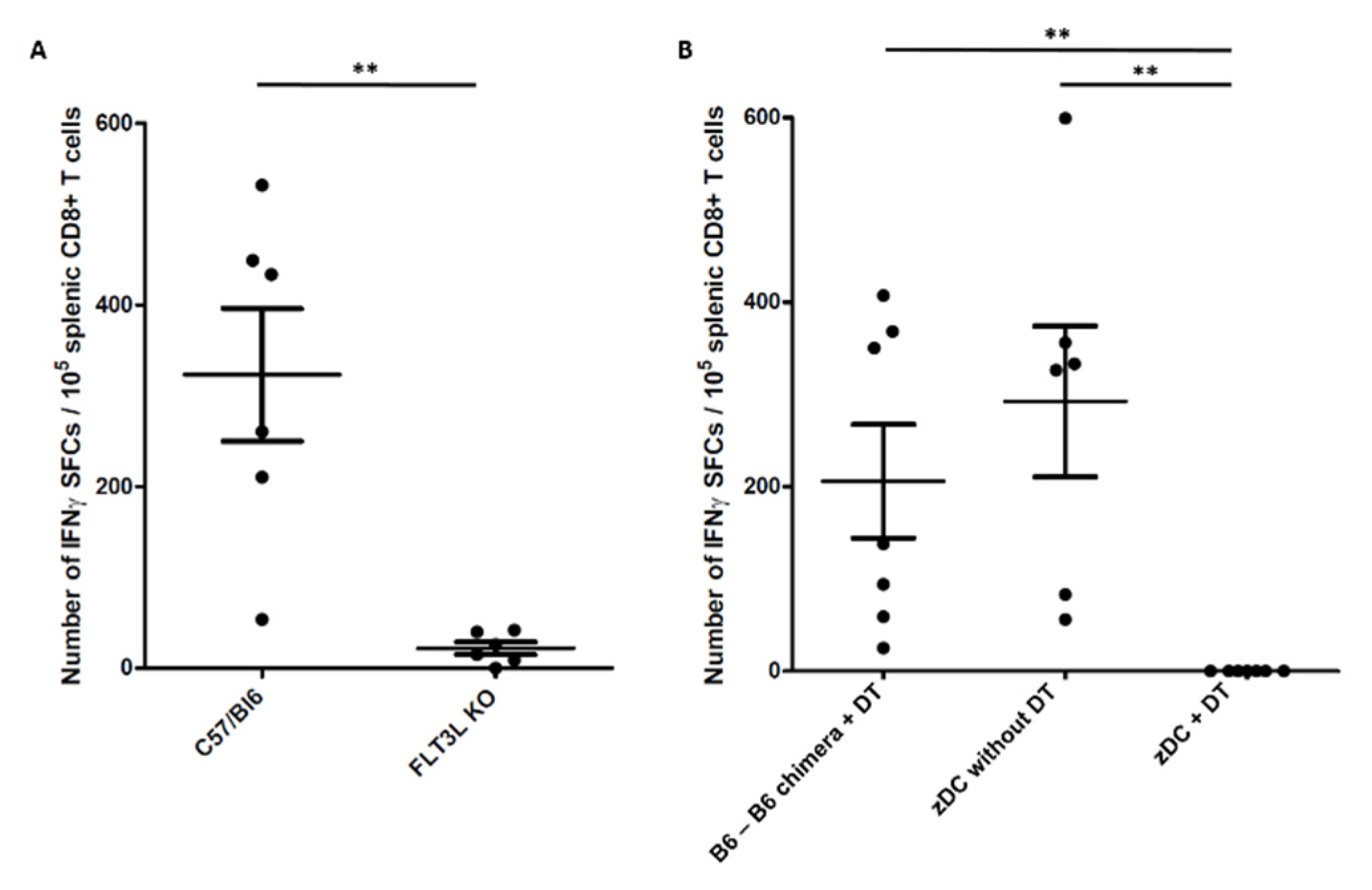
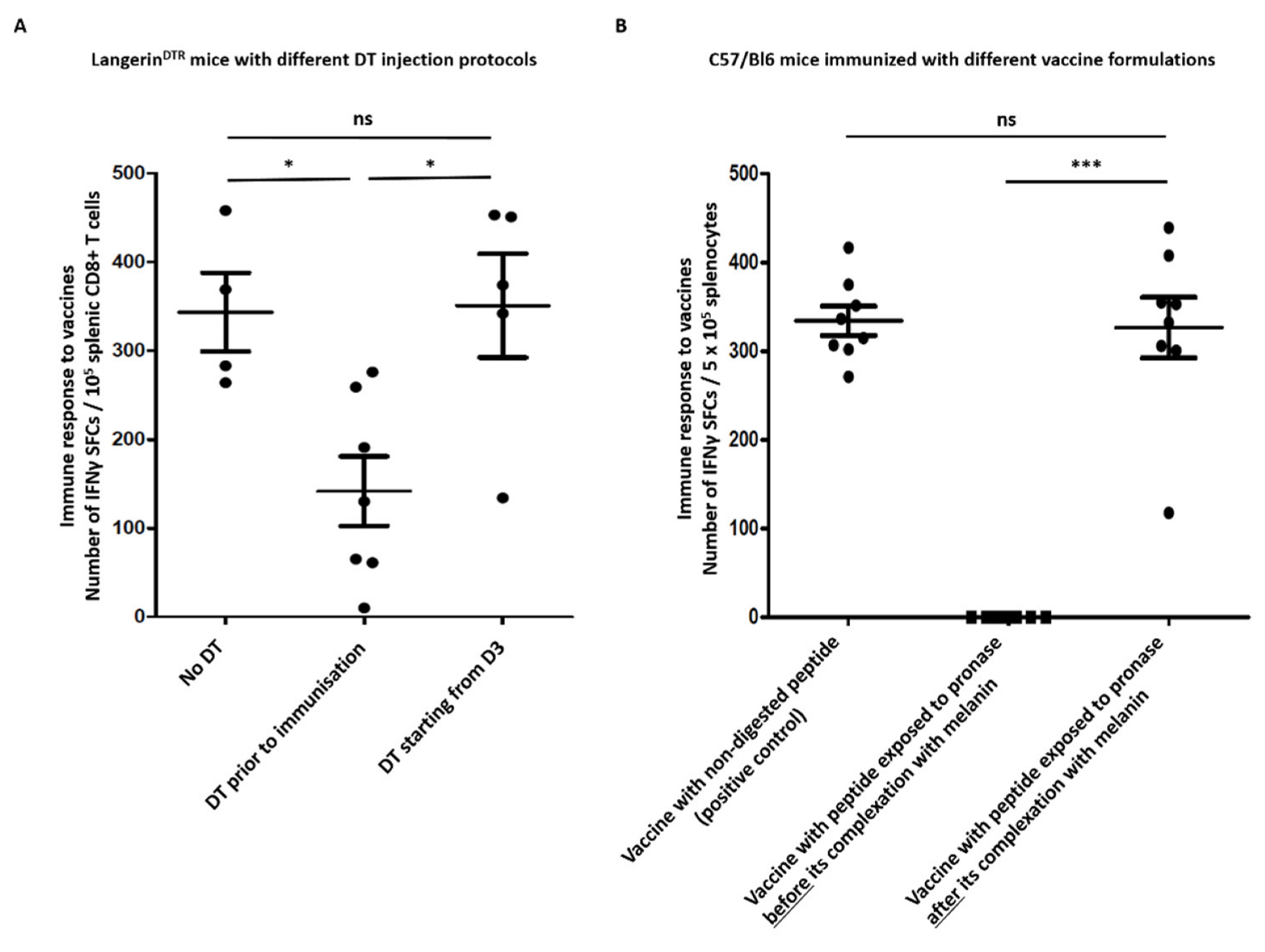
2.2. Conventional Dendritic Cells Are Required for Immune Response to Melanin-Based Vaccine
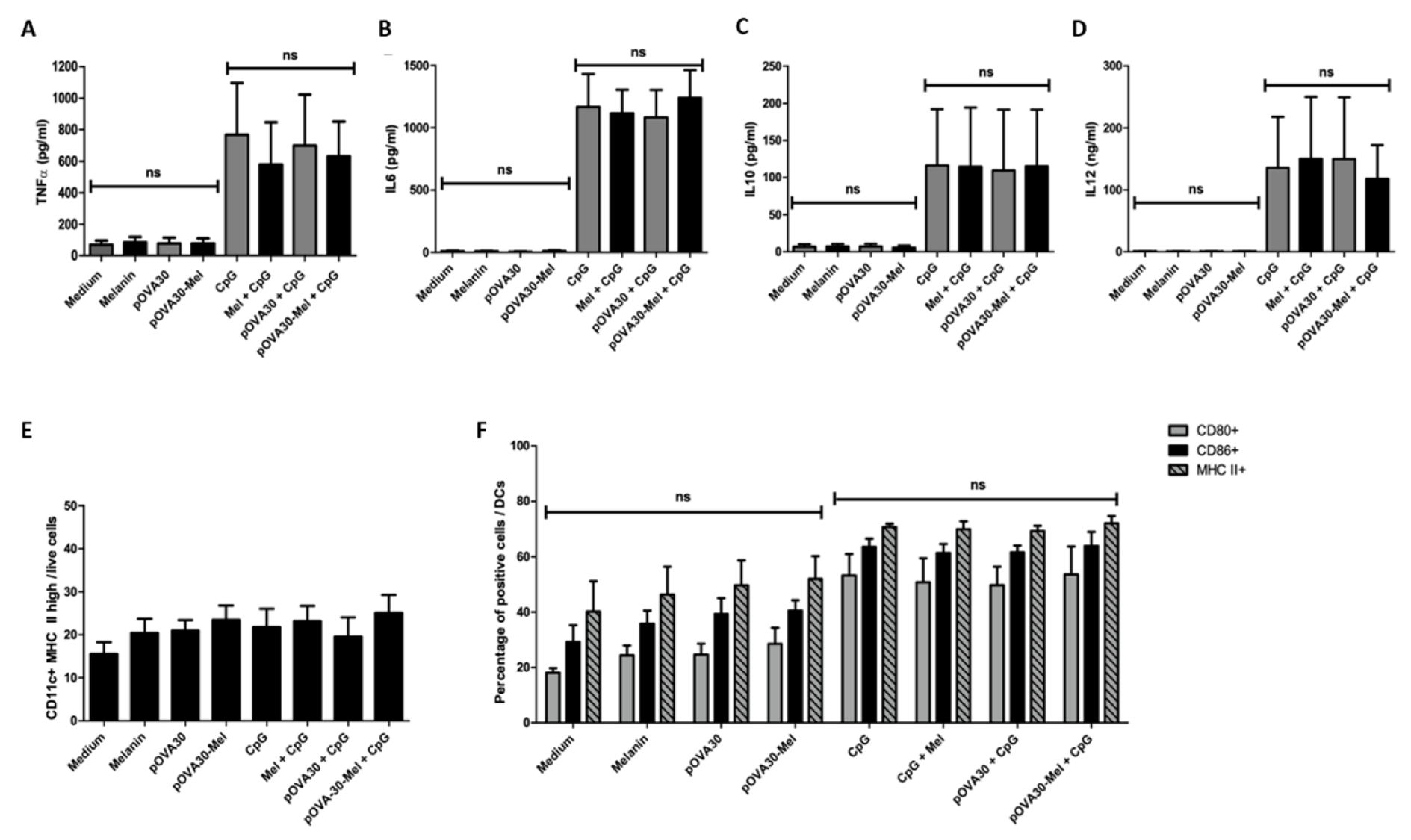
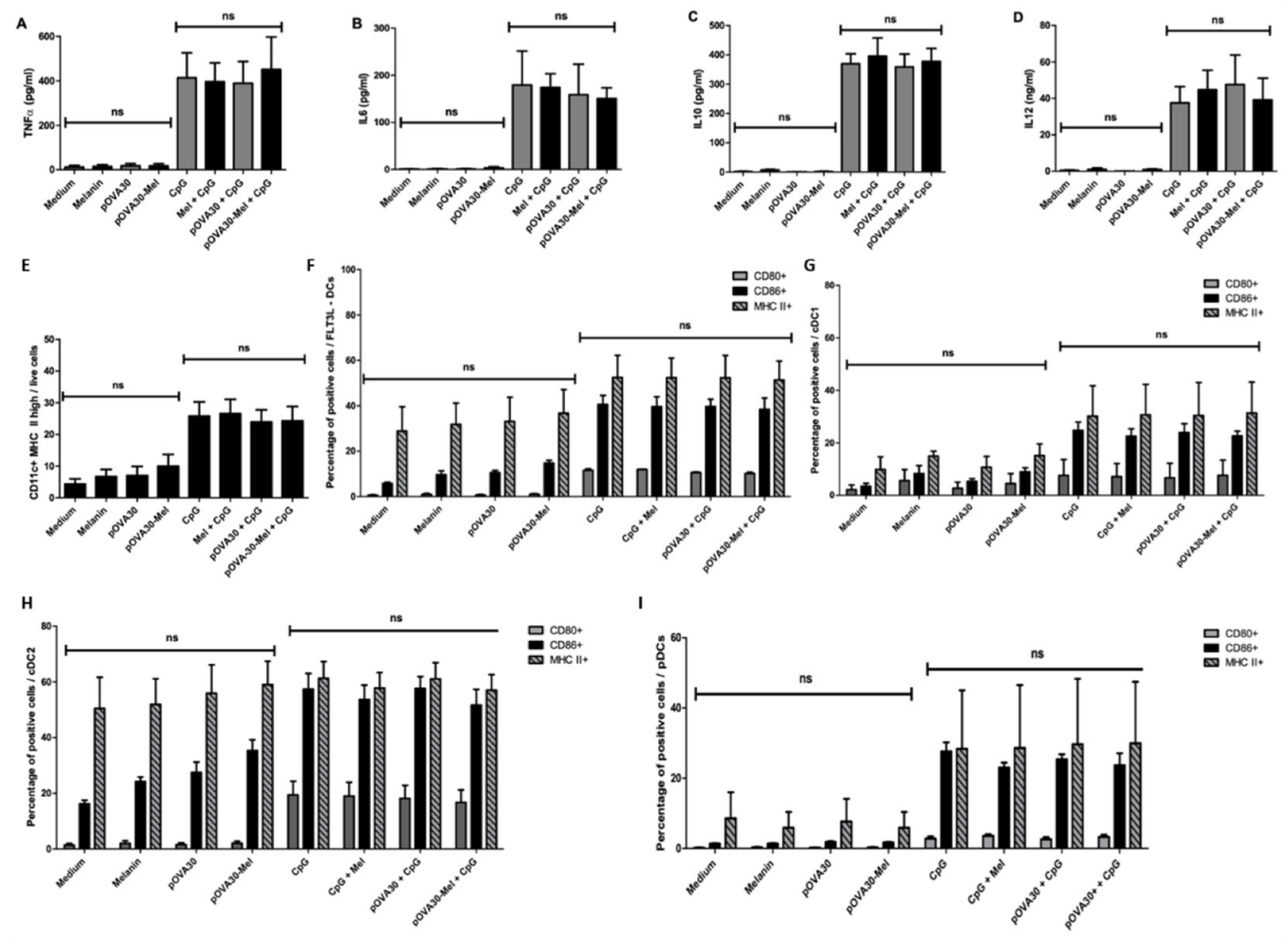
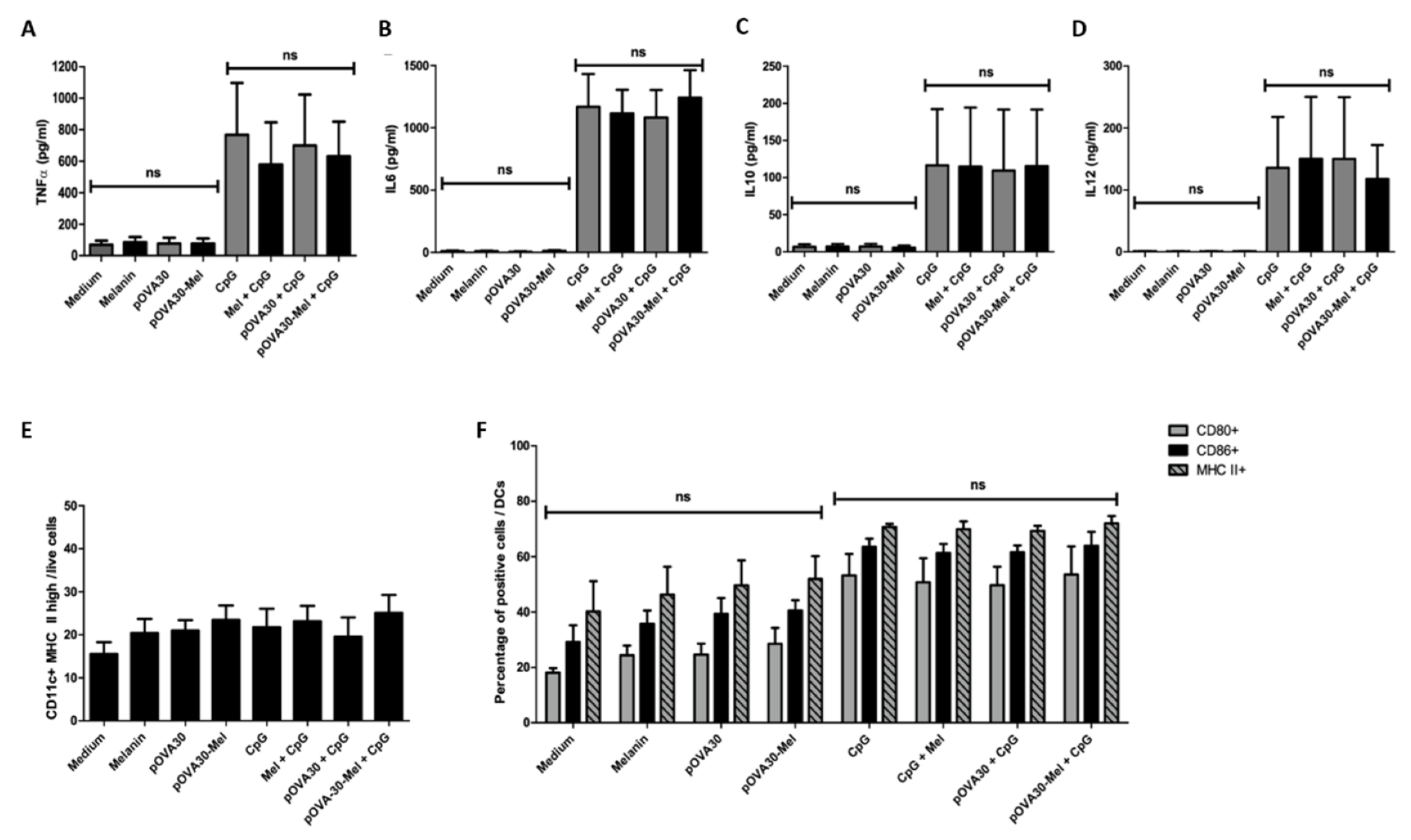
2.3. L-DOPA Melanin Is Not Able to Induce Maturation of Dendritic Cells In Vitro
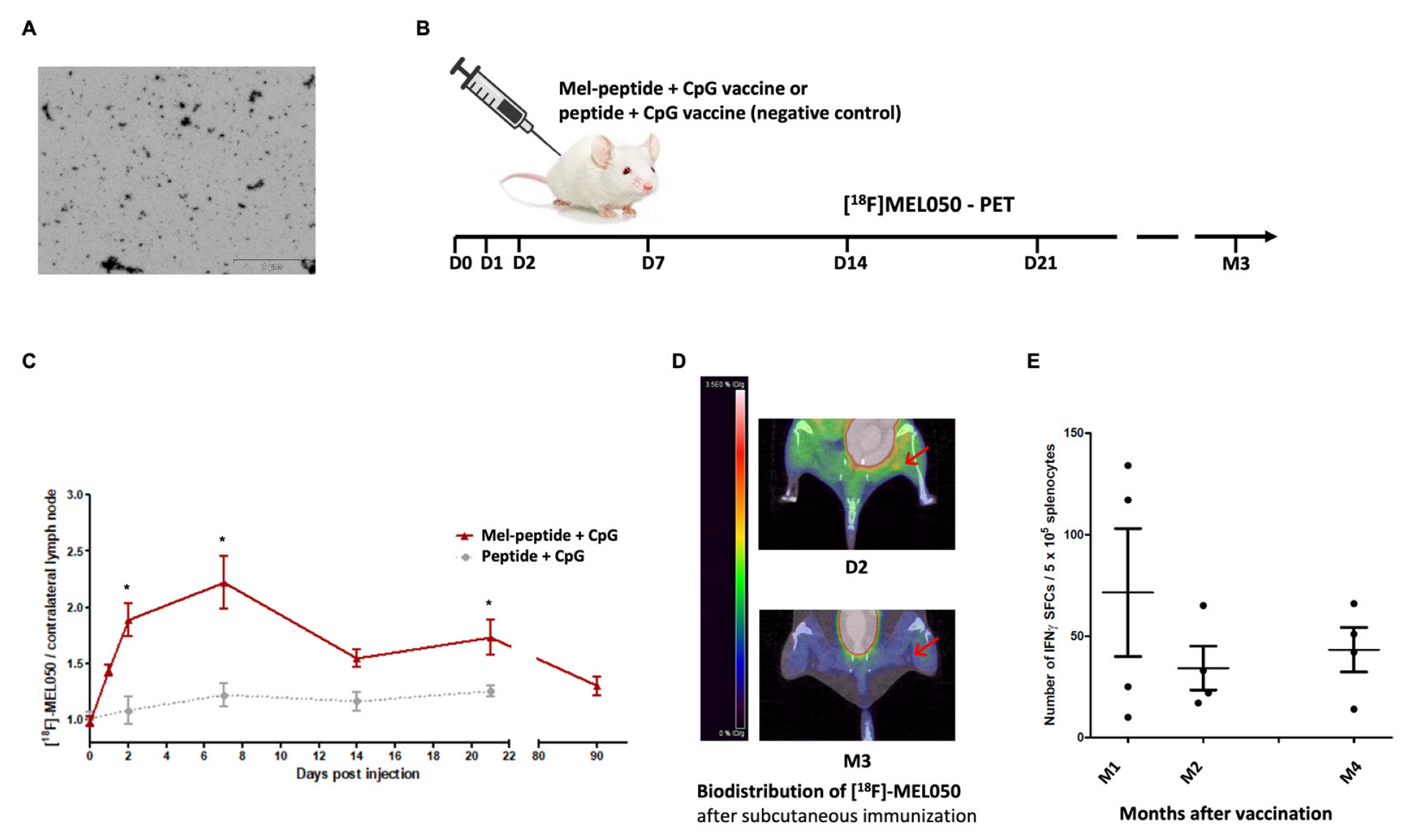
2.4. Melanin-Peptide Nanoaggregates Are Carried by Dermal DCs to the Draining Lymph Node
2.5. L-DOPA Melanin Protects Peptides from Enzymatic Digestion
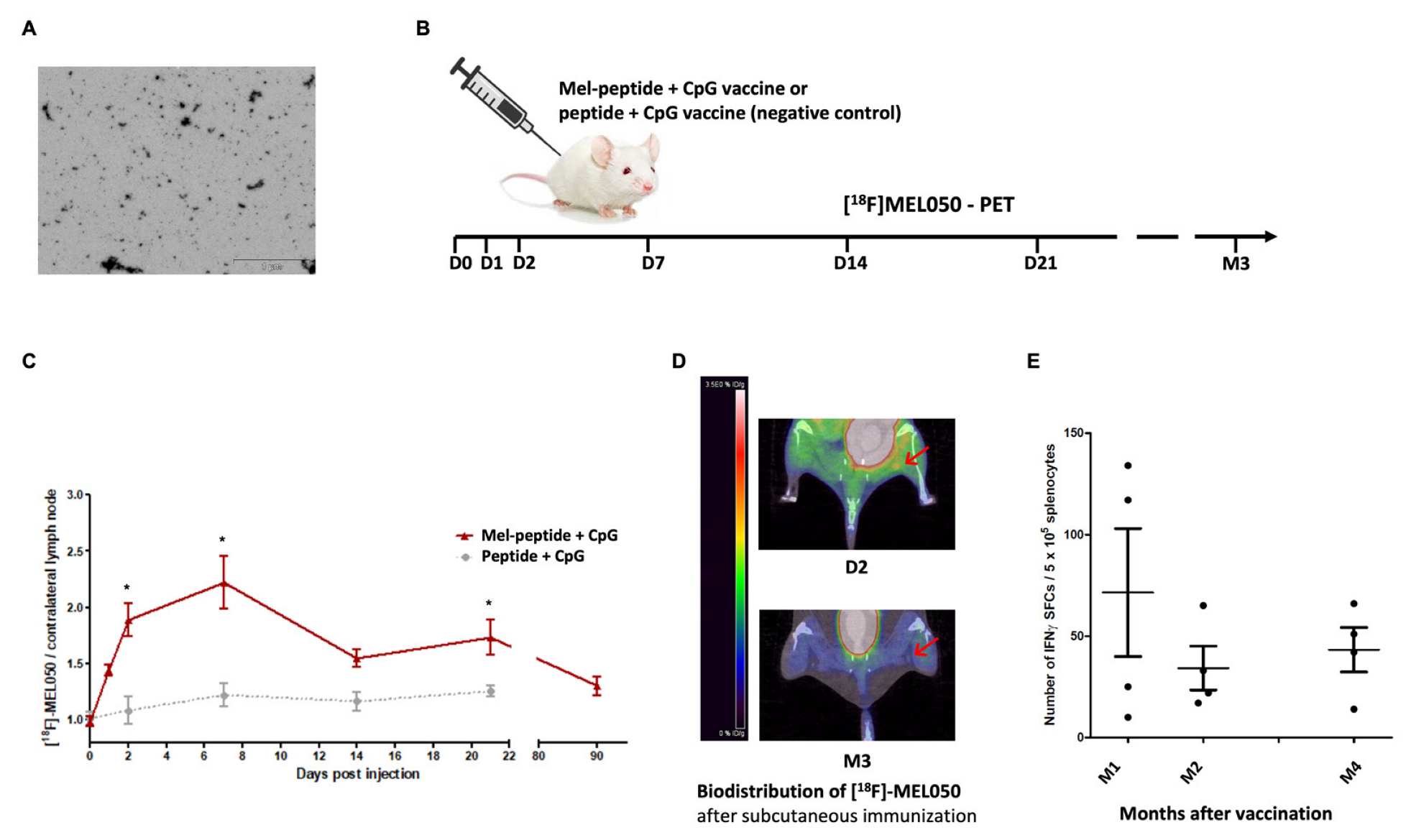
2.6. Melanin-Based Vaccine Induces a Durable Immune Response
3. Materials and Methods
3.1. Peptides and Melanin
3.2. Animals
3.3. Immunization Protocol
3.4. SDS-PAGE
3.5. ELISpot
3.6. Bone Marrow Isolation and Generation of Dendritic Cells
3.7. Incubation of Dendritic Cells with Melanin and Fontana-Masson Staining
3.8. ELISA Analysis
3.9. Flow Cytometry Analysis
3.10. [18F]MEL050 PET/CT Imaging
3.11. Statistics
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cuzzubbo, S.; Mangsbo, S.; Nagarajan, D.; Habra, K.; Pockley, A.G.; McArdle, S.E.B. Cancer vaccines: Adjuvant potency, importance of age, Lifestyle, and treatments. Front. Immunol. 2021, 11, 615240. [Google Scholar] [CrossRef] [PubMed]
- Mougel, A.; Terme, M.; Tanchot, C. Therapeutic Cancer Vaccine and Combinations With Antiangiogenic Therapies and Immune Checkpoint Blockade. Front. Immunol. 2019, 10, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.W.; Hur, S.Y.; Woo, J.W.; Kim, Y.M.; Lim, M.C.; Park, S.Y.; Seo, S.S.; No, J.H.; Kim, B.G.; Lee, J.K.; et al. Pembrolizumab plus GX-188E therapeutic DNA vaccine in patients with HPV-16-positive or HPV-18-positive advanced cervical cancer: Interim results of a single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 1653–1660. [Google Scholar] [CrossRef]
- Kjeldsen, J.W.; Lorentzen, C.L.; Martinenaite, E.; Ellebaek, E.; Donia, M.; Holmstroem, R.B.; Klausen, T.W.; Madsen, C.O.; Ahmed, S.M.; Weis-Banke, S.E.; et al. A phase 1/2 trial of an immune-modulatory vaccine against IDO/PD-L1 in combination with nivolumab in metastatic melanoma. Nat. Med. 2021, 27, 2212–2223. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.; Blanc, C.; Granier, C.; Saldmann, A.; Tanchot, C.; Tartour, E. Therapeutic cancer vaccine: Building the future from lessons of the past. Semin. Immunopathol. 2019, 41, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Irvine, D.J.; Hanson, M.C.; Rakhra, K.; Tokatlian, T. Synthetic nanoparticles for vaccines and immunotherapy. Chem. Rev. 2015, 115, 11109–11146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbell, J.A.; Thomas, S.N.; Swartz, M.A. Materials engineering for immunomodulation. Nature 2009, 462, 449–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speiser, D.E.; Liénard, D.; Rufer, N.; Rubio-Godoy, V.; Rimoldi, D.; Lejeune, F.; Krieg, A.M.; Cerottini, J.C.; Romero, P. Rapid and strong human CD8+ T cell responses to vaccination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J. Clin. Investig. 2005, 115, 739–746. [Google Scholar] [CrossRef]
- Hailemichael, Y.; Dai, Z.; Jaffarzad, N.; Ye, Y.; Medina, M.A.; Huang, X.F.; Dorta-Estremera, S.M.; Greeley, N.R.; Nitti, G.; Peng, W.; et al. Persistent antigen at vaccination sites induces tumor-specific CD8+ T cell sequestration, dysfunction and deletion. Nat. Med. 2013, 19, 465–472. [Google Scholar] [CrossRef]
- Bijker, M.S.; van den Eeden, S.J.; Franken, K.L.; Melief, C.J.; Offringa, R.; van der Burg, S.H. CD8+ CTL priming by exact peptide epitopes in incomplete Freund’s adjuvant induces a vanishing CTL response, whereas long peptides induce sustained CTL reactivity. J. Immunol. 2007, 179, 5033–5040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpentier, A.F.; Geinguenaud, F.; Tran, T.; Sejalon, F.; Martin, A.; Motte, L.; Tartour, E.; Banissi, C. Synthetic melanin bound to subunit vaccine antigens significantly enhances CD8+ T-cell responses. PLoS ONE 2017, 12, e0181403. [Google Scholar] [CrossRef] [Green Version]
- Cuzzubbo, S.; Banissi, C.; Rouchon, M.S.; Tran, T.; Tanchot, C.; Tartour, E.; Carpentier, A.F. The adjuvant effect of melanin is superior to incomplete Freund’s adjuvant in subunit/peptide vaccines in mice. Cancer Immunol. Immunother. 2020, 69, 2501–2512. [Google Scholar] [CrossRef] [PubMed]
- Cuzzubbo, S.; Carpentier, A.F. Applications of melanin and melanin-like nanoparticles in cancer therapy: A review of recent advances. Cancers 2021, 13, 1463. [Google Scholar] [CrossRef]
- Wang, N.; Yang, Y.; Wang, X.; Tian, X.; Qin, W.; Wang, X.; Liang, J.; Zhang, H.; Leng, X. Polydopamine as the Antigen Delivery Nanocarrier for Enhanced Immune Response in Tumor Immunotherapy. ACS Biomater. Sci. Eng. 2019, 5, 2330–2342. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.H.; Zou, M.Z.; Zheng, D.; Peng, S.Y.; Liu, W.; Bai, X.F.; Chen, H.S.; Sun, Y.; Zhou, P.H.; Zhang, X.Z. Nanoparticles from Cuttlefish Ink Inhibit Tumor Growth by Synergizing Immunotherapy and Photothermal Therapy. ACS Nano 2019, 13, 8618–8629. [Google Scholar] [CrossRef]
- El-Obeid, A.; Al-Harbi, S.; Al-Jomah, N.; Hassib, A. Herbal melanin modulates tumor necrosis factor alpha (TNF-alpha), interleukin 6 (IL6) and vascular endothelial growth factor (VEGF) production. Phytomedicine 2006, 13, 324–333. [Google Scholar] [CrossRef]
- Waskow, C.; Liu, K.; Darrasse-Jèze, G.; Guermonprez, P.; Ginhoux, F.; Merad, M.; Shengelia, T.; Yao, K.; Nussenzweig, M. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat. Immunol. 2008, 9, 676–683. [Google Scholar] [CrossRef]
- Allan, R.S.; Waithman, J.; Bedoui, S.; Jones, C.M.; Villadangos, J.A.; Zhan, Y.; Lew, A.M.; Shortman, K.; Heath, W.R.; Carbone, F.R. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity 2006, 25, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Kirkling, M.E.; Cytlak, U.; Lau, C.M.; Lewis, K.L.; Resteu, A.; Khodadadi-Jamayran, A.; Siebel, C.W.; Salmon, H.; Merad, M.; Tsirigos, A.; et al. Notch Signaling Facilitates In Vitro Generation of Cross-Presenting Classical Dendritic Cells. Cell Rep. 2018, 23, 3658–3672. [Google Scholar] [CrossRef] [PubMed]
- van Endert, P. (Ed.) Antigen Processing: Methods and Protocols; Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2013; Volume 960. [Google Scholar] [CrossRef]
- Durai, V.; Bagadia, P.; Briseño, C.G.; Theisen, D.J.; Iwata, A.; Davidson, J.T.; Gargaro, M.; Fremont, D.H.; Murphy, T.L.; Murphy, K.M. Altered compensatory cytokine signaling underlies the discrepancy between Flt3−/− and Flt3l−/− mice. J. Exp. Med. 2018, 215, 1417–1435. [Google Scholar] [CrossRef] [Green Version]
- Garner, M.H.; Garner, W.H.; Gurd, F.R. Recognition of primary sequence variations among sperm whale myoglobin components with successive proteolysis procedures. J. Biol. Chem. 1974, 249, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Webb, A.I.; Dunstone, M.A.; Williamson, N.A.; Price, J.D.; de Kauwe, A.; Chen, W.; Oakley, A.; Perlmutter, P.; McCluskey, J.; Aguilar, M.I.; et al. T cell determinants incorporating beta-amino acid residues are protease resistant and remain immunogenic in vivo. J. Immunol. 2005, 15, 3810–3818. [Google Scholar] [CrossRef] [Green Version]
- Ahonen, C.L.; Doxsee, C.L.; McGurran, S.M.; Riter, T.R.; Wade, W.F.; Barth, R.J.; Vasilakos, J.P.; Noelle, R.J.; Kedl, R.M. Combined TLR and CD40 triggering induces potent CD8+ T cell expansion with variable dependence on type I IFN. J. Exp. Med. 2004, 199, 775–784. [Google Scholar] [CrossRef] [Green Version]
- Meredith, M.M.; Liu, K.; Darrasse-Jèze, G.; Kamphorst, A.O.; Schreiber, H.A.; Guermonprez, P.; Idoyaga, J.; Cheong, C.; Yao, K.H.; Niec, R.E.; et al. Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J. Exp. Med. 2012, 209, 1153–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maubant, S.; Banissi, C.; Beck, S.; Chauvat, A.; Carpentier, A.F. Adjuvant properties of cytosine-phosphateguanosine oligodeoxy-nucleotide in combination with various polycations in an ovalbumin-vaccine model. Nucleic Acid Ther. 2011, 21, 231–240. [Google Scholar] [CrossRef]
- Carpentier, A.; Metellus, P.; Ursu, R.; Zohar, S.; Lafitte, F.; Barrié, M.; Meng, Y.; Richard, M.; Parizot, C.; Laigle-Donadey, F.; et al. Intracerebral administration of CpG oligonucleotide for patients with recurrent glioblastoma, a phase II study. Neuro-Oncol. 2010, 12, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Sathe, P.; Pooley, J.; Vremec, D.; Mintern, J.; Jin, J.O.; Wu, L.; Kwak, J.Y.; Villadangos, J.A.; Shortman, K. The acquisition of antigen cross-presentation function by newly formed dendritic cells. J. Immunol. 2011, 186, 5184–5192. [Google Scholar] [CrossRef] [Green Version]
- Rizzo-Padoin, N.; Chaussard, M.; Vignal, N.; Kotula, E.; Tsoupko-Sitnikov, V.; Vaz, S.; Hontonnou, F.; Liu, W.Q.; Poyet, J.L.; Vidal, M.; et al. [18F]MEL050 as a melanin-targeted PET tracer: Fully automated radiosynthesis and comparison to 18F-FDG for the detection of pigmented melanoma in mice primary subcutaneous tumors and pulmonary metastases. Nucl. Med. Biol. 2016, 43, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Mavridi-Printezi, A.; Guernelli, M.; Menichetti, A.; Montalti, M. Bio-Applications of Multifunctional Melanin Nanoparticles: From Nanomedicine to Nanocosmetics. Nanomaterials 2020, 10, 2276. [Google Scholar] [CrossRef] [PubMed]
- Didierlaurent, A.M.; Collignon, C.; Bourguignon, P.; Wouters, S.; Fierens, K.; Fochesato, M.; Dendouga, N.; Langlet, C.; Malissen, B.; Lambrecht, B.N.; et al. Enhancement of adaptive immunity by the human vaccine adjuvant AS01 depends on activated dendritic cells. J. Immunol. 2014, 193, 1920–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langlet, C.; Tamoutounour, S.; Henri, S.; Luche, H.; Ardouin, L.; Grégoire, C.; Malissen, B.; Guilliams, M. CD64 expression distinguishes monocyte-derived and conventional dendritic cells and reveals their distinct role during intramuscular immunization. J. Immunol. 2012, 188, 1751–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Koker, S.; Van Hoecke, L.; De Beuckelaer, A.; Roose, K.; Deswarte, K.; Willart, M.A.; Bogaert, P.; Naessens, T.; De Geest, B.G.; Saelens, X.; et al. Inflammatory monocytes regulate Th1 oriented immunity to CpG adjuvanted protein vaccines through production of IL-12. Sci. Rep. 2017, 7, 5986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, C.; Vega, M.A.; Marcelo, G.; Del Valle, E.M.M. Polydopamine nanoparticles kill cancer cells. RSC Adv. 2018, 8, 36201–36208. [Google Scholar] [CrossRef] [Green Version]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [CrossRef]
- Reddy, S.T.; van der Vlies, A.J.; Simeoni, E.; Angeli, V.; Randolph, G.J.; O’Neil, C.P.; Lee, L.K.; Swartz, M.A.; Hubbell, J.A. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 2007, 25, 1159–1164. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuzzubbo, S.; Roch, B.; Darrasse-Jèze, G.; Hosten, B.; Leclercq, M.; Vignal, N.; Banissi, C.; Tartour, E.; Carpentier, A.F. Synthetic Melanin Acts as Efficient Peptide Carrier in Cancer Vaccine Strategy. Int. J. Mol. Sci. 2022, 23, 14975. https://doi.org/10.3390/ijms232314975
Cuzzubbo S, Roch B, Darrasse-Jèze G, Hosten B, Leclercq M, Vignal N, Banissi C, Tartour E, Carpentier AF. Synthetic Melanin Acts as Efficient Peptide Carrier in Cancer Vaccine Strategy. International Journal of Molecular Sciences. 2022; 23(23):14975. https://doi.org/10.3390/ijms232314975
Chicago/Turabian StyleCuzzubbo, Stefania, Benoit Roch, Guillaume Darrasse-Jèze, Benoit Hosten, Manon Leclercq, Nicolas Vignal, Claire Banissi, Eric Tartour, and Antoine F. Carpentier. 2022. "Synthetic Melanin Acts as Efficient Peptide Carrier in Cancer Vaccine Strategy" International Journal of Molecular Sciences 23, no. 23: 14975. https://doi.org/10.3390/ijms232314975
APA StyleCuzzubbo, S., Roch, B., Darrasse-Jèze, G., Hosten, B., Leclercq, M., Vignal, N., Banissi, C., Tartour, E., & Carpentier, A. F. (2022). Synthetic Melanin Acts as Efficient Peptide Carrier in Cancer Vaccine Strategy. International Journal of Molecular Sciences, 23(23), 14975. https://doi.org/10.3390/ijms232314975