

Pathogenesis of α-Synuclein in Parkinson’s Disease: From a Neuron-Glia Crosstalk Perspective


Abstract
1. Introduction
1.1. The History and Pathogenesis of Parkinson’s Disease
1.2. Role of Glia in PD

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Subcellular Impacts | Age of Onset | Related Mutations | Clinical Phenotypes |
|---|---|---|---|---|
| BST1 | Immunity system | Late onset (>70) | rs4698412 (SNPs) | Selective vulnerability of DA neurons [50]. |
| HLA | Immunity system | Late onset (>70) | HLADQA1/DQB1/DRB1 (Variant) | Increase expression of MHCII [51]. |
| PINK1 | Mitochondria | Young onset (20–40) | G309D, W437X, L347P, R246X | Early onset of unilateral tremor, bradykinesia, and rigidity [52]. |
| DJ-1 | Mitochondria | Young onset (20–40) | E64D, L166P, M26I, L10P, P158∆ | Slowly progressive Parkinsonism, occasionally with behavioral or psychiatric disturbance [53]. |
| PARK2 | Mitochondria | ~30 on average (range 16–72) | R42P, A46P, K211N, C212Y, C253Y, C289G, C441R | Parkinsonism, often presenting with dystonia, diurnal fluctuations, and sleep benefit; typically responsive to very low doses of L-DOPA [54]. |
| SCNA | Endocytosis/ Autophagy | 38–65 (duplications); 24–48 (triplications) | A30P, E46K, A53T, A53V | Progressive L-DOPA responsive Parkinsonism, associated with cognitive decline, autonomic dysfunction, and dementia; progression is more rapid in SNCA triplication cases [55]. |
| LRRK2 | Endocytosis | 50–70 (range 32–79) | G2019S, R1441C/G/H, Y1699C, I2020T, N1437H | Parkinsonism consistent with sporadic PD; dystonia, gaze palsy, and dementia occasionally develop [56]. |
| VPS35 | Endocytosis | 50–70 (range 34–68) | D620N | DA neuronal loss in SNpc and increase in α-syn levels [17]. |
| GBA | Lysosome | 45–65 | N370S, L444P, K198T, R329C | Cognitive impairment, disease severity, and motor phenotype [57]. |
| ATP13A2 | Lysosome | Young onset (range 20–40) | T517I, A746T, S282C, R980H | Spasticity, dementia, and supranuclear gaze palsy [58]. |
| TMEM175 | Lysosome | Young onset (range 20–40) | M393T, K176E, G311S | Decreased glucocerebrosidase activity, facilitated α-synaggregation [59]. |
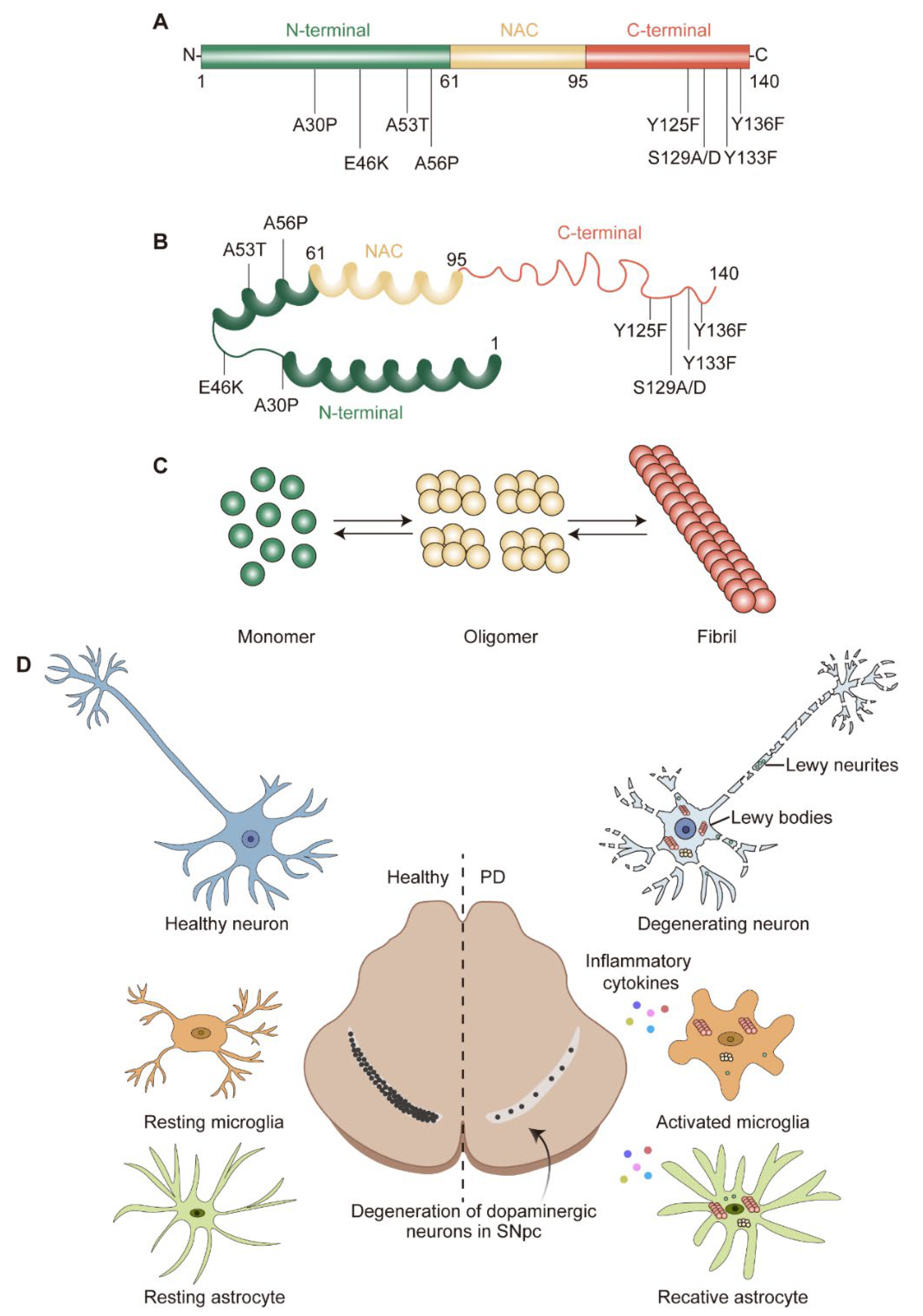
2. α-Syn Structure, Aggregation, and Degradation
2.1. α-Syn Structure and Physiological Function
2.2. α-Syn Misfolding and Aggregation
2.3. α-Syn Degradation
2.3.1. α-Syn Degradation through UPS
2.3.2. α-Syn Degradation through ALP
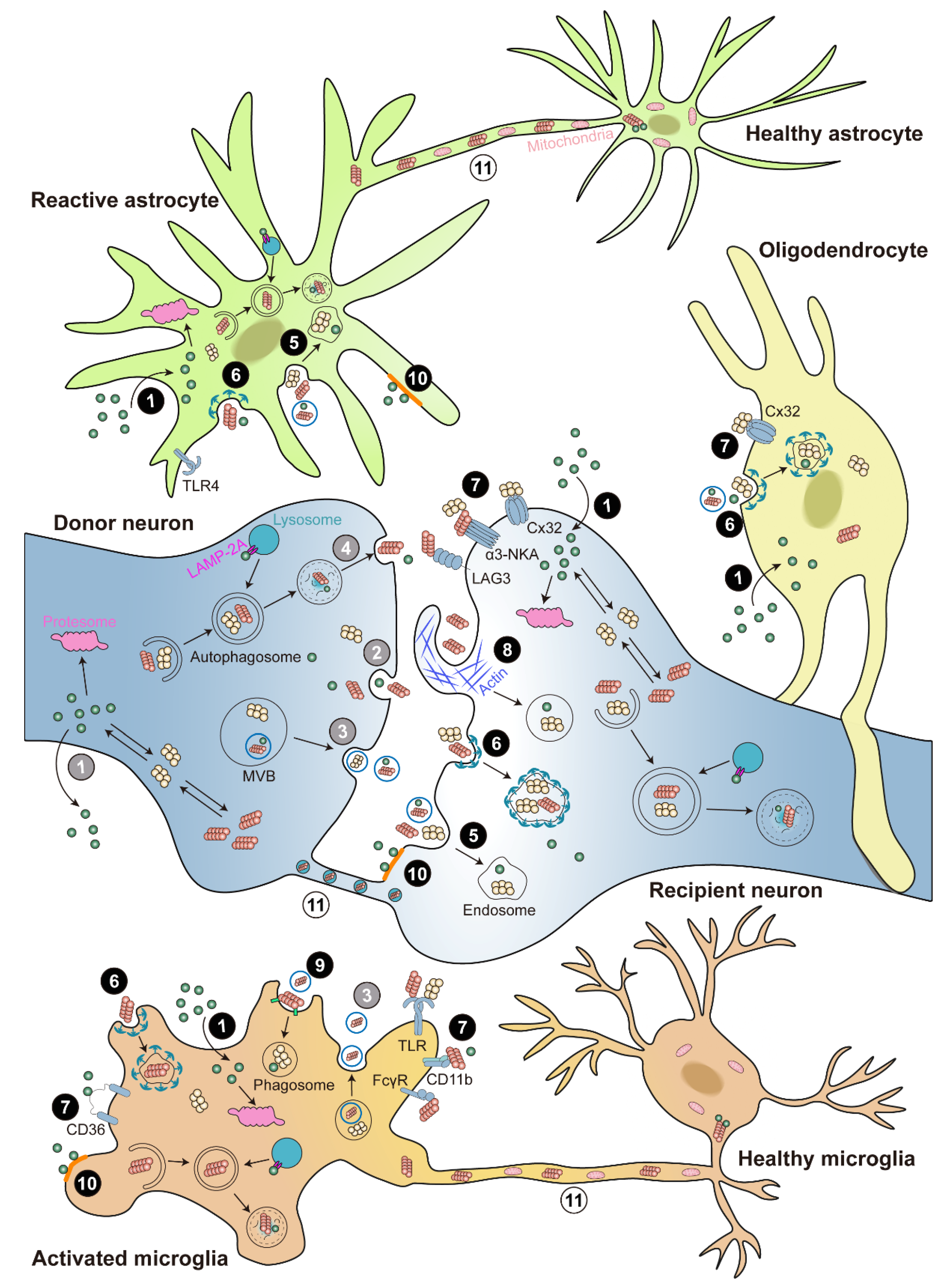
3. α-Syn Transmission and Propagation
3.1. Release of α-Syn
3.2. Uptake of α-Syn
3.3. Neuron-to-Neuron Transmission of α-Syn
3.4. Neuron-to-Glia Transmission of α-Syn
3.4.1. Neuron-to-Microglia Transmission of α-Syn
3.4.2. Neuron-to-Astrocyte Transmission of α-Syn
3.4.3. Neuron-to-Oligodendrocyte Transmission of α-Syn
3.5. An Alternative Way for α-Syn Intercellular Transmission: TNTs
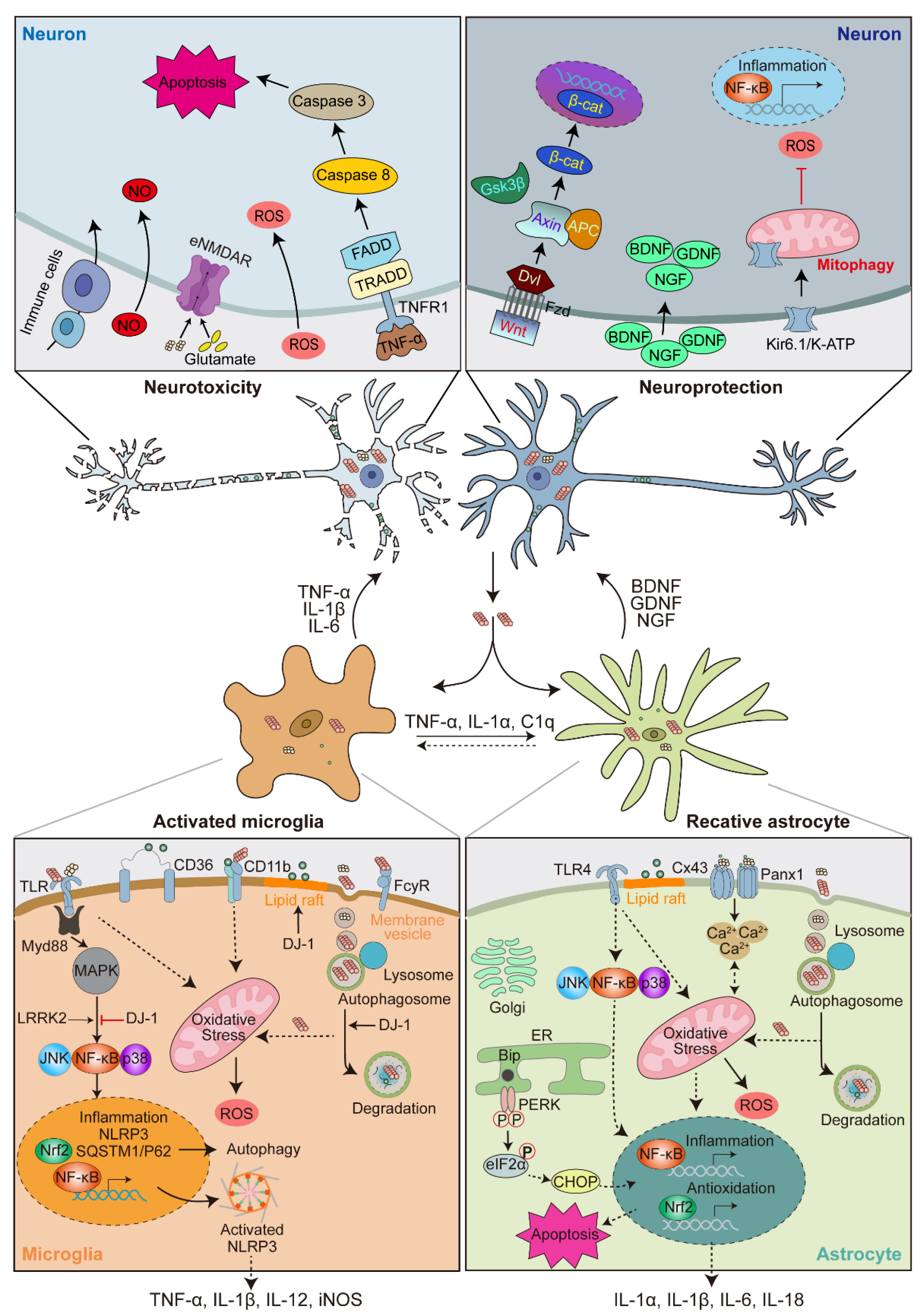
4. α-Syn-Induced Glial Activation
4.1. α-Syn-Induced Microglia Activation
4.2. α-Syn-Induced Astrocyte Activation
4.3. Microglia-Astrocyte Crosstalk
5. Feedback of Glial Activation on DA Neuron Degeneration
5.1. Neuroprotective Role of Activated Glia
5.2. Neurotoxic Role of Activated Glia
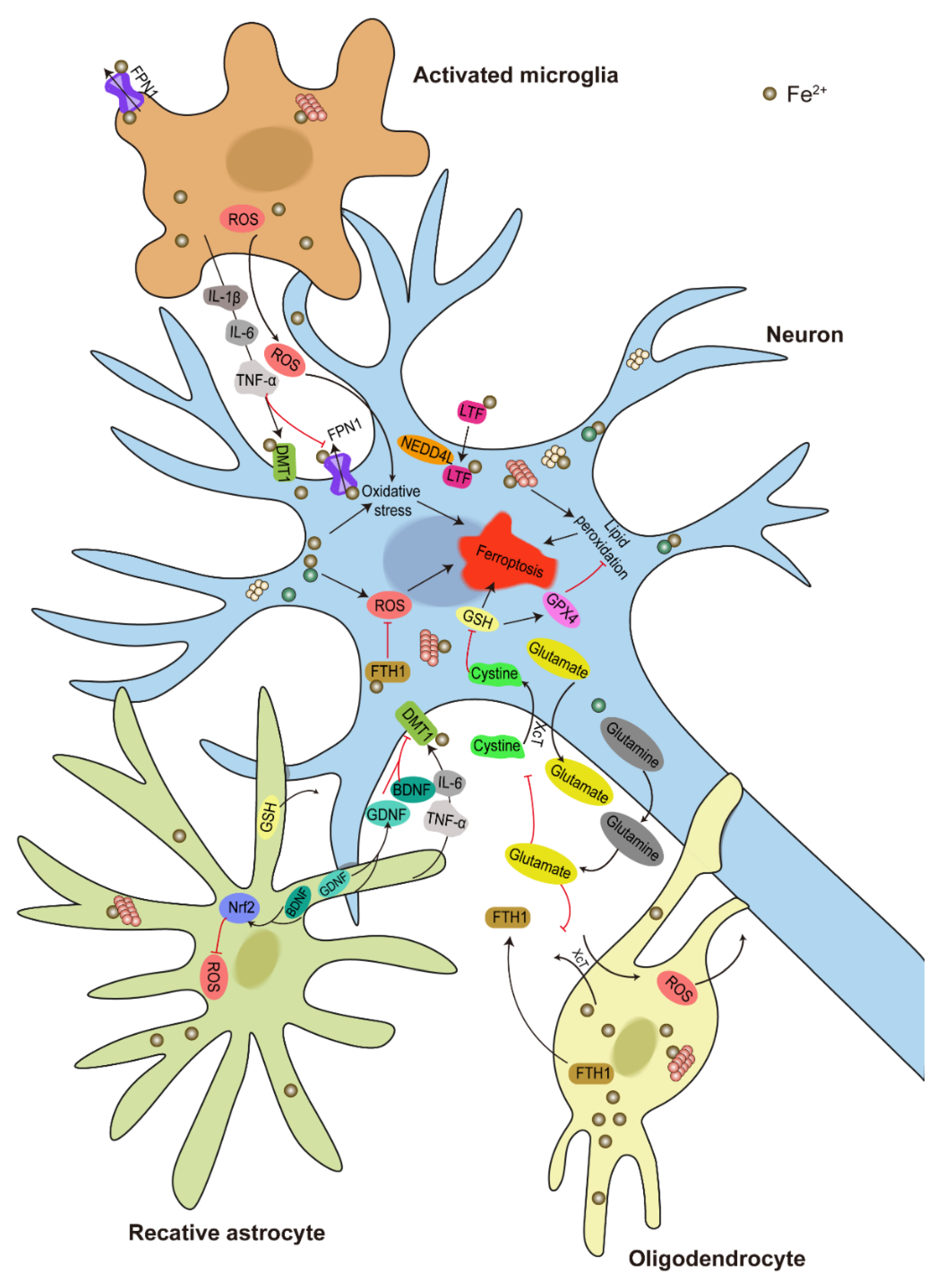
6. Ferroptosis in PD
6.1. Iron Deposition, α-Syn Aggregation, and Ferroptosis in PD
6.2. Molecular Mechanism of DA Neuron Ferroptosis
6.3. Glial Activation in Regulating DA Neuron Ferroptosis
6.4. Iron Transport between Neurons and Glia
7. Prospective of PD Therapy
7.1. Classical Treatments of PD
7.2. Glia as Potential Target for Early PD Treatment
7.3. Ferroptosis Inhibitors in PD Treatment
| Therapy | Mechanism | Drugs or Methods | Weakness or Note |
|---|---|---|---|
| Targeting α-syn | Decrease α-syn expression | Clenbuterol, Salbutamol | A potential to aggravate cardiovascular disease [374]. |
| Inhibit α-syn aggregates | NPT200-11 | Cannot pass through the BBB or might cause even serious poisoning [408]. | |
| Reduce α-syn aggregates | ABBV-0805 | Additional populations and clinical trials are still needed to determine the effects [375]. | |
| Increase α-syn degradation | Nilotinib | The lack of efficacy on motor outcomes indicated that nilotinib had no clinical benefits [409]. | |
| Reduce α-syn spreading | BIIB054 (cinpanemab) | Lack of efficacy data and PK data in participants [410]. | |
| Targeting glia | Inhibit microglia activation | Minocycline Amantadine | Lack of mechanical understanding on microglial activation [411]. |
| Promote anti-inflammation factors in microglia | Cyclin-AMP PPAR-γ agonists | ||
| Deliver chemicals specifically to astrocytes | Peptide-Based Delivery/Viral Delivery | Still unable to specifically deliver drugs to distinct astrocyte subpopulations [412]. | |
| Deliver protect genes such as GDNF to CNS | AAV delivery | Recent preclinical and clinical findings point to significant immune reactions and neuroinflammation associated with vectors [413]. | |
| Astrocyte reprogramming | Astrocyte | Teratoma formation and immune reaction [414]. | |
| Targeting ferroptosis | A medication that chelates iron and is used to treat iron overload | Deferiprone | Waiting for clinical trial in PD patients [415]. |
| Reduce ALOX12 levels and reverses paraquat-induced ferroptosis | Fer-1 | Only show effect in a small part of neurons [338]. |
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parkinson, J. An essay on the shaking palsy, 1817. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Marras, C.; Beck, J.C.; Bower, J.H.; Roberts, E.; Ritz, B.; Ross, G.W.; Abbott, R.D.; Savica, R.; Van Den Eeden, S.K.; Willis, A.W.; et al. Prevalence of Parkinson’s disease across North America. npj Park. Dis. 2018, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- de Rijk, M.C.; Breteler, M.M.; Graveland, G.A.; Ott, A.; Grobbee, D.E.; van der Meche, F.G.; Hofman, A. Prevalence of Parkinson’s disease in the elderly: The Rotterdam Study. Neurology 1995, 45, 2143–2146. [Google Scholar] [CrossRef] [PubMed]
- Barbeau, A. L-dopa therapy in Parkinson’s disease: A critical review of nine years’ experience. Can. Med. Assoc. J. 1969, 101, 59–68. [Google Scholar] [PubMed]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.G.; Burn, D.J.; Ballard, C.G.; Collerton, D.; Jaros, E.; Morris, C.M.; McLaren, A.; Perry, E.K.; Perry, R.; Piggott, M.A. Dementia with Lewy bodies. Semin. Clin. Neuropsychiatry 2003, 8, 46–57. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Braak, H.; Muller, C.M.; Rub, U.; Ackermann, H.; Bratzke, H.; de Vos, R.A.; Del Tredici, K. Pathology associated with sporadic Parkinson’s disease—Where does it end? In Parkinson’s Disease and Related Disorders; Riederer, P., Reichmann, H., Youdim, M.B.H., Gerlach, M., Eds.; Journal of Neural Transmission. Supplementa Series; Springer: Vienna, Austria, 2006; pp. 89–97. [Google Scholar]
- Cookson, M.R. The biochemistry of Parkinson’s disease. Annu. Rev. Biochem. 2005, 74, 29–52. [Google Scholar] [CrossRef]
- Filomeni, G.; Graziani, I.; De Zio, D.; Dini, L.; Centonze, D.; Rotilio, G.; Ciriolo, M.R. Neuroprotection of kaempferol by autophagy in models of rotenone-mediated acute toxicity: Possible implications for Parkinson’s disease. Neurobiol. Aging 2012, 33, 767–785. [Google Scholar] [CrossRef]
- Chen, H.; Ritz, B. The search for environmental causes of Parkinson’s disease: Moving forward. J. Park. Dis. 2018, 8, S9–S17. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Haywood, A.F.; Staveley, B.E. Parkin counteracts symptoms in a Drosophila model of Parkinson’s disease. BMC Neurosci. 2004, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat. Rev. Neurosci. 2010, 11, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.E.; Dodson, M.W.; Jiang, C.G.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martinez, A.; Beavan, M.; Gegg, M.E.; Chau, K.Y.; Whitworth, A.J.; Schapira, A.H. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci. Rep. 2016, 6, 31380. [Google Scholar] [CrossRef] [PubMed]
- Vilarino-Guell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef]
- Meulener, M.; Whitworth, A.J.; Armstrong-Gold, C.E.; Rizzu, P.; Heutink, P.; Wes, P.D.; Pallanck, L.J.; Bonini, N.M. Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson’s disease. Curr. Biol. 2005, 15, 1572–1577. [Google Scholar] [CrossRef]
- Pierce, S.; Coetzee, G.A. Parkinson’s disease-associated genetic variation is linked to quantitative expression of inflammatory genes. PLoS ONE 2017, 12, e0175882. [Google Scholar] [CrossRef]
- Kremer, M.C.; Jung, C.; Batelli, S.; Rubin, G.M.; Gaul, U. The glia of the adult, D.rosophila nervous system. Glia 2017, 65, 606–638. [Google Scholar] [CrossRef]
- Losada-Perez, M. Glia: From ‘just glue’ to essential players in complex nervous systems: A comparative view from flies to mammals. J. Neurogenet. 2018, 32, 78–91. [Google Scholar] [CrossRef]
- Fields, R.D.; Stevens-Graham, B. New insights into neuron-glia communication. Science 2002, 298, 556–562. [Google Scholar] [CrossRef]
- Wilton, D.K.; Dissing-Olesen, L.; Stevens, B. Neuron-Glia Signaling in Synapse Elimination. Annu. Rev. Neurosci. 2019, 42, 107–127. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; He, Y.; Xiao, X.; Yu, T.M.; Chen, C.; Gao, Z.; Ho, M.S. Glial cells in neuronal development: Recent advances and insights from Drosophila melanogaster. Neurosci. Bull. 2014, 30, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wang, H.; Chen, C.; Zhang, S.; Wang, M.; Senapati, B.; Li, S.; Yi, S.; Wang, L.; Zhang, M.; et al. Glia-derived temporal signals orchestrate neurogenesis in the Drosophila mushroom body. Proc. Natl. Acad. Sci. USA 2021, 118, e2020098118. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A. The mystery and magic of glia: A perspective on their roles in health and disease. Neuron 2008, 60, 430–440. [Google Scholar] [CrossRef]
- Chotard, C.; Salecker, I. Neurons and glia: Team players in axon guidance. Trends Neurosci. 2004, 27, 655–661. [Google Scholar] [CrossRef]
- Bainton, R.J.; Tsai, L.T.; Schwabe, T.; DeSalvo, M.; Gaul, U.; Heberlein, U. Moody encodes two GPCRs that regulate cocaine behaviors and blood-brain barrier permeability in Drosophila. Cell 2005, 123, 145–156. [Google Scholar] [CrossRef]
- Blauth, K.; Banerjee, S.; Bhat, M.A. Axonal ensheathment and intercellular barrier formation in Drosophila. Int. Rev. Cell Mol. Biol. 2010, 283, 93–128. [Google Scholar]
- Eroglu, C.; Barres, B.A. Regulation of synaptic connectivity by glia. Nature 2010, 468, 223–231. [Google Scholar] [CrossRef]
- Stork, T.; Bernardos, R.; Freeman, M.R. Analysis of glial cell development and function in Drosophila. Cold Spring Harb. Protoc. 2012, 2012, pdb-top067587. [Google Scholar] [CrossRef]
- Zuchero, J.B.; Barres, B.A. Glia in mammalian development and disease. Development 2015, 142, 3805–3809. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, H.; Yi, S.; Zhang, S.; Ho, M.S. A LRRK2/dLRRK-mediated lysosomal pathway that contributes to glial cell death and DA neuron survival. Traffic 2022, 23, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Bandopadhyay, R.; Kingsbury, A.E.; Cookson, M.R.; Reid, A.R.; Evans, I.M.; Hope, A.D.; Pittman, A.M.; Lashley, T.; Canet-Aviles, R.; Miller, D.W.; et al. The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain 2004, 127, 420–430. [Google Scholar] [CrossRef]
- Mullett, S.J.; Hamilton, R.L.; Hinkle, D.A. DJ-1 immunoreactivity in human brain astrocytes is dependent on infarct presence and infarct age. Neuropathology 2009, 29, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Miklossy, J.; Arai, T.; Guo, J.P.; Klegeris, A.; Yu, S.; McGeer, E.G.; McGeer, P.L. LRRK2 expression in normal and pathologic human brain and in human cell lines. J. Neuropathol. Exp. Neurol. 2006, 65, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Kam, T.I.; Hinkle, J.T.; Dawson, T.M.; Dawson, V.L. Microglia and astrocyte dysfunction in parkinson’s disease. Neurobiol. Dis. 2020, 144, 105028. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Koyanagi, S.; Kusunose, N.; Okada, R.; Wu, Z.; Tozaki-Saitoh, H.; Ukai, K.; Kohsaka, S.; Inoue, K.; Ohdo, S.; et al. The intrinsic microglial molecular clock controls synaptic strength via the circadian expression of cathepsin S. Sci. Rep. 2013, 3, 2744. [Google Scholar] [CrossRef]
- Le, W.; Wu, J.; Tang, Y. Protective Microglia and Their Regulation in Parkinson’s Disease. Front. Mol. Neurosci. 2016, 9, 89. [Google Scholar] [CrossRef]
- Allaman, I.; Belanger, M.; Magistretti, P.J. Astrocyte-neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011, 34, 76–87. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Kimelberg, H.K. Functions of mature mammalian astrocytes: A current view. Neuroscientist 2010, 16, 79–106. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.M.; Stevens, C.H. Glia: Initiators and progressors of pathology in Parkinson’s disease. Mov. Disord. 2011, 26, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Sacino, A.N.; Brooks, M.; McKinney, A.B.; Thomas, M.A.; Shaw, G.; Golde, T.E.; Giasson, B.I. Brain injection of alpha-synuclein induces multiple proteinopathies, gliosis, and a neuronal injury marker. J. Neurosci. 2014, 34, 12368–12378. [Google Scholar] [CrossRef] [PubMed]
- Hishikawa, N.; Hashizume, Y.; Yoshida, M.; Sobue, G. Widespread occurrence of argyrophilic glial inclusions in Parkinson’s disease. Neuropathol. Appl. Neurobiol. 2001, 27, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Doorn, K.J.; Moors, T.; Drukarch, B.; van de Berg, W.; Lucassen, P.J.; van Dam, A.M. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol. Commun. 2014, 2, 90. [Google Scholar] [PubMed]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1361. [Google Scholar] [CrossRef]
- Kannarkat, G.T.; Cook, D.A.; Lee, J.K.; Chang, J.; Chung, J.; Sandy, E.; Paul, K.C.; Ritz, B.; Bronstein, J.; Factor, S.A.; et al. Common Genetic Variant Association with Altered HLA Expression, Synergy with Pyrethroid Exposure, and Risk for Parkinson’s Disease: An Observational and Case-Control Study. npj Park. Dis. 2015, 1, 15002. [Google Scholar] [CrossRef]
- Hedrich, K.; Hagenah, J.; Djarmati, A.; Hiller, A.; Lohnau, T.; Lasek, K.; Grunewald, A.; Hilker, R.; Steinlechner, S.; Boston, H.; et al. Clinical spectrum of homozygous and heterozygous PINK1 mutations in a large German family with Parkinson disease: Role of a single hit? Arch. Neurol. 2006, 63, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Repici, M.; Giorgini, F. DJ-1 in Parkinson’s Disease: Clinical Insights and Therapeutic Perspectives. J. Clin. Med. 2019, 8, 1377. [Google Scholar] [CrossRef] [PubMed]
- Ueno, S.I.; Hatano, T.; Okuzumi, A.; Saiki, S.; Oji, Y.; Mori, A.; Koinuma, T.; Fujimaki, M.; Takeshige-Amano, H.; Kondo, A.; et al. Nonmercaptalbumin as an oxidative stress marker in Parkinson’s and PARK2 disease. Ann. Clin. Transl. Neur. 2020, 7, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, C.C.; Lange, J.; Forland, M.G.G.; Macleod, A.D.; Alves, G.; Maple-Grodem, J. A systematic review of associations between common SNCA variants and clinical heterogeneity in Parkinson’s disease. npj Park. Dis. 2021, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Liong, C.; Alcalay, R.N. GBA-Associated Parkinson’s Disease and Other Synucleinopathies. Curr. Neurol. Neurosci. 2018, 18, 48. [Google Scholar] [CrossRef]
- Park, J.S.; Blair, N.F.; Sue, C.M. The role of ATP13A2 in Parkinson’s disease: Clinical phenotypes and molecular mechanisms. Mov. Disord. 2015, 30, 770–779. [Google Scholar] [CrossRef]
- Li, C.Y.; Ou, R.W.; Chen, Y.P.; Gu, X.J.; Wei, Q.Q.; Cao, B.; Zhang, L.Y.; Hou, Y.B.; Liu, K.C.; Chen, X.P.; et al. Mutation analysis of TMEM family members for early-onset Parkinson’s disease in Chinese population. Neurobiol. Aging 2021, 101, e291–e299. [Google Scholar] [CrossRef]
- Venda, L.L.; Cragg, S.J.; Buchman, V.L.; Wade-Martins, R. Alpha-Synuclein and dopamine at the crossroads of Parkinson’s disease. Trends Neurosci. 2010, 33, 559–568. [Google Scholar] [CrossRef]
- Jao, C.C.; Hegde, B.G.; Chen, J.; Haworth, I.S.; Langen, R. Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. USA 2008, 105, 19666–19671. [Google Scholar] [CrossRef]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and dynamics of micelle-bound human α-synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef] [PubMed]
- Oueslati, A.; Fournier, M.; Lashuel, H.A. Role of post-translational modifications in modulating the structure, function and toxicity of α-synuclein: Implications for Parkinson’s disease pathogenesis and therapies. Prog. Brain Res. 2010, 183, 115–145. [Google Scholar]
- Jakes, R.; Spillantini, M.G.; Goedert, M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994, 345, 27–32. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of alpha-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Nakai, M.; Fujita, M.; Waragai, M.; Sugama, S.; Wei, J.; Akatsu, H.; Ohtaka-Maruyama, C.; Okado, H.; Hashimoto, M. Expression of α-synuclein, a presynaptic protein implicated in Parkinson’s disease, in erythropoietic lineage. Biochem. Biophys. Res. Commun. 2007, 358, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-W.; Yang, R.; Guo, J.-C.; Ren, H.-M.; Zha, X.-L.; Cheng, J.-S.; Cai, D.-F. Localization of α-synuclein to mitochondria within midbrain of mice. NeuroReport 2007, 18, 1543–1546. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; van Haastert, E.S.; Eikelenboom, P.; de Vos, R.A.I.; Rozemuller, J.M.; Scheper, W. Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711. [Google Scholar] [CrossRef]
- Gosavi, N.; Lee, H.J.; Lee, J.S.; Patel, S.; Lee, S.J. Golgi fragmentation occurs in the cells with prefibrillar alpha-synuclein aggregates and precedes the formation of fibrillar inclusion. J. Biol. Chem. 2002, 277, 48984–48992. [Google Scholar] [CrossRef]
- Lee, H.J.; Khoshaghideh, F.; Patel, S.; Lee, S.J. Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J. Neurosci. 2004, 24, 1888–1896. [Google Scholar] [CrossRef]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Yavich, L.; Tanila, H.; Vepsalainen, S.; Jakala, P. Role of alpha-synuclein in presynaptic dopamine recruitment. J. Neurosci. 2004, 24, 11165–11170. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Physiological functions of mitochondrial fusion. Ann. N. Y. Acad. Sci. 2010, 1201, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Belal, C.; Ameli, N.J.; El Kommos, A.; Bezalel, S.; Al’Khafaji, A.M.; Mughal, M.R.; Mattson, M.P.; Kyriazis, G.A.; Tyrberg, B.; Chan, S.L. The homocysteine-inducible endoplasmic reticulum (ER) stress protein Herp counteracts mutant alpha-synuclein-induced ER stress via the homeostatic regulation of ER-resident calcium release channel proteins. Hum. Mol. Genet. 2012, 21, 963–977. [Google Scholar] [CrossRef]
- Fan, J.; Hu, Z.; Zeng, L.; Lu, W.; Tang, X.; Zhang, J.; Li, T. Golgi apparatus and neurodegenerative diseases. Int. J. Dev. Neurosci. 2008, 26, 523–534. [Google Scholar] [CrossRef]
- Fujita, Y.; Ohama, E.; Takatama, M.; Al-Sarraj, S.; Okamoto, K. Fragmentation of Golgi apparatus of nigral neurons with α-synuclein-positive inclusions in patients with Parkinson’s disease. Acta Neuropathol. 2006, 112, 261–265. [Google Scholar] [CrossRef]
- Parsian, A.; Racette, B.; Zhang, Z.H.; Chakraverty, S.; Rundle, M.; Goate, A.; Perlmutter, J.S. Mutation, sequence analysis, and association studies of alpha-synuclein in Parkinson’s disease. Neurology 1998, 51, 1757–1759. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H. Autosomal dominant Parkinson’s disease and alpha-Synuclein. Ann. Neurol. 1998, 44, S63–S64. [Google Scholar] [CrossRef]
- Morgan, J. A seat at the table for people with Parkinson’s disease. Lancet Neurol. 2015, 14, 1077–1078. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.-Y. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Perovic, I.; Chittuluru, J.; Kaganovich, A.; Nguyen, L.T.; Liao, J.; Auclair, J.R.; Johnson, D.; Landeru, A.; Simorellis, A.K.; et al. A soluble alpha-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. USA 2011, 108, 17797–17802. [Google Scholar] [CrossRef] [PubMed]
- Fauvet, B.; Mbefo, M.K.; Fares, M.B.; Desobry, C.; Michael, S.; Ardah, M.T.; Tsika, E.; Coune, P.; Prudent, M.; Lion, N.; et al. Alpha-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 2012, 287, 15345–15364. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. Alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Sharon, R.; Bar-Joseph, I.; Frosch, M.P.; Walsh, D.M.; Hamilton, J.A.; Selkoe, D.J. The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron 2003, 37, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Waxman, E.A.; Mazzulli, J.R.; Giasson, B.I. Characterization of hydrophobic residue requirements for α-synuclein fibrillization. Biochemistry 2009, 48, 9427–9436. [Google Scholar] [CrossRef] [PubMed]
- Du, H.N.; Tang, L.; Luo, X.Y.; Li, H.T.; Hu, J.; Zhou, J.W.; Hu, H.Y. A peptide motif consisting of glycine, alanine, and valine is required for the fibrillization and cytotoxicity of human alpha-synuclein. Biochemistry 2003, 42, 8870–8878. [Google Scholar] [CrossRef]
- Narhi, L.; Wood, S.J.; Steavenson, S.; Jiang, Y.; Wu, G.M.; Anafi, D.; Kaufman, S.A.; Martin, F.; Sitney, K.; Denis, P.; et al. Both familial Parkinson’s disease mutations accelerate alpha-synuclein aggregation. J. Biol. Chem. 1999, 274, 9843–9846. [Google Scholar] [CrossRef]
- Jo, E.; Fuller, N.; Rand, R.P.; St George-Hyslop, P.; Fraser, P.E. Defective membrane interactions of familial Parkinson’s disease mutant A30P alpha-synuclein. J. Mol. Biol. 2002, 315, 799–807. [Google Scholar] [CrossRef]
- Choi, W.; Zibaee, S.; Jakes, R.; Serpell, L.C.; Davletov, B.; Crowther, R.A.; Goedert, M. Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS Lett. 2004, 576, 363–368. [Google Scholar] [CrossRef]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q.; a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Poyhonen, M.; Paetau, A. Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1–2180.e5. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Hirano, M.; Stoessl, A.J.; Imamichi, Y.; Ikeda, A.; Li, Y.; Funayama, M.; Yamada, I.; Nakamura, Y.; Sossi, V.; et al. Homozygous alpha-synuclein p.A53V in familial Parkinson’s disease. Neurobiol. Aging 2017, 57, 248.e7–248.e12. [Google Scholar] [CrossRef]
- Karpinar, D.P.; Balija, M.B.G.; Kügler, S.; Opazo, F.; Rezaei-Ghaleh, N.; Wender, N.; Kim, H.Y.; Taschenberger, G.; Falkenburger, B.H.; Heise, H. Pre-fibrillar α-synuclein variants with impaired β-structure increase neurotoxicity in Parkinson’s disease models. EMBO J. 2009, 28, 3256–3268. [Google Scholar] [CrossRef]
- Negro, A.; Brunati, A.M.; Donella-Deana, A.; Massimino, M.L.; Pinna, L.A. Multiple phosphorylation of alpha-synuclein by protein tyrosine kinase Syk prevents eosin-induced aggregation. FASEB J. 2002, 16, 210–212. [Google Scholar] [CrossRef]
- Mavroeidi, P.; Xilouri, M. Neurons and Glia Interplay in alpha-Synucleinopathies. Int. J. Mol. Sci. 2021, 22, 4994. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Sudhof, T.C. Cell Biology and Pathophysiology of alpha-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef]
- Tenreiro, S.; Eckermann, K.; Outeiro, T.F. Protein phosphorylation in neurodegeneration: Friend or foe? Front. Mol. Neurosci. 2014, 7, 42. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.; Shen, J.; Takio, K.; Iwatsubo, T. Alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Takahashi, M.; Kanuka, H.; Fujiwara, H.; Koyama, A.; Hasegawa, M.; Miura, M.; Iwatsubo, T. Phosphorylation of alpha-synuclein characteristic of synucleinopathy lesions is recapitulated in alpha-synuclein transgenic Drosophila. Neurosci. Lett. 2003, 336, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Feany, M.B. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, S.; Reimao-Pinto, M.M.; Antas, P.; Rino, J.; Wawrzycka, D.; Macedo, D.; Rosado-Ramos, R.; Amen, T.; Waiss, M.; Magalhaes, F.; et al. Phosphorylation modulates clearance of alpha-synuclein inclusions in a yeast model of Parkinson’s disease. PLoS Genet. 2014, 10, e1004302. [Google Scholar] [CrossRef]
- Ghanem, S.S.; Majbour, N.K.; Vaikath, N.N.; Ardah, M.T.; Erskine, D.; Jensen, N.M.; Fayyad, M.; Sudhakaran, I.P.; Vasili, E.; Melachroinou, K.; et al. Alpha-Synuclein phosphorylation at serine 129 occurs after initial protein deposition and inhibits seeded fibril formation and toxicity. Proc. Natl. Acad. Sci. USA 2022, 119, e2109617119. [Google Scholar] [CrossRef] [PubMed]
- Bertoncini, C.W.; Jung, Y.S.; Fernandez, C.O.; Hoyer, W.; Griesinger, C.; Jovin, T.M.; Zweckstetter, M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 1430–1435. [Google Scholar] [CrossRef] [PubMed]
- Paleologou, K.E.; Schmid, A.W.; Rospigliosi, C.C.; Kim, H.Y.; Lamberto, G.R.; Fredenburg, R.; Lansbury, P.T., Jr.; Fernandez, C.; Eliezer, D.; Zweckstetter, M.; et al. Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of alpha-synuclein. J. Biol. Chem. 2008, 283, 16895–16905. [Google Scholar] [CrossRef]
- Chen, L.; Periquet, M.; Wang, X.; Negro, A.; McLean, P.J.; Hyman, B.T.; Feany, M.B. Tyrosine and serine phosphorylation of α-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J. Clin. Investig. 2009, 119, 3257–3265. [Google Scholar] [CrossRef][Green Version]
- Tofaris, G.K.; Goedert, M.; Spillantini, M.G. The Transcellular Propagation and Intracellular Trafficking of alpha-Synuclein. Cold Spring Harb. Perspect. Med. 2017, 7, a024380. [Google Scholar] [CrossRef]
- McNaught, K.S.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C.W. Altered proteasomal function in sporadic Parkinson’s disease. Exp. Neurol. 2003, 179, 38–46. [Google Scholar] [CrossRef]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, J.H.; Paik, S.R.; Lee, S.J. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int. J. Biochem. Cell Biol. 2008, 40, 1835–1849. [Google Scholar] [CrossRef]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef]
- McNaught, K.S.P.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Shimura, H.; Schlossmacher, M.C.; Hattori, N.; Frosch, M.P.; Trockenbacher, A.; Schneider, R.; Mizuno, Y.; Kosik, K.S.; Selkoe, D.J. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: Implications for Parkinson’s disease. Science 2001, 293, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [PubMed]
- Rott, R.; Szargel, R.; Haskin, J.; Shani, V.; Shainskaya, A.; Manov, I.; Liani, E.; Avraham, E.; Engelender, S. Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 2008, 283, 3316–3328. [Google Scholar] [CrossRef]
- Lee, J.T.; Wheeler, T.C.; Li, L.; Chin, L.S. Ubiquitination of alpha-synuclein by Siah-1 promotes alpha-synuclein aggregation and apoptotic cell death. Hum. Mol. Genet. 2008, 17, 906–917. [Google Scholar] [CrossRef]
- Waxman, E.A.; Giasson, B.I. Specificity and regulation of casein kinase-mediated phosphorylation of alpha-synuclein. J. Neuropathol. Exp. Neurol. 2008, 67, 402–416. [Google Scholar] [CrossRef]
- Tofaris, G.K.; Layfield, R.; Spillantini, M.G. Alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 2001, 509, 22–26. [Google Scholar] [CrossRef]
- Machiya, Y.; Hara, S.; Arawaka, S.; Fukushima, S.; Sato, H.; Sakamoto, M.; Koyama, S.; Kato, T. Phosphorylated α-synuclein at Ser-129 is targeted to the proteasome pathway in a ubiquitin-independent manner. J. Biol. Chem. 2010, 285, 40732–40744. [Google Scholar] [CrossRef]
- Arawaka, S.; Sato, H.; Sasaki, A.; Koyama, S.; Kato, T. Mechanisms underlying extensive Ser129-phosphorylation in alpha-synuclein aggregates. Acta Neuropathol. Commun. 2017, 5, 48. [Google Scholar] [CrossRef]
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 2018, 557, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol. Aging 2010, 31, 953–968. [Google Scholar] [CrossRef] [PubMed]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Cantuti-Castelvetri, I.; Fan, Z.; Rockenstein, E.; Masliah, E.; Hyman, B.T.; McLean, P.J.; Unni, V.K. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J. Neurosci. 2011, 31, 14508–14520. [Google Scholar] [CrossRef] [PubMed]
- Bedford, L.; Hay, D.; Devoy, A.; Paine, S.; Powe, D.G.; Seth, R.; Gray, T.; Topham, I.; Fone, K.; Rezvani, N.; et al. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J. Neurosci. 2008, 28, 8189–8198. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in, Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type α-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef]
- Yu, W.H.; Dorado, B.; Figueroa, H.Y.; Wang, L.; Planel, E.; Cookson, M.R.; Clark, L.N.; Duff, K.E. Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric α-synuclein. Am. J. Pathol. 2009, 175, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Tanik, S.A.; Schultheiss, C.E.; Volpicelli-Daley, L.A.; Brunden, K.R.; Lee, V.M. Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 2013, 288, 15194–15210. [Google Scholar] [CrossRef]
- Gao, J.; Perera, G.; Bhadbhade, M.; Halliday, G.M.; Dzamko, N. Autophagy activation promotes clearance of alpha-synuclein inclusions in fibril-seeded human neural cells. J. Biol. Chem. 2019, 294, 14241–14256. [Google Scholar] [CrossRef]
- Klucken, J.; Poehler, A.-M.; Ebrahimi-Fakhari, D.; Schneider, J.; Nuber, S.; Rockenstein, E.; Schlötzer-Schrehardt, U.; Hyman, B.T.; McLean, P.J.; Masliah, E. Alpha-synuclein aggregation involves a bafilomycin A1-sensitive autophagy pathway. Autophagy 2012, 8, 754–766. [Google Scholar] [CrossRef]
- Chen, L.; Xie, Z.; Turkson, S.; Zhuang, X. A53T human alpha-synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration. J. Neurosci. 2015, 35, 890–905. [Google Scholar] [CrossRef]
- Dice, J.F.; Chiang, H.L.; Spencer, E.P.; Backer, J.M. Regulation of catabolism of microinjected ribonuclease A. Identification of residues 7-11 as the essential pentapeptide. J. Biol. Chem. 1986, 261, 6853–6859. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef]
- Huang, C.; Cheng, H.; Hao, S.; Zhou, H.; Zhang, X.; Gao, J.; Sun, Q.H.; Hu, H.; Wang, C.C. Heat shock protein 70 inhibits alpha-synuclein fibril formation via interactions with diverse intermediates. J. Mol. Biol. 2006, 364, 323–336. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Autophagy and Alpha-Synuclein: Relevance to Parkinson’s Disease and Related Synucleopathies. Mov. Disord. 2016, 31, 178–192. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Spiro, A.S.; Furuta, A.; Cooper, A.; Garner, B.; Kabuta, T.; Halliday, G.M. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov. Disord. 2015, 30, 1639–1647. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Polissidis, A.; Chrysanthou-Piterou, M.; Kloukina, I.; Stefanis, L. Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy 2016, 12, 2230–2247. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.-C.; Follenzi, A. Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. J. Clin. Investig. 2008, 118, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Rothaug, M.; Stroobants, S.; Schweizer, M.; Peters, J.; Zunke, F.; Allerding, M.; D’Hooge, R.; Saftig, P.; Blanz, J. LAMP-2 deficiency leads to hippocampal dysfunction but normal clearance of neuronal substrates of chaperone-mediated autophagy in a mouse model for Danon disease. Acta Neuropathol. Commun. 2015, 3, 6. [Google Scholar] [CrossRef]
- Prusiner, S.B. Molecular biology of prion diseases. Science 1991, 252, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Bjorklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Recasens, A.; Dehay, B.; Bove, J.; Carballo-Carbajal, I.; Dovero, S.; Perez-Villalba, A.; Fernagut, P.O.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from, Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2014, 75, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils seed the formation of, L.ewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef]
- Paumier, K.; Luk, K.C.; Manfredsson, F.P.; Kanaan, N.M.; Lipton, J.W.; Collier, T.; Steece-Collier, K.; Kemp, C.J.; Celano, S.; Schulz, E.; et al. Intrastriatal injection of pre-formed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol. Dis. 2015, 82, 185–199. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef]
- Borghi, R.; Marchese, R.; Negro, A.; Marinelli, L.; Forloni, G.; Zaccheo, D.; Abbruzzese, G.; Tabaton, M. Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neurosci. Lett. 2000, 287, 65–67. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.; Salem, S.A.; Paleologou, K.E.; Cooper, L.J.; Fullwood, N.J.; Gibson, M.J.; Curran, M.D.; Court, J.A.; Mann, D.M.; Ikeda, S.; et al. Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003, 17, 1945–1947. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Patel, S.; Lee, S.J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.Y.; Park, S.M.; Lee, C.H.; Um, J.W.; Lee, H.J.; Kim, J.; Oh, Y.J.; Lee, S.T.; Paik, S.R.; Chung, K.C. Proteolytic cleavage of extracellular secreted {alpha}-synuclein via matrix metalloproteinases. J. Biol. Chem. 2005, 280, 25216–25224. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef]
- Shi, M.; Liu, C.; Cook, T.J.; Bullock, K.M.; Zhao, Y.; Ginghina, C.; Li, Y.; Aro, P.; Dator, R.; He, C.; et al. Plasma exosomal alpha-synuclein is likely CNS-derived and increased in Parkinson’s disease. Acta Neuropathol. 2014, 128, 639–650. [Google Scholar] [CrossRef]
- Jang, A.; Lee, H.J.; Suk, J.E.; Jung, J.W.; Kim, K.P.; Lee, S.J. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J. Neurochem. 2010, 113, 1263–1274. [Google Scholar]
- Ejlerskov, P.; Rasmussen, I.; Nielsen, T.T.; Bergstrom, A.L.; Tohyama, Y.; Jensen, P.H.; Vilhardt, F. Tubulin polymerization-promoting protein (TPPP/p25alpha) promotes unconventional secretion of alpha-synuclein through exophagy by impairing autophagosome-lysosome fusion. J. Biol. Chem. 2013, 288, 17313–17335. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, J.P.; Shi, M.; Quinn, T.; Bradner, J.; Beyer, R.; Chen, S.; Zhang, J. Rab11a and HSP90 regulate recycling of extracellular alpha-synuclein. J. Neurosci. 2009, 29, 1480–1485. [Google Scholar] [CrossRef]
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Oikawa, T.; Arai, T.; Akiyama, H.; Mann, D.; Hasegawa, M. Prion-like spreading of pathological alpha-synuclein in brain. Brain 2013, 136, 1128–1138. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Luk, K.C.; Lee, V.M. Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc. 2014, 9, 2135–2146. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, S.J. Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun. 2008, 372, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.; Angot, E.; Bergstrom, A.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. Alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Investig. 2011, 121, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Konno, M.; Hasegawa, T.; Baba, T.; Miura, E.; Sugeno, N.; Kikuchi, A.; Fiesel, F.C.; Sasaki, T.; Aoki, M.; Itoyama, Y.; et al. Suppression of dynamin GTPase decreases alpha-synuclein uptake by neuronal and oligodendroglial cells: A potent therapeutic target for synucleinopathy. Mol. Neurodegener. 2012, 7, 38. [Google Scholar] [CrossRef]
- Song, L.; He, Y.; Ou, J.; Zhao, Y.; Li, R.; Cheng, J.; Lin, C.H.; Ho, M.S. Auxilin Underlies Progressive Locomotor Deficits and Dopaminergic Neuron Loss in a Drosophila Model of Parkinson’s Disease. Cell Rep. 2017, 18, 1132–1143. [Google Scholar] [CrossRef]
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.H.; et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353, aah3374. [Google Scholar] [CrossRef]
- Shrivastava, A.N.; Redeker, V.; Fritz, N.; Pieri, L.; Almeida, L.G.; Spolidoro, M.; Liebmann, T.; Bousset, L.; Renner, M.; Lena, C.; et al. Alpha-synuclein assemblies sequester neuronal alpha3-Na+/K+-ATPase and impair Na+ gradient. EMBO J. 2015, 34, 2408–2423. [Google Scholar] [CrossRef]
- Reyes, J.F.; Sackmann, C.; Hoffmann, A.; Svenningsson, P.; Winkler, J.; Ingelsson, M.; Hallbeck, M. Binding of alpha-synuclein oligomers to Cx32 facilitates protein uptake and transfer in neurons and oligodendrocytes. Acta Neuropathol. 2019, 138, 23–47. [Google Scholar] [CrossRef]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.P.; Gleeson, P.A. Macropinocytosis: An endocytic pathway for internalising large gulps. Immunol. Cell Biol. 2011, 89, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Moll, T.; Marshall, J.N.G.; Soni, N.; Zhang, S.; Cooper-Knock, J.; Shaw, P.J. Membrane lipid raft homeostasis is directly linked to neurodegeneration. Essays Biochem. 2021, 65, 999–1011. [Google Scholar]
- Fortin, D.; Troyer, M.D.; Nakamura, K.; Kubo, S.; Anthony, M.D.; Edwards, R.H. Lipid rafts mediate the synaptic localization of alpha-synuclein. J. Neurosci. 2004, 24, 6715–6723. [Google Scholar] [CrossRef] [PubMed]
- Kubo, S.; Nemani, V.M.; Chalkley, R.J.; Anthony, M.D.; Hattori, N.; Mizuno, Y.; Edwards, R.H.; Fortin, D.L. A combinatorial code for the interaction of alpha-synuclein with membranes. J. Biol. Chem. 2005, 280, 31664–31672. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, Y.; Wang, Y.; Fong, H.; Murray, T.M.; Zhang, J. Identification of proteins involved in microglial endocytosis of alpha-synuclein. J. Proteome Res. 2007, 6, 3614–3627. [Google Scholar] [CrossRef]
- Bliederhaeuser, C.; Grozdanov, V.; Speidel, A.; Zondler, L.; Ruf, W.P.; Bayer, H.; Kiechle, M.; Feiler, M.S.; Freischmidt, A.; Brenner, D.; et al. Age-dependent defects of alpha-synuclein oligomer uptake in microglia and monocytes. Acta Neuropathol. 2016, 131, 379–391. [Google Scholar] [CrossRef]
- Ho, M.S. Microglia in Parkinson’s Disease. In Neuroglia in Neurodegenerative Diseases; Verkhratsky, A., Ho, M.S., Zorec, R., Parpura, V., Eds.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; Volume 1175, pp. 335–353. [Google Scholar]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef]
- Park, J.Y.; Kim, K.S.; Lee, S.B.; Ryu, J.S.; Chung, K.; Choo, Y.; Jou, I.; Kim, J.; Park, S.M. On the mechanism of internalization of alpha-synuclein into microglia: Roles of ganglioside, G.M1 and lipid raft. J. Neurochem. 2009, 110, 400–411. [Google Scholar] [CrossRef]
- Nash, Y.; Schmukler, E.; Trudler, D.; Pinkas-Kramarski, R.; Frenkel, D. DJ-1 deficiency impairs autophagy and reduces alpha-synuclein phagocytosis by microglia. J. Neurochem. 2017, 143, 584–594. [Google Scholar] [CrossRef]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chu, C.H.; Stewart, T.; Ginghina, C.; Wang, Y.; Nie, H.; Guo, M.; Wilson, B.; Hong, J.S.; Zhang, J. Alpha-Synuclein, a chemoattractant, directs microglial migration via H2O2-dependent Lyn phosphorylation. Proc. Natl. Acad. Sci. USA 2015, 112, E1926–E1935. [Google Scholar] [PubMed]
- Hou, L.; Bao, X.; Zang, C.; Yang, H.; Sun, F.; Che, Y.; Wu, X.; Li, S.; Zhang, D.; Wang, Q. Integrin CD11b mediates alpha-synuclein-induced activation of NADPH oxidase through a Rho-dependent pathway. Redox Biol. 2018, 14, 600–608. [Google Scholar] [CrossRef]
- Hou, L.; Qu, X.; Qiu, X.; Huang, R.; Zhao, X.; Wang, Q. Integrin CD11b mediates locus coeruleus noradrenergic neurodegeneration in a mouse Parkinson’s disease model. J. Neuroinflamm. 2020, 17, 148. [Google Scholar] [CrossRef]
- Kim, S.; Cho, S.H.; Kim, K.Y.; Shin, K.Y.; Kim, H.S.; Park, C.H.; Chang, K.A.; Lee, S.H.; Cho, D.; Suh, Y.H. Alpha-synuclein induces migration of BV-2 microglial cells by up-regulation of CD44 and MT1-MMP. J. Neurochem. 2009, 109, 1483–1496. [Google Scholar] [CrossRef]
- Lee, E.J.; Woo, M.S.; Moon, P.G.; Baek, M.C.; Choi, I.Y.; Kim, W.K.; Junn, E.; Kim, H.S. Alpha-synuclein activates microglia by inducing the expressions of matrix metalloproteinases and the subsequent activation of protease-activated receptor-1. J. Immunol. 2010, 185, 615–623. [Google Scholar] [CrossRef]
- Cao, S.; Standaert, D.G.; Harms, A.S. The gamma chain subunit of Fc receptors is required for alpha-synuclein-induced pro-inflammatory signaling in microglia. J. Neuroinflamm. 2012, 9, 259. [Google Scholar] [CrossRef]
- Zhang, W.; Dallas, S.; Zhang, D.; Guo, J.P.; Pang, H.; Wilson, B.; Miller, D.S.; Chen, B.; Zhang, W.; McGeer, P.L.; et al. Microglial PHOX and Mac-1 are essential to the enhanced dopaminergic neurodegeneration elicited by A30P and A53T mutant alpha-synuclein. Glia 2007, 55, 1178–1188. [Google Scholar] [CrossRef]
- Jiang, T.; Hoekstra, J.; Heng, X.; Kang, W.; Ding, J.; Liu, J.; Chen, S.; Zhang, J. P2X7 receptor is critical in alpha-synuclein—Mediated microglial NADPH oxidase activation. Neurobiol. Aging 2015, 36, 2304–2318. [Google Scholar] [CrossRef]
- Panicker, N.; Sarkar, S.; Harischandra, D.S.; Neal, M.; Kam, T.I.; Jin, H.; Saminathan, H.; Langley, M.; Charli, A.; Samidurai, M.; et al. Fyn kinase regulates misfolded alpha-synuclein uptake and NLRP3 inflammasome activation in microglia. J. Exp. Med. 2019, 216, 1411–1430. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.P.; Chartrand, K.; Reynolds, A.; Vitvitsky, V.; Banerjee, R.; Gendelman, H.E. Ion channel blockade attenuates aggregated alpha synuclein induction of microglial reactive oxygen species: Relevance for the pathogenesis of Parkinson’s disease. J. Neurochem. 2007, 100, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released alpha-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef] [PubMed]
- Bussi, C.; Peralta Ramos, J.; Arroyo, D.S.; Gallea, J.I.; Ronchi, P.; Kolovou, A.; Wang, J.M.; Florey, O.; Celej, M.S.; Schwab, Y.; et al. Alpha-synuclein fibrils recruit TBK1 and OPTN to lysosomal damage sites and induce autophagy in microglial cells. J. Cell Sci. 2018, 131, jcs226241. [Google Scholar] [CrossRef]
- Guo, M.; Wang, J.; Zhao, Y.; Feng, Y.; Han, S.; Dong, Q.; Cui, M.; Tieu, K. Microglial exosomes facilitate alpha-synuclein transmission in Parkinson’s disease. Brain 2020, 143, 1476–1497. [Google Scholar] [CrossRef]
- Chang, C.; Lang, H.; Geng, N.; Wang, J.; Li, N.; Wang, X. Exosomes of BV-2 cells induced by alpha-synuclein: Important mediator of neurodegeneration in PD. Neurosci. Lett. 2013, 548, 190–195. [Google Scholar] [CrossRef]
- Lee, H.J.; Suk, J.E.; Patrick, C.; Bae, E.J.; Cho, J.H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.J. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef]
- Cavaliere, F.; Cerf, L.; Dehay, B.; Ramos-Gonzalez, P.; De Giorgi, F.; Bourdenx, M.; Bessede, A.; Obeso, J.A.; Matute, C.; Ichas, F.; et al. In vitro alpha-synuclein neurotoxicity and spreading among neurons and astrocytes using Lewy body extracts from Parkinson disease brains. Neurobiol. Dis. 2017, 103, 101–112. [Google Scholar] [CrossRef]
- Loria, F.; Vargas, J.Y.; Bousset, L.; Syan, S.; Salles, A.; Melki, R.; Zurzolo, C. Alpha-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol. 2017, 134, 789–808. [Google Scholar] [CrossRef]
- di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Munoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-specific iPSC-derived astrocytes contribute to non-cell-autonomous neurodegeneration in Parkinson’s disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef]
- Hua, J.; Yin, N.; Xu, S.; Chen, Q.; Tao, T.; Zhang, J.; Ding, J.; Fan, Y.; Hu, G. Enhancing the Astrocytic Clearance of Extracellular alpha-Synuclein Aggregates by Ginkgolides Attenuates Neural Cell Injury. Cell. Mol. Neurobiol. 2019, 39, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, Z.A.; Brooks, M.M.T.; Hudson, V., 3rd; Rutherford, N.J.; Golde, T.E.; Giasson, B.I.; Chakrabarty, P. Intrastriatal injection of alpha-synuclein can lead to widespread synucleinopathy independent of neuroanatomic connectivity. Mol. Neurodegener. 2017, 12, 40. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, Z.A.; Xia, Y.; Funk, C.; Riffe, C.J.; Rutherford, N.J.; Ceballos Diaz, C.; Sacino, A.N.; Price, N.D.; Golde, T.E.; Giasson, B.I.; et al. Motor neuron loss and neuroinflammation in a model of alpha-synuclein-induced neurodegeneration. Neurobiol. Dis. 2018, 120, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Vargas, M.R.; Johnson, D.A.; Johnson, J.A. Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J. Neurosci. 2012, 32, 17775–17787. [Google Scholar] [CrossRef] [PubMed]
- Erustes, A.G.; Stefani, F.Y.; Terashima, J.Y.; Stilhano, R.S.; Monteforte, P.T.; da Silva Pereira, G.J.; Han, S.W.; Calgarotto, A.K.; Hsu, Y.T.; Ureshino, R.P.; et al. Overexpression of alpha-synuclein in an astrocyte cell line promotes autophagy inhibition and apoptosis. J. Neurosci. Res. 2018, 96, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; McGeer, P.L.; McGeer, E.G. Lewy bodies in Parkinson’s disease are recognized by antibodies to complement proteins. Acta Neuropathol. 1992, 84, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Hayashi, S.; Yoshimoto, M.; Kudo, H.; Takahashi, H. NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 2000, 99, 14–20. [Google Scholar] [CrossRef]
- Takagi, S.; Hayakawa, N.; Kimoto, H.; Kato, H.; Araki, T. Damage to oligodendrocytes in the striatum after MPTP neurotoxicity in mice. J. Neural Transm. 2007, 114, 1553–1557. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Langston, J.W.; Forno, L.S.; Tetrud, J.; Reeves, A.G.; Kaplan, J.A.; Karluk, D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann. Neurol. 1999, 46, 598–605. [Google Scholar] [CrossRef]
- Bove, J.; Prou, D.; Perier, C.; Przedborski, S. Toxin-induced models of Parkinson’s disease. NeuroRx 2005, 2, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.H.; Halliday, G.M.; Kim, W.S. Exploring myelin dysfunction in multiple system atrophy. Exp. Neurobiol. 2014, 23, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Kisos, H.; Pukass, K.; Ben-Hur, T.; Richter-Landsberg, C.; Sharon, R. Increased neuronal alpha-synuclein pathology associates with its accumulation in oligodendrocytes in mice modeling alpha-synucleinopathies. PLoS ONE 2012, 7, e46817. [Google Scholar] [CrossRef] [PubMed]
- Pukass, K.; Richter-Landsberg, C. Oxidative stress promotes uptake, accumulation, and oligomerization of extracellular alpha-synuclein in oligodendrocytes. J. Mol. Neurosci. 2014, 52, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.F.; Rey, N.L.; Bousset, L.; Melki, R.; Brundin, P.; Angot, E. Alpha-synuclein transfers from neurons to oligodendrocytes. Glia 2014, 62, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Gurke, S.; Barroso, J.F.; Gerdes, H.H. The art of cellular communication: Tunneling nanotubes bridge the divide. Histochem. Cell Biol. 2008, 129, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef]
- Zhu, D.; Tan, K.S.; Zhang, X.; Sun, A.Y.; Sun, G.Y.; Lee, J.C. Hydrogen peroxide alters membrane and cytoskeleton properties and increases intercellular connections in astrocytes. J. Cell Sci. 2005, 118, 3695–3703. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Y.; Zhang, J.; Tu, J.; Wang, X.J.; Su, X.D.; Wang, L.; Zhang, Y. Tunneling-nanotube direction determination in neurons and astrocytes. Cell Death Dis. 2012, 3, e438. [Google Scholar] [CrossRef]
- Abounit, S.; Bousset, L.; Loria, F.; Zhu, S.; de, C. haumont, F.; Pieri, L.; Olivo-Marin, J.C.; Melki, R.; Zurzolo, C. Tunneling nanotubes spread fibrillar alpha-synuclein by intercellular trafficking of lysosomes. EMBO J. 2016, 35, 2120–2138. [Google Scholar] [CrossRef]
- Dieriks, B.V.; Park, T.I.; Fourie, C.; Faull, R.L.; Dragunow, M.; Curtis, M.A. Alpha-synuclein transfer through tunneling nanotubes occurs in, S.H-SY5Y cells and primary brain pericytes from Parkinson’s disease patients. Sci. Rep. 2017, 7, 42984. [Google Scholar] [CrossRef] [PubMed]
- Rostami, J.; Holmqvist, S.; Lindstrom, V.; Sigvardson, J.; Westermark, G.T.; Ingelsson, M.; Bergstrom, J.; Roybon, L.; Erlandsson, A. Human Astrocytes Transfer Aggregated Alpha-Synuclein via Tunneling Nanotubes. J. Neurosci. 2017, 37, 11835–11853. [Google Scholar] [CrossRef] [PubMed]
- Scheiblich, H.; Dansokho, C.; Mercan, D.; Schmidt, S.V.; Bousset, L.; Wischhof, L.; Eikens, F.; Odainic, A.; Spitzer, J.; Griep, A.; et al. Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell 2021, 184, 5089–5106. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Guajardo, V.; Febbraro, F.; Kirik, D.; Romero-Ramos, M. Microglia acquire distinct activation profiles depending on the degree of alpha-synuclein neuropathology in a rAAV based model of Parkinson’s disease. PLoS ONE 2010, 5, e8784. [Google Scholar] [CrossRef] [PubMed]
- Barkholt, P.; Sanchez-Guajardo, V.; Kirik, D.; Romero-Ramos, M. Long-term polarization of microglia upon alpha-synuclein overexpression in nonhuman primates. Neuroscience 2012, 208, 85–96. [Google Scholar] [CrossRef]
- Watson, M.B.; Richter, F.; Lee, S.K.; Gabby, L.; Wu, J.; Masliah, E.; Effros, R.B.; Chesselet, M.F. Regionally-specific microglial activation in young mice over-expressing human wildtype alpha-synuclein. Exp. Neurol. 2012, 237, 318–334. [Google Scholar] [CrossRef]
- Hoenen, C.; Gustin, A.; Birck, C.; Kirchmeyer, M.; Beaume, N.; Felten, P.; Grandbarbe, L.; Heuschling, P.; Heurtaux, T. Alpha-Synuclein Proteins Promote Pro-Inflammatory Cascades in Microglia: Stronger Effects of the A53T Mutant. PLoS ONE 2016, 11, e0162717. [Google Scholar] [CrossRef]
- Gu, X.L.; Long, C.X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef]
- Su, X.; Federoff, H.J.; Maguire-Zeiss, K.A. Mutant alpha-synuclein overexpression mediates early proinflammatory activity. Neurotox. Res. 2009, 16, 238–254. [Google Scholar] [CrossRef]
- Sherer, T.B.; Betarbet, R.; Kim, J.H.; Greenamyre, J.T. Selective microglial activation in the rat rotenone model of Parkinson’s disease. Neurosci. Lett. 2003, 341, 87–90. [Google Scholar] [CrossRef]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Rey, N.L.; Tyson, T.; Esquibel, C.; Meyerdirk, L.; Schulz, E.; Pierce, S.; Burmeister, A.R.; Madaj, Z.; Steiner, J.A.; et al. Microglia affect alpha-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Mol. Neurodegener. 2019, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Theodore, S.; Cao, S.; McLean, P.J.; Standaert, D.G. Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Y.; Koprich, J.B.; Siddiqi, H.; Isacson, O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J. Neurosci. 2009, 29, 3365–3373. [Google Scholar] [CrossRef]
- Shavali, S.; Combs, C.K.; Ebadi, M. Reactive macrophages increase oxidative stress and alpha-synuclein nitration during death of dopaminergic neuronal cells in co-culture: Relevance to Parkinson’s disease. Neurochem. Res. 2006, 31, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.M.; Kotzbauer, P.T.; Uryu, K.; Leight, S.; Trojanowski, J.Q.; Lee, V.M. Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J. Neurosci. 2008, 28, 7687–7698. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease—A double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef]
- Liu, C.Y.; Wang, X.; Liu, C.; Zhang, H.L. Pharmacological Targeting of Microglial Activation: New Therapeutic Approach. Front. Cell. Neurosci. 2019, 13, 514. [Google Scholar] [CrossRef]
- Boche, D.; Perry, V.H.; Nicoll, J.A. Review: Activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef]
- Pisanu, A.; Lecca, D.; Mulas, G.; Wardas, J.; Simbula, G.; Spiga, S.; Carta, A.R. Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-gamma agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson’s disease. Neurobiol. Dis. 2014, 71, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Zhao, Y.F. Aging modulates microglia phenotypes in neuroinflammation of MPTP-PD mice. Exp. Gerontol. 2018, 111, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Harms, A.S.; Delic, V.; Thome, A.D.; Bryant, N.; Liu, Z.; Chandra, S.; Jurkuvenaite, A.; West, A.B. Alpha-Synuclein fibrils recruit peripheral immune cells in the rat brain prior to neurodegeneration. Acta Neuropathol. Commun. 2017, 5, 85. [Google Scholar] [CrossRef]
- Wilms, H.; Rosenstiel, P.; Romero-Ramos, M.; Arlt, A.; Schafer, H.; Seegert, D.; Kahle, P.J.; Odoy, S.; Claasen, J.H.; Holzknecht, C.; et al. Suppression of MAP kinases inhibits microglial activation and attenuates neuronal cell death induced by alpha-synuclein protofibrils. Int. J. Immunopathol. Pharmacol. 2009, 22, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Klegeris, A.; Pelech, S.; Giasson, B.I.; Maguire, J.; Zhang, H.; McGeer, E.G.; McGeer, P.L. Alpha-synuclein activates stress signaling protein kinases in, T.HP-1 cells and microglia. Neurobiol. Aging 2008, 29, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.G.; Beraud, D.; Davenport, C.; Cheng, K.; Yin, H.; Maguire-Zeiss, K.A. Activation of MyD88-dependent TLR1/2 signaling by misfolded alpha-synuclein, a protein linked to neurodegenerative disorders. Sci. Signal. 2015, 8, ra45. [Google Scholar] [CrossRef]
- Jin, J.; Shie, F.S.; Liu, J.; Wang, Y.; Davis, J.; Schantz, A.M.; Montine, K.S.; Montine, T.J.; Zhang, J. Prostaglandin E2 receptor subtype 2 (EP2) regulates microglial activation and associated neurotoxicity induced by aggregated alpha-synuclein. J. Neuroinflamm. 2007, 4, 2. [Google Scholar] [CrossRef]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, M.; Du, R.H.; Qiao, C.; Jiang, C.Y.; Zhang, K.Z.; Ding, J.H.; Hu, G. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol. Neurodegener. 2016, 11, 28. [Google Scholar] [CrossRef]
- Li, Y.; Xia, Y.; Yin, S.; Wan, F.; Hu, J.; Kou, L.; Sun, Y.; Wu, J.; Zhou, Q.; Huang, J.; et al. Targeting Microglial alpha-Synuclein/TLRs/NF-kappaB/NLRP3 Inflammasome Axis in Parkinson’s Disease. Front. Immunol. 2021, 12, 719807. [Google Scholar] [CrossRef]
- Moehle, M.S.; Webber, P.J.; Tse, T.; Sukar, N.; Standaert, D.G.; DeSilva, T.M.; Cowell, R.M.; West, A.B. LRRK2 inhibition attenuates microglial inflammatory responses. J. Neurosci. 2012, 32, 1602–1611. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Yang, M.; Choi, D.; Kim, J.H.; Kim, H.S.; Seol, W.; Choi, S.; Jou, I.; Kim, E.Y.; Joe, E.H. Impaired inflammatory responses in murine Lrrk2-knockdown brain microglia. PLoS ONE 2012, 7, e34693. [Google Scholar] [CrossRef] [PubMed]
- Daher, J.P.; Volpicelli-Daley, L.A.; Blackburn, J.P.; Moehle, M.S.; West, A.B. Abrogation of alpha-synuclein-mediated dopaminergic neurodegeneration in, L.RRK2-deficient rats. Proc. Natl. Acad. Sci. USA 2014, 111, 9289–9294. [Google Scholar] [CrossRef]
- Beraud, D.; Hathaway, H.A.; Trecki, J.; Chasovskikh, S.; Johnson, D.A.; Johnson, J.A.; Federoff, H.J.; Shimoji, M.; Mhyre, T.R.; Maguire-Zeiss, K.A. Microglial activation and antioxidant responses induced by the Parkinson’s disease protein alpha-synuclein. J. Neuroimmune. Pharmacol. 2013, 8, 94–117. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.G.; Sahin, G.; Rabano, A.; Kirik, D.; Cuadrado, A. Alpha-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 3173–3192. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, R.; Avila, M.; Gonzalez, J.; El-Bacha, R.S.; Baez, E.; Garcia-Segura, L.M.; Jurado Coronel, J.C.; Capani, F.; Cardona-Gomez, G.P.; Barreto, G.E. Astrocytic modulation of blood brain barrier: Perspectives on Parkinson’s disease. Front. Cell. Neurosci. 2014, 8, 211. [Google Scholar] [CrossRef]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef]
- Song, Y.J.; Halliday, G.M.; Holton, J.L.; Lashley, T.; O’Sullivan, S.S.; McCann, H.; Lees, A.J.; Ozawa, T.; Williams, D.R.; Lockhart, P.J.; et al. Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. J. Neuropathol. Exp. Neurol. 2009, 68, 1073–1083. [Google Scholar] [CrossRef]
- Miklossy, J.; Doudet, D.D.; Schwab, C.; Yu, S.; McGeer, E.G.; McGeer, P.L. Role of ICAM-1 in persisting inflammation in Parkinson disease and MPTP monkeys. Exp. Neurol. 2006, 197, 275–283. [Google Scholar] [CrossRef]
- Henning, J.; Strauss, U.; Wree, A.; Gimsa, J.; Rolfs, A.; Benecke, R.; Gimsa, U. Differential astroglial activation in 6-hydroxydopamine models of Parkinson’s disease. Neurosci. Res. 2008, 62, 246–253. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M. Neuron-Astrocyte Interactions in Parkinson’s Disease. Cells 2020, 9, 2623. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Klegeris, A.; Giasson, B.I.; Zhang, H.; Maguire, J.; Pelech, S.; McGeer, P.L. Alpha-synuclein and its disease-causing mutants induce ICAM-1 and IL-6 in human astrocytes and astrocytoma cells. FASEB J. 2006, 20, 2000–2008. [Google Scholar] [CrossRef] [PubMed]
- Chavarria, C.; Rodriguez-Bottero, S.; Quijano, C.; Cassina, P.; Souza, J.M. Impact of monomeric, oligomeric and fibrillar alpha-synuclein on astrocyte reactivity and toxicity to neurons. Biochem. J. 2018, 475, 3153–3169. [Google Scholar] [CrossRef] [PubMed]
- Rannikko, E.H.; Weber, S.S.; Kahle, P.J. Exogenous alpha-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 2015, 16, 57. [Google Scholar] [CrossRef]
- Diaz, E.F.; Labra, V.C.; Alvear, T.F.; Mellado, L.A.; Inostroza, C.A.; Oyarzun, J.E.; Salgado, N.; Quintanilla, R.A.; Orellana, J.A. Connexin 43 hemichannels and pannexin-1 channels contribute to the alpha-synuclein-induced dysfunction and death of astrocytes. Glia 2019, 67, 1598–1619. [Google Scholar]
- Koob, A.O.; Paulino, A.D.; Masliah, E. GFAP reactivity, apolipoprotein, E. redistribution and cholesterol reduction in human astrocytes treated with alpha-synuclein. Neurosci. Lett. 2010, 469, 11–14. [Google Scholar] [CrossRef]
- Liu, M.; Qin, L.; Wang, L.; Tan, J.; Zhang, H.; Tang, J.; Shen, X.; Tan, L.; Wang, C. Alphasynuclein induces apoptosis of astrocytes by causing dysfunction of the endoplasmic reticulumGolgi compartment. Mol. Med. Rep. 2018, 18, 322–332. [Google Scholar]
- Chen, P.C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar] [CrossRef]
- Williamson, T.P.; Johnson, D.A.; Johnson, J.A. Activation of the Nrf2-ARE pathway by siRNA knockdown of Keap1 reduces oxidative stress and provides partial protection from MPTP-mediated neurotoxicity. Neurotoxicology 2012, 33, 272–279. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.; Kim, J.H.; Song, G.J.; Seo, M.; Hwang, E.M.; Suk, K. Astrocytic Orosomucoid-2 Modulates Microglial Activation and Neuroinflammation. J. Neurosci. 2017, 37, 2878–2894. [Google Scholar] [CrossRef] [PubMed]
- Rohl, C.; Sievers, J. Microglia is activated by astrocytes in trimethyltin intoxication. Toxicol. Appl. Pharmacol. 2005, 204, 36–45. [Google Scholar] [CrossRef]
- Sawada, M.; Imamura, K.; Nagatsu, T. Role of cytokines in inflammatory process in Parkinson’s disease. In Parkinson’s Disease and Related Disorders; Riederer, P., Reichmann, H., Youdim, M.B.H., Gerlach, M., Eds.; Journal of Neural Transmission. Supplementa; Springer: Vienna, Austria, 2006; pp. 373–381. [Google Scholar]
- Vilhardt, F.; Plastre, O.; Sawada, M.; Suzuki, K.; Wiznerowicz, M.; Kiyokawa, E.; Trono, D.; Krause, K.H. The HIV-1 Nef protein and phagocyte NADPH oxidase activation. J. Biol. Chem. 2002, 277, 42136–42143. [Google Scholar] [CrossRef]
- Nagatsu, T.; Sawada, M. Cellular and molecular mechanisms of Parkinson’s disease: Neurotoxins, causative genes, and inflammatory cytokines. Cell. Mol. Neurobiol. 2006, 26, 781–802. [Google Scholar] [CrossRef]
- Huang, J.; Li, C.; Shang, H. Astrocytes in Neurodegeneration: Inspiration From Genetics. Front. Neurosci. 2022, 16, 882316. [Google Scholar] [CrossRef]
- Wilson, H.; Dervenoulas, G.; Pagano, G.; Tyacke, R.J.; Polychronis, S.; Myers, J.; Gunn, R.N.; Rabiner, E.A.; Nutt, D.; Politis, M. Imidazoline 2 binding sites reflecting astroglia pathology in Parkinson’s disease: An in vivo11C-BU99008 PET study. Brain 2019, 142, 3116–3128. [Google Scholar] [CrossRef]
- Batchelor, P.E.; Porritt, M.J.; Martinello, P.; Parish, C.L.; Liberatore, G.T.; Donnan, G.A.; Howells, D.W. Macrophages and Microglia Produce Local Trophic Gradients That Stimulate Axonal Sprouting Toward but Not beyond the Wound Edge. Mol. Cell. Neurosci. 2002, 21, 436–453. [Google Scholar] [CrossRef]
- Muller, H.W.; Junghans, U.; Kappler, J. Astroglial neurotrophic and neurite-promoting factors. Pharmacol. Ther. 1995, 65, 1–18. [Google Scholar] [CrossRef]
- Cao, Q.; Luo, S.; Yao, W.; Qu, Y.; Wang, N.; Hong, J.; Murayama, S.; Zhang, Z.; Chen, J.; Hashimoto, K.; et al. Suppression of abnormal alpha-synuclein expression by activation of BDNF transcription ameliorates Parkinson’s disease-like pathology. Mol. Ther. Nucleic Acids 2022, 29, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ding, J.; Wang, X.; Gu, C.; He, Y.; Li, Y.; Fan, H.; Xie, Q.; Qi, X.; Wang, Z.; et al. Transfer of neuron-derived alpha-synuclein to astrocytes induces neuroinflammation and blood-brain barrier damage after methamphetamine exposure: Involving the regulation of nuclear receptor-associated protein 1. Brain Behav. Immun. 2022, 106, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Sun, J.; Zhao, M.; Hu, J.; Wang, X.; Du, G.; Chen, N.H. Overexpression of alpha-synuclein down-regulates BDNF expression. Cell. Mol. Neurobiol. 2010, 30, 939–946. [Google Scholar] [CrossRef]
- Decressac, M.; Kadkhodaei, B.; Mattsson, B.; Laguna, A.; Perlmann, T.; Bjorklund, A. Alpha-Synuclein-induced down-regulation of Nurr1 disrupts GDNF signaling in nigral dopamine neurons. Sci. Transl. Med. 2012, 4, 163ra156. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Komure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Brain-derived growth factor and nerve growth factor concentrations are decreased in the substantia nigra in Parkinson’s disease. Neurosci. Lett. 1999, 270, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T.; Mogi, M.; Ichinose, H.; Togari, A. Changes in cytokines and neurotrophins in Parkinson’s disease. In Advances in Research on Neurodegeneration; Riederer, P., Calne, D.B., Horowski, R., Mizuno, Y., Olanow, C.W., Poewe, W., Youdim, M.B.H., Eds.; Journal of Neural Transmission. Supplementa; Springer: Vienna, Austria, 2000; pp. 277–290. [Google Scholar]
- Chauhan, N.B.; Siegel, G.J.; Lee, J.M. Depletion of glial cell line-derived neurotrophic factor in substantia nigra neurons of Parkinson’s disease brain. J. Chem. Neuroanat. 2001, 21, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Tomac, A.; Lindqvist, E.; Lin, L.F.; Ogren, S.O.; Young, D.; Hoffer, B.J.; Olson, L. Protection and repair of the nigrostriatal dopaminergic system by GDNF in vivo. Nature 1995, 373, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, P.B.; Jenkins, B.G.; Schulz, J.B.; Finkelstein, S.P.; Matthews, R.T.; Rosen, B.R.; Beal, M.F. NGF, BDNF and NT-5, but not NT-3 protect against MPP+ toxicity and oxidative stress in neonatal animals. Brain Res. 1996, 713, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, G.A.; Cass, W.A.; Huettl, P.; Brock, S.; Zhang, Z.; Gash, D.M. GDNF improves dopamine function in the substantia nigra but not the putamen of unilateral MPTP-lesioned rhesus monkeys. Brain Res. 1999, 817, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.P.; Cass, W.A. GDNF reduces oxidative stress in a 6-hydroxydopamine model of Parkinson’s disease. Neurosci. Lett. 2007, 412, 259–263. [Google Scholar] [CrossRef]
- Bowenkamp, K.E.; Lapchak, P.A.; Hoffer, B.J.; Miller, P.J.; Bickford, P.C. Intracerebroventricular glial cell line-derived neurotrophic factor improves motor function and supports nigrostriatal dopamine neurons in bilaterally 6-hydroxydopamine lesioned rats. Exp. Neurol. 1997, 145, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Lapchak, P.A.; Miller, P.J.; Collins, F.; Jiao, S. Glial cell line-derived neurotrophic factor attenuates behavioural deficits and regulates nigrostriatal dopaminergic and peptidergic markers in 6-hydroxydopamine-lesioned adult rats: Comparison of intraventricular and intranigral delivery. Neuroscience 1997, 78, 61–72. [Google Scholar] [CrossRef] [PubMed]
- L’Episcopo, F.; Serapide, M.F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Pluchino, S.; Marchetti, B. A Wnt1 regulated Frizzled-1/beta-Catenin signaling pathway as a candidate regulatory circuit controlling mesencephalic dopaminergic neuron-astrocyte crosstalk: Therapeutical relevance for neuron survival and neuroprotection. Mol. Neurodegener. 2011, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Cossetti, C.; D’Adamo, P.; Zardini, E.; Andreoni, L.; Ihekwaba, A.E.; et al. Reactive astrocytes and Wnt/beta-catenin signaling link nigrostriatal injury to repair in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Neurobiol. Dis. 2011, 41, 508–527. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.Y.; Biswas, S.; Li, J.; Mao, C.J.; Chechneva, O.; Chen, J.; Li, K.; Li, J.; Zhang, J.R.; Liu, C.F.; et al. Human iPSCs derived astrocytes rescue rotenone-induced mitochondrial dysfunction and dopaminergic neurodegeneration in vitro by donating functional mitochondria. Transl. Neurodegener. 2020, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.L.; Sun, T.; Lu, M.; Ding, J.H.; Du, R.H.; Hu, G. Kir6.1/K-ATP channel on astrocytes protects against dopaminergic neurodegeneration in the MPTP mouse model of Parkinson’s disease via promoting mitophagy. Brain Behav. Immun. 2019, 81, 509–522. [Google Scholar] [CrossRef]
- Morales, I.; Puertas-Avendano, R.; Sanchez, A.; Perez-Barreto, A.; Rodriguez-Sabate, C.; Rodriguez, M. Astrocytes and retrograde degeneration of nigrostriatal dopaminergic neurons in Parkinson’s disease: Removing axonal debris. Transl. Neurodegener. 2021, 10, 43. [Google Scholar] [CrossRef]
- Bido, S.; Muggeo, S.; Massimino, L.; Marzi, M.J.; Giannelli, S.G.; Melacini, E.; Nannoni, M.; Gambare, D.; Bellini, E.; Ordazzo, G.; et al. Microglia-specific overexpression of alpha-synuclein leads to severe dopaminergic neurodegeneration by phagocytic exhaustion and oxidative toxicity. Nat. Commun. 2021, 12, 6237. [Google Scholar] [CrossRef]
- Xia, Y.; Zhang, G.; Han, C.; Ma, K.; Guo, X.; Wan, F.; Kou, L.; Yin, S.; Liu, L.; Huang, J.; et al. Microglia as modulators of exosomal alpha-synuclein transmission. Cell Death Dis. 2019, 10, 174. [Google Scholar] [CrossRef]
- Tanji, K.; Imaizumi, T.; Yoshida, H.; Mori, F.; Yoshimoto, M.; Satoh, K.; Wakabayashi, K. Expression of alpha-synuclein in a human glioma cell line and its up-regulation by interleukin-1beta. NeuroReport 2001, 12, 1909–1912. [Google Scholar] [CrossRef]
- Trudler, D.; Sanz-Blasco, S.; Eisele, Y.S.; Ghatak, S.; Bodhinathan, K.; Akhtar, M.W.; Lynch, W.P.; Pina-Crespo, J.C.; Talantova, M.; Kelly, J.W.; et al. Alpha-Synuclein Oligomers Induce Glutamate Release from Astrocytes and Excessive Extrasynaptic NMDAR Activity in Neurons, Thus Contributing to Synapse Loss. J. Neurosci. 2021, 41, 2264–2273. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.; Mouatt-Prigent, A.; Faucheux, B.A.; Agid, Y.; Hirsch, E.C. FADD. A link between TNF family receptors and caspases in Parkinson’s disease. Neurology 2002, 58, 308–310. [Google Scholar] [CrossRef] [PubMed]
- Boka, G.; Anglade, P.; Wallach, D.; Javoy-Agid, F.; Agid, Y.; Hirsch, E.C. Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson’s disease. Neurosci. Lett. 1994, 172, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Nagatsu, T. Brain beta 2-microglobulin levels are elevated in the striatum in Parkinson’s disease. J. Neural Transm. Park. Dis. Dement. Sect. 1995, 9, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Mizuta, I.; Mizuta, E.; Yamasaki, S.; Ohta, M.; Kaji, R.; Kuno, S. Tumor necrosis factor gene polymorphisms in patients with sporadic Parkinson’s disease. Neurosci. Lett. 2001, 311, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Komure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Caspase activities and tumor necrosis factor receptor, R.1 (p55) level are elevated in the substantia nigra from parkinsonian brain. J. Neural Transm. 2000, 107, 335–341. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Breidert, T.; Rousselet, E.; Hunot, S.; Hartmann, A.; Michel, P.P. The role of glial reaction and inflammation in Parkinson’s disease. Ann. N. Y. Acad. Sci. 2003, 991, 214–228. [Google Scholar] [CrossRef]
- Hartmann, A.; Troadec, J.D.; Hunot, S.; Kikly, K.; Faucheux, B.A.; Mouatt-Prigent, A.; Ruberg, M.; Agid, Y.; Hirsch, E.C. Caspase-8 is an effector in apoptotic death of dopaminergic neurons in Parkinson’s disease, but pathway inhibition results in neuronal necrosis. J. Neurosci. 2001, 21, 2247–2255. [Google Scholar] [CrossRef]
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: Implications for Parkinson’s disease. FASEB J. 2002, 16, 1474–1476. [Google Scholar] [CrossRef]
- Sriram, K.; Miller, D.B.; O’Callaghan, J.P. Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: Role of tumor necrosis factor-alpha. J. Neurochem. 2006, 96, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Deficiency of, T.NF receptors suppresses microglial activation and alters the susceptibility of brain regions to, M.PTP-induced neurotoxicity: Role of TNF-alpha. FASEB J. 2006, 20, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.M.; Jiang, J.; Wilson, B.; Zhang, W.; Hong, J.S.; Liu, B. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: Relevance to Parkinson’s disease. J. Neurochem. 2002, 81, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.C.; Teismann, P.; Tieu, K.; Vila, M.; Jackson-Lewis, V.; Ischiropoulos, H.; Przedborski, S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 6145–6150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, T.; Qin, L.; Gao, H.M.; Wilson, B.; Ali, S.F.; Zhang, W.; Hong, J.S.; Liu, B. Neuroprotective effect of dextromethorphan in the MPTP Parkinson’s disease model: Role of NADPH oxidase. FASEB J. 2004, 18, 589–591. [Google Scholar] [CrossRef] [PubMed]
- Hunot, S.; Dugas, N.; Faucheux, B.; Hartmann, A.; Tardieu, M.; Debre, P.; Agid, Y.; Dugas, B.; Hirsch, E.C. FcepsilonRII/CD23 is expressed in Parkinson’s disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-alpha in glial cells. J. Neurosci. 1999, 19, 3440–3447. [Google Scholar] [CrossRef]
- Vila, M.; Jackson-Lewis, V.; Guegan, C.; Wu, D.C.; Teismann, P.; Choi, D.K.; Tieu, K.; Przedborski, S. The role of glial cells in Parkinson’s disease. Curr. Opin. Neurol. 2001, 14, 483–489. [Google Scholar] [CrossRef]
- Qureshi, G.A.; Baig, S.; Bednar, I.; Sodersten, P.; Forsberg, G.; Siden, A. Increased cerebrospinal fluid concentration of nitrite in Parkinson’s disease. NeuroReport 1995, 6, 1642–1644. [Google Scholar] [CrossRef]
- Good, P.F.; Hsu, A.; Werner, P.; Perl, D.P.; Olanow, C.W. Protein nitration in Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1998, 57, 338–342. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Q.; Yu, R.; Zhang, Q.; Zhang, Z.; Li, H.; Ren, C.; Yang, R.; Niu, H. Minocycline inhibition of microglial rescues nigrostriatal dopaminergic neurodegeneration caused by mutant alpha-synuclein overexpression. Aging 2020, 12, 14232–14243. [Google Scholar] [CrossRef]
- Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Tieu, K.; Teismann, P.; Vadseth, C.; Choi, D.K.; Ischiropoulos, H.; Przedborski, S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J. Neurosci. 2002, 22, 1763–1771. [Google Scholar] [CrossRef] [PubMed]
- Mahoney-Sanchez, L.; Bouchaoui, H.; Ayton, S.; Devos, D.; Duce, J.A.; Devedjian, J.C. Ferroptosis and its potential role in the physiopathology of Parkinson’s Disease. Prog. Neurobiol. 2021, 196, 101890. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.L.; Yuan, L.; Li, W.; Li, J.Y. Ferroptosis in Parkinson’s disease: Glia-neuron crosstalk. Trends Mol. Med. 2022, 28, 258–269. [Google Scholar] [CrossRef]
- Castellani, R.J.; Siedlak, S.L.; Perry, G.; Smith, M.A. Sequestration of iron by Lewy bodies in Parkinson’s disease. Acta Neuropathol. 2000, 100, 111–114. [Google Scholar] [CrossRef]
- Guo, J.J.; Yue, F.; Song, D.Y.; Bousset, L.; Liang, X.; Tang, J.; Yuan, L.; Li, W.; Melki, R.; Tang, Y.; et al. Intranasal administration of alpha-synuclein preformed fibrils triggers microglial iron deposition in the substantia nigra of Macaca fascicularis. Cell Death Dis. 2021, 12, 81. [Google Scholar] [CrossRef]
- Golts, N.; Snyder, H.; Frasier, M.; Theisler, C.; Choi, P.; Wolozin, B. Magnesium inhibits spontaneous and iron-induced aggregation of alpha-synuclein. J. Biol. Chem. 2002, 277, 16116–16123. [Google Scholar] [CrossRef]
- Febbraro, F.; Giorgi, M.; Caldarola, S.; Loreni, F.; Romero-Ramos, M. α-Synuclein expression is modulated at the translational level by iron. NeuroReport 2012, 23, 576–580. [Google Scholar] [CrossRef]
- Cahill, C.M.; Lahiri, D.K.; Huang, X.; Rogers, J.T. Amyloid precursor protein and alpha synuclein translation, implications for iron and inflammation in neurodegenerative diseases. Biochim. Biophys. Acta 2009, 1790, 615–628. [Google Scholar] [CrossRef]
- Angelova, P.R.; Choi, M.L.; Berezhnov, A.V.; Horrocks, M.H.; Hughes, C.D.; De, S.; Rodrigues, M.; Yapom, R.; Little, D.; Dolt, K.S.; et al. Alpha synuclein aggregation drives ferroptosis: An interplay of iron, calcium and lipid peroxidation. Cell Death Differ. 2020, 27, 2781–2796. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; He, L.; Wang, T.; Hua, W.; Qin, H.; Wang, J.; Wang, L.; Gu, W.; Li, T.; Li, N.; et al. Activation of p62-Keap1-Nrf2 Pathway Protects 6-Hydroxydopamine-Induced Ferroptosis in Dopaminergic Cells. Mol. Neurobiol. 2020, 57, 4628–4641. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Eguchi, Y.; Imagawa, Y.; Akai, S.; Mochizuki, H.; Tsujimoto, Y. MPP plus induces necrostatin-1-and ferrostatin-1-sensitive necrotic death of neuronal SH-SY5Y cells. Cell Death Discov. 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gele, P.; Petrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Yan, F.; Deng, R.; Gu, R.; Zhang, X.; Bai, J. Thioredoxin-1 Rescues MPP(+)/MPTP-Induced Ferroptosis by Increasing Glutathione Peroxidase 4. Mol. Neurobiol. 2021, 58, 3187–3197. [Google Scholar] [CrossRef]
- Vallerga, C.L.; Zhang, F.; Fowdar, J.; McRae, A.F.; Qi, T.; Nabais, M.F.; Zhang, Q.; Kassam, I.; Henders, A.K.; Wallace, L.; et al. Analysis of DNA methylation associates the cystine-glutamate antiporter SLC7A11 with risk of Parkinson’s disease. Nat. Commun. 2020, 11, 1238. [Google Scholar] [CrossRef]
- Li, Y.; Maher, P.; Schubert, D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron 1997, 19, 453–463. [Google Scholar] [CrossRef]
- Di Domenico, F.; Tramutola, A.; Butterfield, D.A. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic. Biol. Med. 2017, 111, 253–261. [Google Scholar] [CrossRef]
- Cao, J.; Chen, X.B.; Jiang, L.; Lu, B.; Yuan, M.; Zhu, D.F.; Zhu, H.; He, Q.J.; Yang, B.; Ying, M.D. DJ-1 suppresses ferroptosis through preserving the activity of S-adenosyl homocysteine hydrolase. Nat. Commun. 2020, 11, 1251. [Google Scholar] [CrossRef]
- Oakley, A.E.; Collingwood, J.F.; Dobson, J.; Love, G.; Perrott, H.R.; Edwardson, J.A.; Elstner, M.; Morris, C.M. Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology 2007, 68, 1820–1825. [Google Scholar] [CrossRef]
- Mahoney-Sanchez, L.; Bouchaoui, H.; Boussaad, I.; Jonneaux, A.; Timmerman, K.; Berdeaux, O.; Ayton, S.; Kruger, R.; Duce, J.A.; Devos, D.; et al. Alpha synuclein determines ferroptosis sensitivity in dopaminergic neurons via modulation of ether-phospholipid membrane composition. Cell Rep. 2022, 40, 111231. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Liu, J.; Kang, R.; Tang, D. NEDD4L-mediated LTF protein degradation limits ferroptosis. Biochem. Biophys. Res. Commun. 2020, 531, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Lu, J.; Hao, X.Q.; Li, H.; Zhang, G.Y.; Liu, X.L.; Li, X.R.; Zhao, C.P.; Kuang, W.H.; Chen, D.F.; et al. FTH1 Inhibits Ferroptosis Through Ferritinophagy in the 6-OHDA Model of Parkinson’s Disease. Neurotherapeutics 2020, 17, 1796–1812. [Google Scholar] [CrossRef] [PubMed]
- Bi, M.; Du, X.; Jiao, Q.; Liu, Z.; Jiang, H. Alpha-Synuclein Regulates Iron Homeostasis via Preventing Parkin-Mediated DMT1 Ubiquitylation in Parkinson’s Disease Models. ACS Chem. Neurosci. 2020, 11, 1682–1691. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Conrad, M. Selenium and GPX4, a vital symbiosis. Free Radic. Biol. Med. 2018, 127, 153–159. [Google Scholar] [CrossRef]
- She, X.; Lan, B.; Tian, H.M.; Tang, B. Cross Talk between Ferroptosis and Cerebral Ischemia. Front. Neurosci.-Switz. 2020, 14, 776. [Google Scholar] [CrossRef]
- Kaidery, N.A.; Ahuja, M.; Thomas, B. Crosstalk between Nrf2 signaling and mitochondrial function in Parkinson’s disease. Mol. Cell. Neurosci. 2019, 101, 103413. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Burkhart, A.; Skjorringe, T.; Johnsen, K.B.; Siupka, P.; Thomsen, L.B.; Nielsen, M.S.; Thomsen, L.L.; Moos, T. Expression of Iron-Related Proteins at the Neurovascular Unit Supports Reduction and Reoxidation of Iron for Transport Through the Blood-Brain Barrier. Mol. Neurobiol. 2016, 53, 7237–7253. [Google Scholar] [CrossRef]
- Cui, Z.; Zhong, Z.; Yang, Y.; Wang, B.; Sun, Y.; Sun, Q.; Yang, G.Y.; Bian, L. Ferrous Iron Induces Nrf2 Expression in Mouse Brain Astrocytes to Prevent Neurotoxicity. J. Biochem. Mol. Toxicol. 2016, 30, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Ratan, R.R. The chemical biology of ferroptosis in the central nervous system. Cell Chem. Biol. 2020, 27, 479–498. [Google Scholar] [CrossRef] [PubMed]
- Skjorringe, T.; Burkhart, A.; Johnsen, K.B.; Moos, T. Divalent metal transporter 1 (DMT1) in the brain: Implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front. Mol. Neurosci. 2015, 8, 19. [Google Scholar] [PubMed]
- Belov Kirdajova, D.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-Triggered Glutamate Excitotoxicity From the Perspective of Glial Cells. Front. Cell. Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef]
- Stephenson, E.; Nathoo, N.; Mahjoub, Y.; Dunn, J.F.; Yong, V.W. Iron in multiple sclerosis: Roles in neurodegeneration and repair. Nat. Rev. Neurol. 2014, 10, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.L.; van der Pol, S.M.; Baron, W.; McCord, J.M.; de Vries, H.E.; van Horssen, J. Protandim Protects Oligodendrocytes against an Oxidative Insult. Antioxidants 2016, 5, 30. [Google Scholar] [CrossRef]
- Mukherjee, C.; Kling, T.; Russo, B.; Miebach, K.; Kess, E.; Schifferer, M.; Pedro, L.D.; Weikert, U.; Fard, M.K.; Kannaiyan, N.; et al. Oligodendrocytes Provide Antioxidant Defense Function for Neurons by Secreting Ferritin Heavy Chain. Cell Metab. 2020, 32, 259–272.e210. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, B.; Tao, K.; Yang, H.; Wang, Y.; Zhou, T.; Yang, Y.; Yuan, L.; Liu, X.; Duan, Y. Iron accumulation and microglia activation contribute to substantia nigra hyperechogenicity in the 6-OHDA-induced rat model of Parkinson’s disease. Park. Relat. Disord. 2017, 36, 76–82. [Google Scholar] [CrossRef]
- Novgorodov, S.A.; Voltin, J.R.; Gooz, M.A.; Li, L.; Lemasters, J.J.; Gudz, T.I. Acid sphingomyelinase promotes mitochondrial dysfunction due to glutamate-induced regulated necrosis. J. Lipid Res. 2018, 59, 312–329. [Google Scholar] [CrossRef]
- Kroner, A.; Greenhalgh, A.D.; Zarruk, J.G.; Dos Santos, R.P.; Gaestel, M.; David, S. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron 2014, 83, 1098–1116. [Google Scholar] [CrossRef]
- Wang, J.; Song, N.; Jiang, H.; Wang, J.; Xie, J.X. Pro-inflammatory cytokines modulate iron regulatory protein 1 expression and iron transportation through reactive oxygen/nitrogen species production in ventral mesencephalic neurons. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Nehru, B. Apocyanin, a Microglial NADPH Oxidase Inhibitor Prevents Dopaminergic Neuronal Degeneration in Lipopolysaccharide-Induced Parkinson’s Disease Model. Mol. Neurobiol. 2016, 53, 3326–3337. [Google Scholar] [CrossRef]
- Xu, H.M.; Wang, Y.C.; Song, N.; Wang, J.; Jiang, H.; Xie, J.X. New Progress on the Role of Glia in Iron Metabolism and Iron-Induced Degeneration of Dopamine Neurons in Parkinson’s Disease. Front. Mol. Neurosci. 2018, 10, 455. [Google Scholar] [CrossRef] [PubMed]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef]
- Truman-Rosentsvit, M.; Berenbaum, D.; Spektor, L.; Cohen, L.A.; Belizowsky-Moshe, S.; Lifshitz, L.; Ma, J.; Li, W.; Kesselman, E.; Abutbul-Ionita, I.; et al. Ferritin is secreted via 2 distinct nonclassical vesicular pathways. Blood 2018, 131, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.W.; Amante, J.J.; Chhoy, P.; Elaimy, A.L.; Liu, H.; Zhu, L.J.; Baer, C.E.; Dixon, S.J.; Mercurio, A.M. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev. Cell 2019, 51, 575–586.e4. [Google Scholar] [CrossRef]
- Burtey, A.; Wagner, M.; Hodneland, E.; Skaftnesmo, K.O.; Schoelermann, J.; Mondragon, I.R.; Espedal, H.; Golebiewska, A.; Niclou, S.P.; Bjerkvig, R.; et al. Intercellular transfer of transferrin receptor by a contact-, Rab8-dependent mechanism involving tunneling nanotubes. FASEB J. 2015, 29, 4695–4712. [Google Scholar] [CrossRef]
- Kaufman, S.K.; Diamond, M.I. Prion-like propagation of protein aggregation and related therapeutic strategies. Neurotherapeutics 2013, 10, 371–382. [Google Scholar] [CrossRef]
- Mittal, S.; Bjornevik, K.; Im, D.S.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. β2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson’s disease. Science 2017, 357, 891–898. [Google Scholar] [CrossRef]
- Magistrelli, L.; Comi, C. Beta2-Adrenoceptor Agonists in Parkinson’s Disease and Other Synucleinopathies. J. Neuroimmune Pharmacol. 2020, 15, 74–81. [Google Scholar] [CrossRef]
- Nordstrom, E.; Eriksson, F.; Sigvardson, J.; Johannesson, M.; Kasrayan, A.; Jones-Kostalla, M.; Appelkvist, P.; Soderberg, L.; Nygren, P.; Blom, M.; et al. ABBV-0805, a novel antibody selective for soluble aggregated alpha-synuclein, prolongs lifespan and prevents buildup of alpha-synuclein pathology in mouse models of Parkinson’s disease. Neurobiol. Dis. 2021, 161, 105543. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, V.; Fagerqvist, T.; Nordstrom, E.; Eriksson, F.; Lord, A.; Tucker, S.; Andersson, J.; Johannesson, M.; Schell, H.; Kahle, P.J.; et al. Immunotherapy targeting alpha-synuclein protofibrils reduced pathology in (Thy-1)-h[A30P] alpha-synuclein mice. Neurobiol. Dis. 2014, 69, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Agarwal, P. Depression and Anxiety in Parkinson Disease. Clin. Geriatr. Med. 2020, 36, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Miocinovic, S.; Somayajula, S.; Chitnis, S.; Vitek, J.L. History, applications, and mechanisms of deep brain stimulation. JAMA Neurol. 2013, 70, 163–171. [Google Scholar] [CrossRef]
- Okun, M.S. Deep-Brain Stimulation for Parkinson’s Disease. N. Engl. J. Med. 2012, 367, 1529–1538. [Google Scholar] [CrossRef]
- Bartus, R.T.; Weinberg, M.S.; Samulski, R.J. Parkinson’s disease gene therapy: Success by design meets failure by efficacy. Mol. Ther. 2014, 22, 487–497. [Google Scholar] [CrossRef]
- Barker, R.A.; Drouin-Ouellet, J.; Parmar, M. Cell-based therapies for Parkinson disease-past insights and future potential. Nat. Rev. Neurol. 2015, 11, 492–503. [Google Scholar] [CrossRef]
- Lindvall, O. Clinical translation of stem cell transplantation in Parkinson’s disease. J. Intern. Med. 2016, 279, 30–40. [Google Scholar] [CrossRef]
- Allan, L.E.; Petit, G.H.; Brundin, P. Cell transplantation in Parkinson’s disease: Problems and perspectives. Curr. Opin. Neurol. 2010, 23, 426–432. [Google Scholar] [CrossRef]
- Beattie, R.; Hippenmeyer, S. Mechanisms of radial glia progenitor cell lineage progression. FEBS Lett. 2017, 591, 3993–4008. [Google Scholar] [CrossRef]
- Chou, C.H.; Fan, H.C.; Hueng, D.Y. Potential of Neural Stem Cell-Based Therapy for Parkinson’s Disease. Park. Dis. 2015, 2015, 571475. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.E.; Lozano, A.M. Parkinson’s disease: First of two parts. N. Engl. J. Med. 1998, 339, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- De Virgilio, A.; Greco, A.; Fabbrini, G.; Inghilleri, M.; Rizzo, M.I.; Gallo, A.; Conte, M.; Rosato, C.; Ciniglio Appiani, M.; de Vincentiis, M. Parkinson’s disease: Autoimmunity and neuroinflammation. Autoimmun. Rev. 2016, 15, 1005–1011. [Google Scholar] [CrossRef]
- Gao, H.M.; Hong, J.S. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Badanjak, K.; Fixemer, S.; Smajic, S.; Skupin, A.; Grunewald, A. The Contribution of Microglia to Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4676. [Google Scholar] [CrossRef] [PubMed]
- Dobbs, R.J.; Charlett, A.; Purkiss, A.G.; Dobbs, S.M.; Weller, C.; Peterson, D.W. Association of circulating TNF-alpha and IL-6 with ageing and parkinsonism. Acta Neurol. Scand. 1999, 100, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Borsche, M.; Konig, I.R.; Delcambre, S.; Petrucci, S.; Balck, A.; Bruggemann, N.; Zimprich, A.; Wasner, K.; Pereira, S.L.; Avenali, M.; et al. Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain 2020, 143, 3041–3051. [Google Scholar] [CrossRef] [PubMed]
- Menza, M.; Dobkin, R.D.; Marin, H.; Mark, M.H.; Gara, M.; Bienfait, K.; Dicke, A.; Kusnekov, A. The role of inflammatory cytokines in cognition and other non-motor symptoms of Parkinson’s disease. Psychosomatics 2010, 51, 474–479. [Google Scholar]
- Lindqvist, D.; Kaufman, E.; Brundin, L.; Hall, S.; Surova, Y.; Hansson, O. Non-motor symptoms in patients with Parkinson’s disease-correlations with inflammatory cytokines in serum. PLoS ONE 2012, 7, e47387. [Google Scholar] [CrossRef]
- Cronin, A.; Grealy, M. Neuroprotective and Neuro-restorative Effects of Minocycline and Rasagiline in a Zebrafish 6-Hydroxydopamine Model of Parkinson’s Disease. Neuroscience 2017, 367, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Arellano, H.F.; Jimenez-Del-Rio, M.; Velez-Pardo, C. Minocycline protects, rescues and prevents knockdown transgenic parkin Drosophila against paraquat/iron toxicity: Implications for autosomic recessive juvenile parkinsonism. Neurotoxicology 2017, 60, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, Y.; Mo, M.; Song, C.; Wang, X.; Chen, S.; Liu, Y. Minocycline Protects against Rotenone-Induced Neurotoxicity Correlating with Upregulation of Nurr1 in a Parkinson’s Disease Rat Model. BioMed Res. Int. 2019, 2019, 6843265. [Google Scholar] [CrossRef] [PubMed]
- Boger, H.A.; Middaugh, L.D.; Granholm, A.C.; McGinty, J.F. Minocycline restores striatal tyrosine hydroxylase in GDNF heterozygous mice but not in methamphetamine-treated mice. Neurobiol. Dis. 2009, 33, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Investigators, N.N.-P. A randomized, double-blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology 2006, 66, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Investigators, N.N.-P. A pilot clinical trial of creatine and minocycline in early Parkinson disease: 18-month results. Clin. Neuropharmacol. 2008, 31, 141–150. [Google Scholar]
- Yang, L.; Sugama, S.; Chirichigno, J.W.; Gregorio, J.; Lorenzl, S.; Shin, D.H.; Browne, S.E.; Shimizu, Y.; Joh, T.H.; Beal, M.F.; et al. Minocycline enhances MPTP toxicity to dopaminergic neurons. J. Neurosci. Res. 2003, 74, 278–285. [Google Scholar] [CrossRef]
- Diguet, E.; Fernagut, P.O.; Wei, X.; Du, Y.; Rouland, R.; Gross, C.; Bezard, E.; Tison, F. Deleterious effects of minocycline in animal models of Parkinson’s disease and Huntington’s disease. Eur. J. Neurosci. 2004, 19, 3266–3276. [Google Scholar] [CrossRef]
- Churchill, M.J.; Cantu, M.A.; Kasanga, E.A.; Moore, C.; Salvatore, M.F.; Meshul, C.K. Glatiramer Acetate Reverses Motor Dysfunction and the Decrease in Tyrosine Hydroxylase Levels in a Mouse Model of Parkinson’s Disease. Neuroscience 2019, 414, 8–27. [Google Scholar] [CrossRef]
- Rivetti di Val Cervo, P.; Romanov, R.A.; Spigolon, G.; Masini, D.; Martin-Montanez, E.; Toledo, E.M.; La Manno, G.; Feyder, M.; Pifl, C.; Ng, Y.H.; et al. Induction of functional dopamine neurons from human astrocytes in vitro and mouse astrocytes in a Parkinson’s disease model. Nat. Biotechnol. 2017, 35, 444–452. [Google Scholar] [CrossRef]
- Santos, R.; Vadodaria, K.C.; Jaeger, B.N.; Mei, A.; Lefcochilos-Fogelquist, S.; Mendes, A.P.D.; Erikson, G.; Shokhirev, M.; Randolph-Moore, L.; Fredlender, C.; et al. Differentiation of Inflammation-Responsive Astrocytes from Glial Progenitors Generated from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 8, 1757–1769. [Google Scholar] [CrossRef] [PubMed]
- Krencik, R.; Zhang, S.C. Directed differentiation of functional astroglial subtypes from human pluripotent stem cells. Nat. Protoc. 2011, 6, 1710–1717. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garcon, G.; Rouaix, N.; et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Peña-Díaz, S.; Ventura, S. One ring is sufficient to inhibit α-synuclein aggregation. Neural Regen. Res. 2022, 17, 508. [Google Scholar] [PubMed]
- Xie, X.; Yuan, P.; Kou, L.; Chen, X.; Li, J.; Li, Y. Nilotinib in Parkinson’s disease: A systematic review and meta-analysis. Front. Aging Neurosci. 2022, 14, 996217. [Google Scholar] [CrossRef]
- Kuchimanchi, M.; Monine, M.; Kandadi Muralidharan, K.; Woodward, C.; Penner, N. Phase II Dose Selection for Alpha Synuclein-Targeting Antibody Cinpanemab (BIIB054) Based on Target Protein Binding Levels in the Brain. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 515–522. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Federoff, H.J. Targeting Microglial Activation States as a Therapeutic Avenue in Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 176. [Google Scholar] [CrossRef]
- Valori, C.F.; Possenti, A.; Brambilla, L.; Rossi, D. Challenges and Opportunities of Targeting Astrocytes to Halt Neurodegenerative Disorders. Cells 2021, 10, 2019. [Google Scholar] [CrossRef]
- Perez, B.A.; Shutterly, A.; Chan, Y.K.; Byrne, B.J.; Corti, M. Management of Neuroinflammatory Responses to AAV-Mediated Gene Therapies for Neurodegenerative Diseases. Brain Sci. 2020, 10, 119. [Google Scholar] [CrossRef]
- Wei, Z.Y.D.; Shetty, A.K. Treating Parkinson’s disease by astrocyte reprogramming: Progress and challenges. Sci. Adv. 2021, 7, eabg3198. [Google Scholar] [CrossRef]
- Sun, Y.Y.; Pham, A.N.; Waite, T.D. Mechanism Underlying the Effectiveness of Deferiprone in Alleviating Parkinson’s Disease Symptoms. ACS Chem. Neurosci. 2018, 9, 1118–1127. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yi, S.; Wang, L.; Wang, H.; Ho, M.S.; Zhang, S. Pathogenesis of α-Synuclein in Parkinson’s Disease: From a Neuron-Glia Crosstalk Perspective. Int. J. Mol. Sci. 2022, 23, 14753. https://doi.org/10.3390/ijms232314753
Yi S, Wang L, Wang H, Ho MS, Zhang S. Pathogenesis of α-Synuclein in Parkinson’s Disease: From a Neuron-Glia Crosstalk Perspective. International Journal of Molecular Sciences. 2022; 23(23):14753. https://doi.org/10.3390/ijms232314753
Chicago/Turabian StyleYi, Shuanglong, Linfang Wang, Honglei Wang, Margaret S. Ho, and Shiping Zhang. 2022. "Pathogenesis of α-Synuclein in Parkinson’s Disease: From a Neuron-Glia Crosstalk Perspective" International Journal of Molecular Sciences 23, no. 23: 14753. https://doi.org/10.3390/ijms232314753
APA StyleYi, S., Wang, L., Wang, H., Ho, M. S., & Zhang, S. (2022). Pathogenesis of α-Synuclein in Parkinson’s Disease: From a Neuron-Glia Crosstalk Perspective. International Journal of Molecular Sciences, 23(23), 14753. https://doi.org/10.3390/ijms232314753