
Regulation of P-Glycoprotein in the Brain



{kind=link}
Abstract
1. Introduction
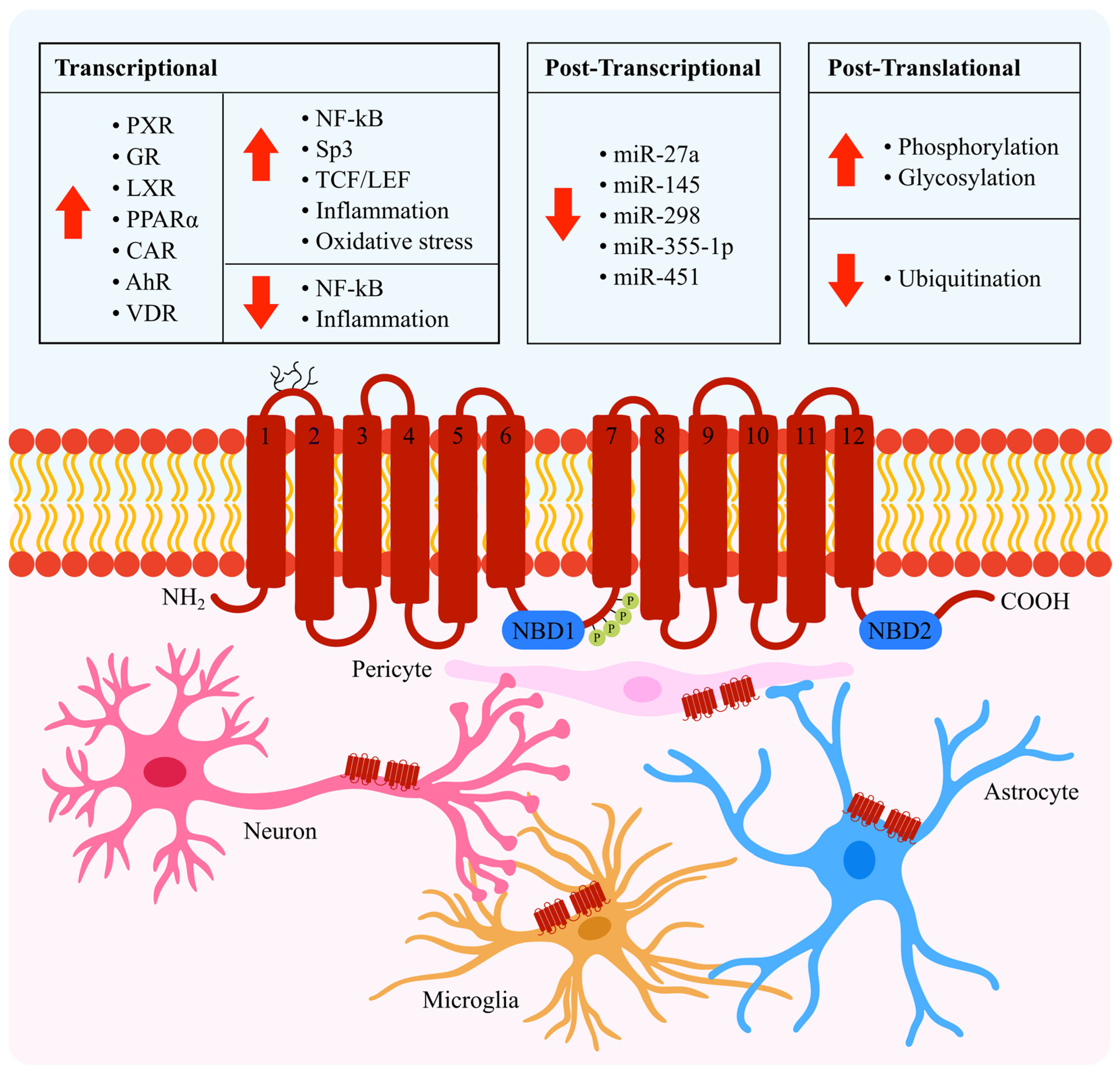
2. Transcriptional Regulation
2.1. NF-κB
2.2. Sp3
2.3. TCF/LEF
2.4. Nuclear Receptors
2.4.1. Pregnane X Receptor (PXR) and Glucocorticoid Receptor (GR)
2.4.2. Liver X Receptor (LXR)
2.4.3. Peroxisome Proliferator Activated Receptor (PPAR)
2.4.4. Constitutive Androstane Receptor (CAR)
2.4.5. Aryl Hydrocarbon Receptor (AhR)
2.4.6. Vitamin D Receptor (VDR)
2.5. Inflammation
2.6. Oxidative Stress
3. Post-Transcriptional Regulation
4. Post-Translational Regulation
4.1. Phosphorylation and Glycosylation
4.2. Ubiquitination
5. Indirect Regulation
6. Significance and Implications of Modulating P-gp
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H. P-Glycoprotein, a gatekeeper in the blood–brain barrier. Adv. Drug Deliv. Rev. 1999, 36, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Qosa, H.; Miller, D.S.; Pasinelli, P.; Trotti, D. Regulation of ABC efflux transporters at blood-brain barrier in health and neurological disorders. Brain Res. 2015, 1628 Pt B, 298–316. [Google Scholar] [CrossRef]
- Miller, D.S.; Bauer, B.; Hartz, A.M. Modulation of P-glycoprotein at the blood-brain barrier: Opportunities to improve central nervous system pharmacotherapy. Pharmacol. Rev. 2008, 60, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Katayama, K.; Noguchi, K.; Sugimoto, Y. Regulations of P-Glycoprotein/ABCB1/MDR1 in Human Cancer Cells. New J. Sci. 2014, 2014, 476974. [Google Scholar] [CrossRef]
- Ambudkar, S.V.; Kimchi-Sarfaty, C.; Sauna, Z.E.; Gottesman, M.M. P-glycoprotein: From genomics to mechanism. Oncogene 2003, 22, 7468–7485. [Google Scholar] [CrossRef]
- Bernstein, H.-G.; Hölzl, G.; Dobrowolny, H.; Hildebrandt, J.; Trübner, K.; Krohn, M.; Bogerts, B.; Pahnke, J. Vascular and extravascular distribution of the ATP-binding cassette transporters ABCB1 and ABCC1 in aged human brain and pituitary. Mech. Ageing Dev. 2014, 141–142, 12–21. [Google Scholar] [CrossRef]
- Bendayan, R.; Ronaldson, P.T.; Gingras, D.; Bendayan, M. In situ localization of P-glycoprotein (ABCB1) in human and rat brain. J. Histochem. Cytochem. 2006, 54, 1159–1167. [Google Scholar] [CrossRef]
- Chai, A.B.; Hartz, A.M.S.; Gao, X.; Yang, A.; Callaghan, R.; Gelissen, I.C. New Evidence for P-gp-Mediated Export of Amyloid-β Peptides in Molecular, Blood-Brain Barrier and Neuronal Models. Int. J. Mol. Sci. 2020, 22, 246. [Google Scholar] [CrossRef]
- Lee, G.; Schlichter, L.; Bendayan, M.; Bendayan, R. Functional expression of P-glycoprotein in rat brain microglia. J. Pharmacol. Exp. Ther. 2001, 299, 204–212. [Google Scholar]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Park, R.; Kook, S.Y.; Park, J.C.; Mook-Jung, I. Aβ1-42 reduces P-glycoprotein in the blood-brain barrier through RAGE-NF-κB signaling. Cell Death Dis. 2014, 5, e1299. [Google Scholar] [CrossRef]
- Miller, D.S. Regulation of ABC transporters at the blood–brain barrier. Clin. Pharmacol. Ther. 2015, 97, 395–403. [Google Scholar] [CrossRef]
- Bauer, B.; Hartz, A.M.; Miller, D.S. Tumor necrosis factor alpha and endothelin-1 increase P-glycoprotein expression and transport activity at the blood-brain barrier. Mol. Pharmacol. 2007, 71, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Nwaozuzu, O.M.; Sellers, L.A.; Barrand, M.A. Signalling pathways influencing basal and H2O2-induced P-glycoprotein expression in endothelial cells derived from the blood-brain barrier. J. Neurochem. 2003, 87, 1043–1051. [Google Scholar] [CrossRef]
- Yu, N.; Di, Q.; Liu, H.; Hu, Y.; Jiang, Y.; Yan, Y.K.; Zhang, Y.F.; Zhang, Y.D. Nuclear factor-kappa B activity regulates brain expression of P-glycoprotein in the kainic acid-induced seizure rats. Mediat. Inflamm. 2011, 2011, 670613. [Google Scholar] [CrossRef]
- Wang, X.; Campos, C.R.; Peart, J.C.; Smith, L.K.; Boni, J.L.; Cannon, R.E.; Miller, D.S. Nrf2 upregulates ATP binding cassette transporter expression and activity at the blood-brain and blood-spinal cord barriers. J. Neurosci. 2014, 34, 8585–8593. [Google Scholar] [CrossRef]
- Zhao, J.; Moore, A.N.; Redell, J.B.; Dash, P.K. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J. Neurosci. 2007, 27, 10240–10248. [Google Scholar] [CrossRef]
- Bauer, B.; Hartz, A.M.; Pekcec, A.; Toellner, K.; Miller, D.S.; Potschka, H. Seizure-induced up-regulation of P-glycoprotein at the blood-brain barrier through glutamate and cyclooxygenase-2 signaling. Mol. Pharmacol. 2008, 73, 1444–1453. [Google Scholar] [CrossRef]
- Zibell, G.; Unkrüer, B.; Pekcec, A.; Hartz, A.M.; Bauer, B.; Miller, D.S.; Potschka, H. Prevention of seizure-induced up-regulation of endothelial P-glycoprotein by COX-2 inhibition. Neuropharmacology 2009, 56, 849–855. [Google Scholar] [CrossRef]
- Potschka, H. Modulating P-glycoprotein regulation: Future perspectives for pharmacoresistant epilepsies? Epilepsia 2010, 51, 1333–1347. [Google Scholar] [CrossRef] [PubMed]
- Sui, H.; Fan, Z.Z.; Li, Q. Signal transduction pathways and transcriptional mechanisms of ABCB1/Pgp-mediated multiple drug resistance in human cancer cells. J. Int. Med. Res. 2012, 40, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Hartz, A.M.; Miller, D.S.; Bauer, B. Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-beta in a mouse model of Alzheimer’s disease. Mol. Pharmacol. 2010, 77, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Lam, F.C.; Liu, R.; Lu, P.; Shapiro, A.B.; Renoir, J.-M.; Sharom, F.J.; Reiner, P.B. β-Amyloid efflux mediated by p-glycoprotein. J. Neurochem. 2001, 76, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Chai, A.B.; Leung, G.K.F.; Callaghan, R.; Gelissen, I.C. P-glycoprotein: A role in the export of amyloid-β in Alzheimer’s disease? FEBS J. 2020, 287, 612–625. [Google Scholar] [CrossRef]
- Chen, F.; Ghosh, A.; Hu, M.; Long, Y.; Sun, H.; Kong, L.; Hong, H.; Tang, S. RAGE-NF-κB-PPARγ Signaling is Involved in AGEs-Induced Upregulation of Amyloid-β Influx Transport in an In Vitro BBB Model. Neurotox. Res. 2018, 33, 284–299. [Google Scholar] [CrossRef]
- Gromnicova, R.; Romero, I.; Male, D. Transcriptional Control of the Multi-Drug Transporter ABCB1 by Transcription Factor Sp3 in Different Human Tissues. PLoS ONE 2012, 7, e48189. [Google Scholar] [CrossRef]
- Staud, F.; Ceckova, M.; Micuda, S.; Pavek, P. Expression and function of p-glycoprotein in normal tissues: Effect on pharmacokinetics. Methods Mol. Biol. 2010, 596, 199–222. [Google Scholar]
- Laksitorini, M.D.; Yathindranath, V.; Xiong, W.; Hombach-Klonisch, S.; Miller, D.W. Modulation of Wnt/β-catenin signaling promotes blood-brain barrier phenotype in cultured brain endothelial cells. Sci. Rep. 2019, 9, 19718. [Google Scholar] [CrossRef]
- Liu, L.; Wan, W.; Xia, S.; Kalionis, B.; Li, Y. Dysfunctional Wnt/β-catenin signaling contributes to blood-brain barrier breakdown in Alzheimer’s disease. Neurochem. Int. 2014, 75, 19–25. [Google Scholar] [CrossRef]
- Lim, J.C.; Kania, K.D.; Wijesuriya, H.; Chawla, S.; Sethi, J.K.; Pulaski, L.; Romero, I.A.; Couraud, P.O.; Weksler, B.B.; Hladky, S.B.; et al. Activation of beta-catenin signalling by GSK-3 inhibition increases p-glycoprotein expression in brain endothelial cells. J. Neurochem. 2008, 106, 1855–1865. [Google Scholar]
- Riganti, C.; Salaroglio, I.C.; Pinzòn-Daza, M.L.; Caldera, V.; Campia, I.; Kopecka, J.; Mellai, M.; Annovazzi, L.; Couraud, P.O.; Bosia, A.; et al. Temozolomide down-regulates P-glycoprotein in human blood-brain barrier cells by disrupting Wnt3 signaling. Cell Mol. Life Sci. 2014, 71, 499–516. [Google Scholar] [CrossRef]
- Tirona, R.G.; Kim, R.B. Nuclear receptors and drug disposition gene regulation. J. Pharm. Sci. 2005, 94, 1169–1186. [Google Scholar] [CrossRef]
- Sever, R.; Glass, C.K. Signaling by nuclear receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a016709. [Google Scholar] [CrossRef]
- Geick, A.; Eichelbaum, M.; Burk, O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J. Biol. Chem. 2001, 276, 14581–14587. [Google Scholar] [CrossRef]
- Lemmen, J.; Tozakidis, I.E.; Galla, H.J. Pregnane X receptor upregulates ABC-transporter Abcg2 and Abcb1 at the blood-brain barrier. Brain Res. 2013, 1491, 1–13. [Google Scholar] [CrossRef]
- Bauer, B.; Yang, X.; Hartz, A.M.; Olson, E.R.; Zhao, R.; Kalvass, J.C.; Pollack, G.M.; Miller, D.S. In vivo activation of human pregnane X receptor tightens the blood-brain barrier to methadone through P-glycoprotein up-regulation. Mol. Pharmacol. 2006, 70, 1212–1219. [Google Scholar] [CrossRef]
- Elmeliegy, M.; Vourvahis, M.; Guo, C.; Wang, D.D. Effect of P-glycoprotein (P-gp) Inducers on Exposure of P-gp Substrates: Review of Clinical Drug-Drug Interaction Studies. Clin. Pharmacokinet. 2020, 59, 699–714. [Google Scholar] [CrossRef]
- Brenn, A.; Grube, M.; Jedlitschky, G.; Fischer, A.; Strohmeier, B.; Eiden, M.; Keller, M.; Groschup, M.H.; Vogelgesang, S. St. John’s Wort reduces beta-amyloid accumulation in a double transgenic Alzheimer’s disease mouse model-role of P-glycoprotein. Brain Pathol. 2014, 24, 18–24. [Google Scholar] [CrossRef]
- Molloy, D.W.; Standish, T.I.; Zhou, Q.; Guyatt, G. A multicenter, blinded, randomized, factorial controlled trial of doxycycline and rifampin for treatment of Alzheimer’s disease: The DARAD trial. Int. J. Geriatr. Psychiatry 2013, 28, 463–470. [Google Scholar] [CrossRef]
- Liu, L.; Collier, A.C.; Link, J.M.; Domino, K.B.; Mankoff, D.A.; Eary, J.F.; Spiekerman, C.F.; Hsiao, P.; Deo, A.K.; Unadkat, J.D. Modulation of P-glycoprotein at the Human Blood-Brain Barrier by Quinidine or Rifampin Treatment: A Positron Emission Tomography Imaging Study. Drug Metab. Dispos. 2015, 43, 1795–1804. [Google Scholar] [CrossRef] [PubMed]
- Mealey, K.L.; Barhoumi, R.; Burghardt, R.C.; Safe, S.; Kochevar, D.T. Doxycycline induces expression of P glycoprotein in MCF-7 breast carcinoma cells. Antimicrob. Agents Chemother. 2002, 46, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Narang, V.S.; Fraga, C.; Kumar, N.; Shen, J.; Throm, S.; Stewart, C.F.; Waters, C.M. Dexamethasone increases expression and activity of multidrug resistance transporters at the rat blood-brain barrier. Am. J. Physiol. Cell Physiol. 2008, 295, C440–C450. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.; Baello, S.; Javam, M.; Audette, M.C.; Gibb, W.; Matthews, S.G. Regulation of Multidrug Resistance P-Glycoprotein in the Developing Blood-Brain Barrier: Interplay between Glucocorticoids and Cytokines. J. Neuroendocrinol. 2016, 28, 12360. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Piekarz, R.L.; Hsu, S.I.; DePinho, R.A.; Carrasco, N.; Horwitz, S.B. Structural and functional analysis of the mouse mdr1b gene promoter. J. Biol. Chem. 1991, 266, 2239–2244. [Google Scholar] [CrossRef]
- Baranowski, M. Biological role of liver X receptors. J. Physiol. Pharmacol. 2008, 59 (Suppl. 7), 31–55. [Google Scholar]
- ElAli, A.; Hermann, D.M. Liver X receptor activation enhances blood-brain barrier integrity in the ischemic brain and increases the abundance of ATP-binding cassette transporters ABCB1 and ABCC1 on brain capillary cells. Brain Pathol. 2012, 22, 175–187. [Google Scholar] [CrossRef]
- Saint-Pol, J.; Candela, P.; Boucau, M.C.; Fenart, L.; Gosselet, F. Oxysterols decrease apical-to-basolateral transport of Aß peptides via an ABCB1-mediated process in an in vitro Blood-brain barrier model constituted of bovine brain capillary endothelial cells. Brain Res. 2013, 1517, 1–15. [Google Scholar] [CrossRef]
- Zelcer, N.; Khanlou, N.; Clare, R.; Jiang, Q.; Reed-Geaghan, E.G.; Landreth, G.E.; Vinters, H.V.; Tontonoz, P. Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver x receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 10601. [Google Scholar] [CrossRef]
- Chai, A.B.; Lam, H.H.J.; Kockx, M.; Gelissen, I.C. Apolipoprotein E isoform-dependent effects on the processing of Alzheimer’s amyloid-β. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158980. [Google Scholar] [CrossRef]
- Riddell, D.R.; Zhou, H.; Comery, T.A.; Kouranova, E.; Lo, C.F.; Warwick, H.K.; Ring, R.H.; Kirksey, Y.; Aschmies, S.; Xu, J.; et al. The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol. Cell Neurosci. 2007, 34, 621–628. [Google Scholar] [CrossRef]
- Sodhi, R.K.; Singh, N. Liver X receptors: Emerging therapeutic targets for Alzheimer’s disease. Pharmacol. Res. 2013, 72, 45–51. [Google Scholar] [CrossRef]
- Bernardo-Castro, S.; Sousa, J.A.; Brás, A.; Cecília, C.; Rodrigues, B.; Almendra, L.; Machado, C.; Santo, G.; Silva, F.; Ferreira, L.; et al. Pathophysiology of Blood-Brain Barrier Permeability Throughout the Different Stages of Ischemic Stroke and Its Implication on Hemorrhagic Transformation and Recovery. Front. Neurol. 2020, 11, 594672. [Google Scholar] [CrossRef]
- Lund, E.G.; Peterson, L.B.; Adams, A.D.; Lam, M.-H.N.; Burton, C.A.; Chin, J.; Guo, Q.; Huang, S.; Latham, M.; Lopez, J.C.; et al. Different roles of liver X receptor α and β in lipid metabolism: Effects of an α-selective and a dual agonist in mice deficient in each subtype. Biochem. Pharmacol. 2006, 71, 453–463. [Google Scholar] [CrossRef]
- Wouters, E.; de Wit, N.M.; Vanmol, J.; van der Pol, S.M.A.; van Het Hof, B.; Sommer, D.; Loix, M.; Geerts, D.; Gustafsson, J.A.; Steffensen, K.R.; et al. Liver X Receptor Alpha Is Important in Maintaining Blood-Brain Barrier Function. Front. Immunol. 2019, 10, 1811. [Google Scholar] [CrossRef]
- Bischoff, E.D.; Daige, C.L.; Petrowski, M.; Dedman, H.; Pattison, J.; Juliano, J.; Li, A.C.; Schulman, I.G. Non-redundant roles for LXRalpha and LXRbeta in atherosclerosis susceptibility in low density lipoprotein receptor knockout mice. J. Lipid Res. 2010, 51, 900–906. [Google Scholar]
- Bradley, M.N.; Hong, C.; Chen, M.; Joseph, S.B.; Wilpitz, D.C.; Wang, X.; Lusis, A.J.; Collins, A.; Hseuh, W.A.; Collins, J.L.; et al. Ligand activation of LXRβ reverses atherosclerosis and cellular cholesterol overload in mice lacking LXRα and apoE. J. Clin. Investig. 2007, 117, 2337–2346. [Google Scholar] [CrossRef]
- More, V.R.; Campos, C.R.; Evans, R.A.; Oliver, K.D.; Chan, G.N.; Miller, D.S.; Cannon, R.E. PPAR-α, a lipid-sensing transcription factor, regulates blood-brain barrier efflux transporter expression. J. Cereb. Blood Flow Metab. 2017, 37, 1199–1212. [Google Scholar] [CrossRef]
- Wang, X.; Sykes, D.B.; Miller, D.S. Constitutive androstane receptor-mediated up-regulation of ATP-driven xenobiotic efflux transporters at the blood-brain barrier. Mol. Pharmacol. 2010, 78, 376–383. [Google Scholar] [CrossRef]
- Baldwin, W.S. Phase 0 of the Xenobiotic Response: Nuclear Receptors and Other Transcription Factors as a First Step in Protection from Xenobiotics. Nucl. Recept. Res. 2019, 6, 101447. [Google Scholar] [CrossRef]
- Wang, X.; Hawkins, B.T.; Miller, D.S. Aryl hydrocarbon receptor-mediated up-regulation of ATP-driven xenobiotic efflux transporters at the blood-brain barrier. FASEB J. 2011, 25, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Chow, E.C.; Durk, M.R.; Cummins, C.L.; Pang, K.S. 1Alpha,25-dihydroxyvitamin D3 up-regulates P-glycoprotein via the vitamin D receptor and not farnesoid X receptor in both fxr(−/−) and fxr(+/+) mice and increased renal and brain efflux of digoxin in mice in vivo. J. Pharmacol. Exp. Ther. 2011, 337, 846–859. [Google Scholar] [CrossRef] [PubMed]
- Durk, M.R.; Chan, G.N.; Campos, C.R.; Peart, J.C.; Chow, E.C.; Lee, E.; Cannon, R.E.; Bendayan, R.; Miller, D.S.; Pang, K.S. 1α,25-Dihydroxyvitamin D3-liganded vitamin D receptor increases expression and transport activity of P-glycoprotein in isolated rat brain capillaries and human and rat brain microvessel endothelial cells. J. Neurochem. 2012, 123, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Durk, M.R.; Han, K.; Chow, E.C.; Ahrens, R.; Henderson, J.T.; Fraser, P.E.; Pang, K.S. 1α,25-Dihydroxyvitamin D3 reduces cerebral amyloid-β accumulation and improves cognition in mouse models of Alzheimer’s disease. J. Neurosci. 2014, 34, 7091–7101. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Shin, J.Y.; Lee, Y.S.; Yun, S.P.; Maeng, H.J.; Lee, Y. Brain Endothelial P-Glycoprotein Level Is Reduced in Parkinson’s Disease via a Vitamin D Receptor-Dependent Pathway. Int. J. Mol. Sci. 2020, 21, 8538. [Google Scholar] [CrossRef]
- Roberts, D.J.; Goralski, K.B. A critical overview of the influence of inflammation and infection on P-glycoprotein expression and activity in the brain. Expert Opin. Drug Metab. Toxicol. 2008, 4, 1245–1264. [Google Scholar] [CrossRef]
- Yu, C.; Kastin, A.J.; Tu, H.; Waters, S.; Pan, W. TNF activates P-glycoprotein in cerebral microvascular endothelial cells. Cell Physiol. Biochem. 2007, 20, 853–858. [Google Scholar] [CrossRef]
- Poller, B.; Drewe, J.; Krähenbühl, S.; Huwyler, J.; Gutmann, H. Regulation of BCRP (ABCG2) and P-glycoprotein (ABCB1) by cytokines in a model of the human blood-brain barrier. Cell. Mol. Neurobiol. 2010, 30, 63–70. [Google Scholar] [CrossRef]
- Walther, W.; Stein, U.; Pfeil, D. Gene transfer of human TNFα into glioblastoma cells permits modulation of mdr1 expression and potentiation of chemosensitivity. Int. J. Cancer 1995, 61, 832–839. [Google Scholar] [CrossRef]
- Iqbal, M.; Ho, H.L.; Petropoulos, S.; Moisiadis, V.G.; Gibb, W.; Matthews, S.G. Pro-inflammatory cytokine regulation of P-glycoprotein in the developing blood-brain barrier. PLoS ONE 2012, 7, e43022. [Google Scholar] [CrossRef]
- Fernandez, C.; Buyse, M.; German-Fattal, M.; Gimenez, F. Influence of the pro-inflammatory cytokines on P-glycoprotein expression and functionality. J. Pharm. Pharm. Sci. 2004, 7, 359–371. [Google Scholar]
- Hartz, A.M.; Bauer, B.; Fricker, G.; Miller, D.S. Rapid regulation of P-glycoprotein at the blood-brain barrier by endothelin-1. Mol. Pharmacol. 2004, 66, 387–394. [Google Scholar] [CrossRef]
- Hartz, A.M.; Bauer, B.; Fricker, G.; Miller, D.S. Rapid modulation of P-glycoprotein-mediated transport at the blood-brain barrier by tumor necrosis factor-alpha and lipopolysaccharide. Mol. Pharmacol. 2006, 69, 462–470. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, F.; Chen, Q.; Kang, A.; Lu, M.; Liu, W.; Zang, X.; Wang, G.; Zhang, J. Chronic inflammation up-regulates P-gp in peripheral mononuclear blood cells via the STAT3/Nf-κb pathway in 2,4,6-trinitrobenzene sulfonic acid-induced colitis mice. Sci. Rep. 2015, 5, 13558. [Google Scholar] [CrossRef]
- Alasmari, F.; Ashby, C.R., Jr.; Hall, F.S.; Sari, Y.; Tiwari, A.K. Modulation of the ATP-Binding Cassette B1 Transporter by Neuro-Inflammatory Cytokines: Role in the Pathogenesis of Alzheimer’s Disease. Front. Pharmacol. 2018, 9, 658. [Google Scholar] [CrossRef]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef]
- Amor, S.; Puentes, F.; Baker, D.; van der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef]
- Wu, J.; Hong, H.; Ji, H.; Wang, Y.Y.; Wang, Y.; Li, Y.Q.; Li, W.G.; Long, Y.; Xia, Y.Z. Glutathione depletion upregulates P-glycoprotein expression at the blood-brain barrier in rats. J. Pharm. Pharmacol. 2009, 61, 819–824. [Google Scholar] [CrossRef]
- Hong, H.; Lu, Y.; Ji, Z.N.; Liu, G.Q. Up-regulation of P-glycoprotein expression by glutathione depletion-induced oxidative stress in rat brain microvessel endothelial cells. J. Neurochem. 2006, 98, 1465–1473. [Google Scholar] [CrossRef]
- Felix, R.A.; Barrand, M.A. P-glycoprotein expression in rat brain endothelial cells: Evidence for regulation by transient oxidative stress. J. Neurochem. 2002, 80, 64–72. [Google Scholar] [CrossRef]
- Hartz, A.M.; Bauer, B.; Block, M.L.; Hong, J.S.; Miller, D.S. Diesel exhaust particles induce oxidative stress, proinflammatory signaling, and P-glycoprotein up-regulation at the blood-brain barrier. FASEB J. 2008, 22, 2723–2733. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Rodrigues, V.; Seca, H.; Sousa, D.; Sousa, E.; Lima, R.T.; Vasconcelos, M.H. The network of P-glycoprotein and microRNAs interactions. Int. J. Cancer 2014, 135, 253–263. [Google Scholar] [CrossRef]
- Xie, Y.; Shao, Y.; Deng, X.; Wang, M.; Chen, Y. MicroRNA-298 Reverses Multidrug Resistance to Antiepileptic Drugs by Suppressing MDR1/P-gp Expression in vitro. Front. Neurosci. 2018, 12, 602. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Shao, Y.; Xie, Y.; Feng, Y.; Wu, M.; Wang, M.; Chen, Y. MicroRNA-146a-5p Downregulates the Expression of P-Glycoprotein in Rats with Lithium-Pilocarpine-Induced Status Epilepticus. Biol. Pharm. Bull. 2019, 42, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Requenez-Contreras, J.L.; López-Castillejos, E.S.; Hernández-Flores, R.; Moreno-Eutimio, M.A.; Granados-Riveron, J.T.; Martinez-Ruiz, G.U.; Aquino-Jarquin, G. MiR-138 indirectly regulates the MDR1 promoter by NF-κB/p65 silencing. Biochem. Biophys. Res. Commun. 2017, 484, 648–655. [Google Scholar] [CrossRef]
- Chambers, T.C.; Pohl, J.; Raynor, R.L.; Kuo, J.F. Identification of specific sites in human P-glycoprotein phosphorylated by protein kinase C. J. Biol. Chem. 1993, 268, 4592–4595. [Google Scholar] [CrossRef]
- Vanoye, C.G.; Castro, A.F.; Pourcher, T.; Reuss, L.; Altenberg, G.A. Phosphorylation of P-glycoprotein by PKA and PKC modulates swelling-activated Cl- currents. Am. J. Physiol. 1999, 276, C370–C378. [Google Scholar] [CrossRef]
- Xie, Y.; Burcu, M.; Linn, D.E.; Qiu, Y.; Baer, M.R. Pim-1 kinase protects P-glycoprotein from degradation and enables its glycosylation and cell surface expression. Mol. Pharmacol. 2010, 78, 310–318. [Google Scholar] [CrossRef]
- Gribar, J.J.; Ramachandra, M.; Hrycyna, C.A.; Dey, S.; Ambudkar, S.V. Functional characterization of glycosylation-deficient human P-glycoprotein using a vaccinia virus expression system. J. Membr. Biol. 2000, 173, 203–214. [Google Scholar] [CrossRef]
- Zhang, Z.; Wu, J.Y.; Hait, W.N.; Yang, J.M. Regulation of the stability of P-glycoprotein by ubiquitination. Mol. Pharmacol. 2004, 66, 395–403. [Google Scholar] [CrossRef]
- Foot, N.; Henshall, T.; Kumar, S. Ubiquitination and the Regulation of Membrane Proteins. Physiol. Rev. 2016, 97, 253–281. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Hartz, A.M.S.; Zhong, Y.; Shen, A.N.; Abner, E.L.; Bauer, B. Preventing P-gp Ubiquitination Lowers Aβ Brain Levels in an Alzheimer’s Disease Mouse Model. Front. Aging Neurosci. 2018, 10, 186. [Google Scholar] [CrossRef]
- Hartz, A.M.S.; Zhong, Y.; Wolf, A.; LeVine, H.; Miller, D.S.; Bauer, B. Aβ40 Reduces P-Glycoprotein at the Blood–Brain Barrier through the Ubiquitin–Proteasome Pathway. J. Neurosci. 2016, 36, 1930. [Google Scholar] [CrossRef]
- Katayama, K.; Noguchi, K.; Sugimoto, Y. FBXO15 regulates P-glycoprotein/ABCB1 expression through the ubiquitin--proteasome pathway in cancer cells. Cancer Sci. 2013, 104, 694–702. [Google Scholar] [CrossRef]
- Wertz, I.E.; Wang, X. From Discovery to Bedside: Targeting the Ubiquitin System. Cell Chem. Biol. 2019, 26, 156–177. [Google Scholar] [CrossRef]
- Zhang, X.; Linder, S.; Bazzaro, M. Drug Development Targeting the Ubiquitin-Proteasome System (UPS) for the Treatment of Human Cancers. Cancers 2020, 12, 902. [Google Scholar] [CrossRef]
- Liu, M.; Aneja, R.; Wang, H.; Sun, L.; Dong, X.; Huo, L.; Joshi, H.; Zhou, J. Modulation of multidrug resistance in cancer cells by the E3 ubiquitin ligase seven-in-absentia homologue 1. J. Pathol. 2008, 214, 508–514. [Google Scholar] [CrossRef]
- Zhang, Y.; Qu, X.; Hu, X.; Yang, X.; Hou, K.; Teng, Y.; Zhang, J.; Sada, K.; Liu, Y. Reversal of P-glycoprotein-mediated multi-drug resistance by the E3 ubiquitin ligase Cbl-b in human gastric adenocarcinoma cells. J. Pathol. 2009, 218, 248–255. [Google Scholar] [CrossRef]
- Zhang, Y.; Qu, X.; Teng, Y.; Li, Z.; Xu, L.; Liu, J.; Ma, Y.; Fan, Y.; Li, C.; Liu, S.; et al. Cbl-b inhibits P-gp transporter function by preventing its translocation into caveolae in multiple drug-resistant gastric and breast cancers. Oncotarget 2015, 6, 6737–6748. [Google Scholar] [CrossRef]
- Akkaya, B.G.; Zolnerciks, J.K.; Ritchie, T.K.; Bauer, B.; Hartz, A.M.; Sullivan, J.A.; Linton, K.J. The multidrug resistance pump ABCB1 is a substrate for the ubiquitin ligase NEDD4-1. Mol. Membr. Biol. 2015, 32, 39–45. [Google Scholar] [CrossRef]
- Chai, A.B.; Callaghan, R.; Gelissen, I.C. The Ubiquitin E3 Ligase Nedd4 Regulates the Expression and Amyloid-β Peptide Export Activity of P-Glycoprotein. Int. J. Mol. Sci. 2022, 23, 1019. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.L. P-glycoprotein Inhibition for Optimal Drug Delivery. Drug Target Insights 2013, 7, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.B. Drugs as P-glycoprotein substrates, inhibitors, and inducers. Drug Metab. Rev. 2002, 34, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Cannon, R.E.; Peart, J.C.; Hawkins, B.T.; Campos, C.R.; Miller, D.S. Targeting blood-brain barrier sphingolipid signaling reduces basal P-glycoprotein activity and improves drug delivery to the brain. Proc. Natl. Acad. Sci. USA 2012, 109, 15930–15935. [Google Scholar] [CrossRef] [PubMed]
- Mesev, E.V.; Miller, D.S.; Cannon, R.E. Ceramide 1-Phosphate Increases P-Glycoprotein Transport Activity at the Blood-Brain Barrier via Prostaglandin E2 Signaling. Mol. Pharmacol. 2017, 91, 373–382. [Google Scholar] [CrossRef]
- Hawkins, B.T.; Sykes, D.B.; Miller, D.S. Rapid, reversible modulation of blood-brain barrier P-glycoprotein transport activity by vascular endothelial growth factor. J. Neurosci. 2010, 30, 1417–1425. [Google Scholar] [CrossRef]
- Barakat, S.; Demeule, M.; Pilorget, A.; Régina, A.; Gingras, D.; Baggetto, L.G.; Béliveau, R. Modulation of p-glycoprotein function by caveolin-1 phosphorylation. J. Neurochem. 2007, 101, 1–8. [Google Scholar] [CrossRef]
- Ding, Y.; Zhong, Y.; Baldeshwiler, A.; Abner, E.L.; Bauer, B.; Hartz, A.M.S. Protecting P-glycoprotein at the blood-brain barrier from degradation in an Alzheimer’s disease mouse model. Fluids Barriers CNS 2021, 18, 10. [Google Scholar] [CrossRef]
- Sharom, F.J. Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function. Front. Oncol. 2014, 4, 41. [Google Scholar] [CrossRef]
- Orlowski, S.; Martin, S.; Escargueil, A. P-glycoprotein and ‘lipid rafts’: Some ambiguous mutual relationships (floating on them, building them or meeting them by chance?). Cell. Mol. Life Sci. 2006, 63, 1038–1059. [Google Scholar] [CrossRef] [PubMed]
- Hegedüs, C.; Telbisz, Á.; Hegedűs, T.; Sarkadi, B.; Özvegy-Laczka, C. Lipid Regulation of the ABCB1 and ABCG2 Multidrug Transporters. Adv. Cancer Res. 2015, 125, 97–137. [Google Scholar] [PubMed]
- Kimura, Y.; Kodan, A.; Matsuo, M.; Ueda, K. Cholesterol fill-in model: Mechanism for substrate recognition by ABC proteins. J. Bioenerg. Biomembr. 2007, 39, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Archie, S.R.; Al Shoyaib, A.; Cucullo, L. Blood-Brain Barrier Dysfunction in CNS Disorders and Putative Therapeutic Targets: An Overview. Pharmaceutics 2021, 13, 1779. [Google Scholar] [CrossRef] [PubMed]
- Bartels, A.L.; Willemsen, A.T.; Kortekaas, R.; de Jong, B.M.; de Vries, R.; de Klerk, O.; van Oostrom, J.C.; Portman, A.; Leenders, K.L. Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J. Neural Transm. 2008, 115, 1001–1009. [Google Scholar] [CrossRef]
- Kortekaas, R.; Leenders, K.L.; van Oostrom, J.C.; Vaalburg, W.; Bart, J.; Willemsen, A.T.; Hendrikse, N.H. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef]
- Taggi, V.; Riera Romo, M.; Piquette-Miller, M.; Meyer zu Schwabedissen, H.E.; Neuhoff, S. Transporter Regulation in Critical Protective Barriers: Focus on Brain and Placenta. Pharmaceutics 2022, 14, 1376. [Google Scholar] [CrossRef]
- Kooij, G.; van Horssen, J.; de Lange, E.C.; Reijerkerk, A.; van der Pol, S.M.; van Het Hof, B.; Drexhage, J.; Vennegoor, A.; Killestein, J.; Scheffer, G.; et al. T lymphocytes impair P-glycoprotein function during neuroinflammation. J. Autoimmun. 2010, 34, 416–425. [Google Scholar] [CrossRef]
- Van Assema, D.M.; Lubberink, M.; Boellaard, R.; Schuit, R.C.; Windhorst, A.D.; Scheltens, P.; Lammertsma, A.A.; van Berckel, B.N. P-glycoprotein function at the blood-brain barrier: Effects of age and gender. Mol. Imaging Biol. 2012, 14, 771–776. [Google Scholar] [CrossRef]
- Toornvliet, R.; van Berckel, B.N.; Luurtsema, G.; Lubberink, M.; Geldof, A.A.; Bosch, T.M.; Oerlemans, R.; Lammertsma, A.A.; Franssen, E.J. Effect of age on functional P-glycoprotein in the blood-brain barrier measured by use of (R)-[(11)C]verapamil and positron emission tomography. Clin. Pharmacol. Ther. 2006, 79, 540–548. [Google Scholar] [CrossRef]
- Bartels, A.L.; Kortekaas, R.; Bart, J.; Willemsen, A.T.; de Klerk, O.L.; de Vries, J.J.; van Oostrom, J.C.; Leenders, K.L. Blood-brain barrier P-glycoprotein function decreases in specific brain regions with aging: A possible role in progressive neurodegeneration. Neurobiol. Aging 2009, 30, 1818–1824. [Google Scholar] [CrossRef]
- Abdullahi, W.; Davis, T.P.; Ronaldson, P.T. Functional Expression of P-glycoprotein and Organic Anion Transporting Polypeptides at the Blood-Brain Barrier: Understanding Transport Mechanisms for Improved CNS Drug Delivery? AAPS J. 2017, 19, 931–939. [Google Scholar] [CrossRef]
- Feldmann, M.; Asselin, M.C.; Liu, J.; Wang, S.; McMahon, A.; Anton-Rodriguez, J.; Walker, M.; Symms, M.; Brown, G.; Hinz, R.; et al. P-glycoprotein expression and function in patients with temporal lobe epilepsy: A case-control study. Lancet Neurol. 2013, 12, 777–785. [Google Scholar] [CrossRef]
- Hartz, A.M.S.; Pekcec, A.; Soldner, E.L.B.; Zhong, Y.; Schlichtiger, J.; Bauer, B. P-gp Protein Expression and Transport Activity in Rodent Seizure Models and Human Epilepsy. Mol. Pharm. 2017, 14, 999–1011. [Google Scholar] [CrossRef]
- Jablonski, M.R.; Markandaiah, S.S.; Jacob, D.; Meng, N.J.; Li, K.; Gennaro, V.; Lepore, A.C.; Trotti, D.; Pasinelli, P. Inhibiting drug efflux transporters improves efficacy of ALS therapeutics. Ann. Clin. Transl. Neurol. 2014, 1, 996–1005. [Google Scholar] [CrossRef]
- O’Brien, F.E.; O’Connor, R.M.; Clarke, G.; Dinan, T.G.; Griffin, B.T.; Cryan, J.F. P-glycoprotein Inhibition Increases the Brain Distribution and Antidepressant-Like Activity of Escitalopram in Rodents. Neuropsychopharmacology 2013, 38, 2209–2219. [Google Scholar] [CrossRef]
- De Gooijer, M.C.; de Vries, N.A.; Buckle, T.; Buil, L.C.M.; Beijnen, J.H.; Boogerd, W.; van Tellingen, O. Improved Brain Penetration and Antitumor Efficacy of Temozolomide by Inhibition of ABCB1 and ABCG2. Neoplasia 2018, 20, 710–720. [Google Scholar] [CrossRef]
- Kemper, E.M.; van Zandbergen, A.E.; Cleypool, C.; Mos, H.A.; Boogerd, W.; Beijnen, J.H.; van Tellingen, O. Increased penetration of paclitaxel into the brain by inhibition of P-Glycoprotein. Clin. Cancer Res. 2003, 9, 2849–2855. [Google Scholar]
- Li, J.; Zheng, M.; Shimoni, O.; Banks, W.A.; Bush, A.I.; Gamble, J.R.; Shi, B. Development of Novel Therapeutics Targeting the Blood–Brain Barrier: From Barrier to Carrier. Adv. Sci. 2021, 8, 2101090. [Google Scholar] [CrossRef]
- Phatnani, H.; Maniatis, T. Astrocytes in neurodegenerative disease. Cold Spring Harb. Perspect. Biol. 2015, 7, a020628. [Google Scholar] [CrossRef]
- Kam, T.-I.; Hinkle, J.T.; Dawson, T.M.; Dawson, V.L. Microglia and astrocyte dysfunction in parkinson’s disease. Neurobiol. Dis. 2020, 144, 105028. [Google Scholar] [CrossRef] [PubMed]
- Fakhoury, M. Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. 2018, 16, 508–518. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chai, A.B.; Callaghan, R.; Gelissen, I.C. Regulation of P-Glycoprotein in the Brain. Int. J. Mol. Sci. 2022, 23, 14667. https://doi.org/10.3390/ijms232314667
Chai AB, Callaghan R, Gelissen IC. Regulation of P-Glycoprotein in the Brain. International Journal of Molecular Sciences. 2022; 23(23):14667. https://doi.org/10.3390/ijms232314667
Chicago/Turabian StyleChai, Amanda B., Richard Callaghan, and Ingrid C. Gelissen. 2022. "Regulation of P-Glycoprotein in the Brain" International Journal of Molecular Sciences 23, no. 23: 14667. https://doi.org/10.3390/ijms232314667
APA StyleChai, A. B., Callaghan, R., & Gelissen, I. C. (2022). Regulation of P-Glycoprotein in the Brain. International Journal of Molecular Sciences, 23(23), 14667. https://doi.org/10.3390/ijms232314667