
Recent Advances on P-Glycoprotein (ABCB1) Transporter Modelling with In Silico Methods


Abstract
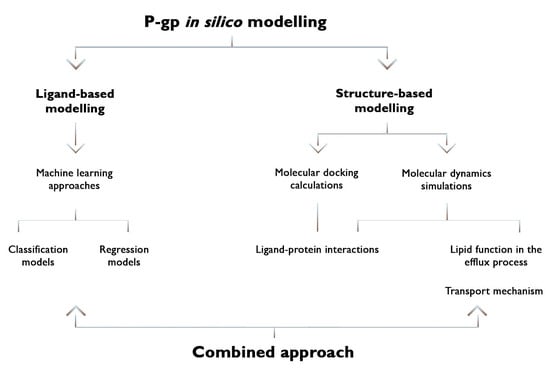
1. Introduction
2. Ligand-Based Models
2.1. Improving Feature Selection
2.2. Reducing Heterogeneity in the Data
2.3. Three-Class Classification Models
2.4. Models Including Other Transporters
3. Structure-Based Approaches
3.1. Homology Modelling and Molecular Docking Studies Involving In Vitro Assays
3.2. Molecular Dynamics Simulations
3.2.1. Role of Membrane Lipids in the Efflux Process
3.2.2. Exploring Ligand Binding Interactions and the Binding Pocket
3.2.3. Experimental Structure of hP-gp
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ou-Yang, S.-S.; Lu, J.-Y.; Kong, X.-Q.; Liang, Z.-J.; Luo, C.; Jiang, H. Computational drug discovery. Acta Pharmacol. Sin. 2012, 33, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Cao, Y.; Fan, W.; Chen, L.; Mo, Y. Computer-aided drug design: Lead discovery and optimization. Comb. Chem. High Throughput Screen. 2012, 15, 328–337. [Google Scholar] [CrossRef]
- Jain, A.N. Virtual screening in lead discovery and optimization. Curr. Opin. Drug Discov. Dev. 2004, 7, 396–403. [Google Scholar]
- Stahura, F.L.; Bajorath, J. New methodologies for ligand-based virtual screening. Curr. Pharm. Des. 2005, 11, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Chai, A.B.; Hartz, A.M.; Gao, X.; Yang, A.; Callaghan, R.; Gelissen, I.C. New evidence for P-gp-mediated export of amyloid-β PEPTIDES in molecular, blood-brain barrier and neuronal models. Int. J. Mol. Sci. 2020, 22, 246. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, R.; Luk, F.; Bebawy, M. Inhibition of the multidrug resistance P-glycoprotein: Time for a change of strategy? Drug Metab. Dispos. 2014, 42, 623–631. [Google Scholar] [CrossRef]
- Jorgensen, W.L. The many roles of computation in drug discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef]
- Prathipati, P.; Dixit, A.; Saxena, A.K. Computer-aided drug design: Integration of structure-based and ligand-based approaches in drug design. Curr. Comput.-Aided Drug Des. 2007, 3, 133–148. [Google Scholar] [CrossRef]
- Favia, A.D. Theoretical and computational approaches to ligand-based drug discovery. Front. Biosci. 2011, 16, 1276–1290. [Google Scholar] [CrossRef]
- Mavromoustakos, T.; Durdagi, S.; Koukoulitsa, C.; Simcic, M.; G Papadopoulos, M.; Hodoscek, M.; Golic Grdadolnik, S. Strategies in the rational drug design. Curr. Med. Chem. 2011, 18, 2517–2530. [Google Scholar] [CrossRef]
- Śledź, P.; Caflisch, A. Protein structure-based drug design: From docking to molecular dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef] [PubMed]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Seelig, A.; Landwojtowicz, E. Structure–activity relationship of P-glycoprotein substrates and modifiers. Eur. J. Pharm. Sci. 2000, 12, 31–40. [Google Scholar] [CrossRef]
- Dearden, J.; Al-Noobi, A.; Scott, A.; Thomson, S. QSAR studies on P-glycoprotein-regulated multidrug resistance and on its reversal by phenothiazines. SAR QSAR Environ. Res. 2003, 14, 447–454. [Google Scholar] [CrossRef]
- Kim, K.H. 3D-QSAR analysis of 2, 4, 5-and 2, 3, 4, 5-substituted imidazoles as potent and nontoxic modulators of P-glycoprotein mediated MDR. Bioorg. Med. Chem. 2001, 9, 1517–1523. [Google Scholar] [CrossRef]
- Pajeva, I.K.; Wiese, M. Pharmacophore model of drugs involved in P-glycoprotein multidrug resistance: Explanation of structural variety (hypothesis). J. Med. Chem. 2002, 45, 5671–5686. [Google Scholar] [CrossRef]
- Tsuruo, T.; Iida, H.; Tsukagoshi, S.; Sakurai, Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res. 1981, 41, 1967–1972. [Google Scholar]
- Moro, S.; Bacilieri, M.; Deflorian, F. Combining ligand-based and structure-based drug design in the virtual screening arena. Expert Opin. Drug Discov. 2007, 2, 37–49. [Google Scholar] [CrossRef]
- Vedani, A.; Briem, H.; Dobler, M.; Dollinger, H.; McMasters, D.R. Multiple-conformation and protonation-state representation in 4D-QSAR: The neurokinin-1 receptor system. J. Med. Chem. 2000, 43, 4416–4427. [Google Scholar] [CrossRef]
- Vedani, A.; Dobler, M. 5D-QSAR: The key for simulating induced fit? J. Med. Chem. 2002, 45, 2139–2149. [Google Scholar] [CrossRef]
- Esposito, C.; Wang, S.; Lange, U.E.; Oellien, F.; Riniker, S. Combining machine learning and molecular dynamics to predict P-glycoprotein substrates. J. Chem. Inf. Model. 2020, 60, 4730–4749. [Google Scholar] [CrossRef]
- Kadioglu, O.; Efferth, T. A Machine Learning-Based Prediction Platform for P-Glycoprotein Modulators and Its Validation by Molecular Docking. Cells 2019, 8, 1286. [Google Scholar] [CrossRef]
- Garcia, G.C.; Garcia-Pedrajas, N. Boosted feature selectors: A case study on prediction P-gp inhibitors and substrates. J. Comput.-Aided Mol. Des. 2018, 32, 1273–1294. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.H.; Tu, Y.S.; Tseng, Y.F.J. PgpRules: A decision tree based prediction server for P-glycoprotein substrates and inhibitors. Bioinformatics 2019, 35, 4193–4195. [Google Scholar] [CrossRef]
- Prachayasittikul, V.; Worachartcheewan, A.; Toropova, A.P.; Toropov, A.A.; Schaduangrat, N.; Prachayasittikul, V.; Nantasenamat, C. Large-scale classification of P-glycoprotein inhibitors using SMILES-based descriptors. Sar Qsar Environ. Res. 2017, 28, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hinge, V.K.; Roy, D.; Kovalenko, A. Prediction of P-glycoprotein inhibitors with machine learning classification models and 3D-RISM-KH theory based solvation energy descriptors. J. Comput.-Aided Mol. Des. 2019, 33, 965–971. [Google Scholar] [CrossRef]
- Broccatelli, F.; Carosati, E.; Neri, A.; Frosini, M.; Goracci, L.; Oprea, T.I.; Cruciani, G. A Novel Approach for Predicting P-Glycoprotein (ABCB1) Inhibition Using Molecular Interaction Fields. J. Med. Chem. 2011, 54, 1740–1751. [Google Scholar] [CrossRef]
- Ohashi, R.; Watanabe, R.; Esaki, T.; Taniguchi, T.; Torimoto-Katori, N.; Watanabe, T.; Ogasawara, Y.; Takahashi, T.; Tsukimoto, M.; Mizuguchi, K. Development of Simplified in Vitro P-Glycoprotein Substrate Assay and in Silico Prediction Models To Evaluate Transport Potential of P-Glycoprotein. Mol. Pharm. 2019, 16, 1851–1863. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Lee, M.H.; Weng, C.F.; Leong, M.K. Theoretical Prediction of the Complex P-Glycoprotein Substrate Efflux Based on the Novel Hierarchical Support Vector Regression Scheme. Molecules 2018, 23, 1820. [Google Scholar] [CrossRef]
- Watanabe, R.; Esaki, T.; Ohashi, R.; Kuroda, M.; Kawashima, H.; Komura, H.; Natsume-Kitatani, Y.; Mizuguchi, K. Development of an In Silico Prediction Model for P-glycoprotein Efflux Potential in Brain Capillary Endothelial Cells toward the Prediction of Brain Penetration. J. Med. Chem. 2021, 64, 2725–2738. [Google Scholar] [CrossRef] [PubMed]
- Mora Lagares, L.; Minovski, N.; Novič, M. Multiclass Classifier for P-Glycoprotein Substrates, Inhibitors, and Non-Active Compounds. Molecules 2019, 24, 2006. [Google Scholar] [CrossRef]
- Benfenati, E.; Manganaro, A.; Gini, G.C. VEGA-QSAR: AI Inside a Platform for Predictive Toxicology. In Proceedings of the PAI@ AI* IA, Turin, Italy, 5 December 2013; pp. 21–28. [Google Scholar]
- Estrada-Tejedor, R.; Ecker, G.F. Predicting drug resistance related to ABC transporters using unsupervised Consensus Self-Organizing Maps. Sci. Rep. 2018, 8, 6803. [Google Scholar] [CrossRef] [PubMed]
- Namasivayam, V.; Silbermann, K.; Wiese, M.; Pahnke, J.; Stefan, S.M. C@PA: Computer-Aided Pattern Analysis to Predict Multitarget ABC Transporter Inhibitors. J. Med. Chem. 2021, 64, 3350–3366. [Google Scholar] [CrossRef] [PubMed]
- Scapin, G. Structural biology and drug discovery. Curr. Pharm. Des. 2006, 12, 2087–2097. [Google Scholar] [CrossRef]
- Verlinde, C.L.; Hol, W.G. Structure-based drug design: Progress, results and challenges. Structure 1994, 2, 577–587. [Google Scholar] [CrossRef]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M. Three decades of P-gp inhibitors: Skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef] [PubMed]
- Klepsch, F.; Ecker, G.F. Impact of the recent mouse P-glycoprotein structure for structure-based ligand design. Mol. Inform. 2010, 29, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Mora Lagares, L.; Minovski, N.; Alfonso, A.Y.C.; Benfenati, E.; Wellens, S.; Culot, M.; Gosselet, F.; Novič, M. Homology Modeling of the Human P-glycoprotein (ABCB1) and Insights into Ligand Binding through Molecular Docking Studies. Int. J. Mol. Sci. 2020, 21, 4058. [Google Scholar] [CrossRef]
- Jin, M.S.; Oldham, M.L.; Zhang, Q.J.; Chen, J. Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 2012, 490, 566–569. [Google Scholar] [CrossRef]
- Marques, S.M.; Supolikova, L.; Molcanova, L.; Smejkal, K.; Bednar, D.; Slaninova, I. Screening of Natural Compounds as P-Glycoprotein Inhibitors against Multidrug Resistance. Biomedicines 2021, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Zeslawska, E.; Tejchman, W.; Kincses, A.; Spengler, G.; Nitek, W.; Zuchowski, G.; Szymanska, E. 5-Arylidenerhodanines as P-gp Modulators: An Interesting Effect of the Carboxyl Group on ABCB1 Function in Multidrug-Resistant Cancer Cells. Int. J. Mol. Sci. 2022, 23, 10812. [Google Scholar] [CrossRef] [PubMed]
- Sagnou, M.; Novikov, F.N.; Ivanova, E.S.; Alexiou, P.; Stroylov, V.S.; Titov, I.Y.; Tatarskiy, V.V.; Vagida, M.S.; Pelecanou, M.; Shtil, A.A.; et al. Novel curcumin derivatives as P-glycoprotein inhibitors: Molecular modeling, synthesis and sensitization of multidrug resistant cells to doxorubicin. Eur. J. Med. Chem. 2020, 198, 112331. [Google Scholar] [CrossRef]
- Syed, S.B.; Arya, H.; Fu, I.H.; Yeh, T.K.; Periyasamy, L.; Hsieh, H.P.; Coumar, M.S. Targeting P-glycoprotein: Investigation of piperine analogs for overcoming drug resistance in cancer. Sci. Rep. 2017, 7, 7972. [Google Scholar] [CrossRef]
- Bortolozzi, R.; Luraghi, A.; Mattiuzzo, E.; Sacchetti, A.; Silvani, A.; Viola, G. Ecdysteroid Derivatives that Reverse P-Glycoprotein-Mediated Drug Resistance. J. Nat. Prod. 2020, 83, 2434–2446. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.C.; Wang, L.; Kang, D.; Zhao, Y.; Wu, S.; Liu, A.L.; Du, G.H. Effects of P-Glycoprotein on the Transport of DL0410, a Potential Multifunctional Anti-Alzheimer Agent. Molecules 2017, 22, 1246. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.B.; Szewczyk, P.; Grimard, V.; Lee, C.W.; Martinez, L.; Doshi, R.; Caya, A.; Villaluz, M.; Pardon, E.; Cregger, C.; et al. Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc. Natl. Acad. Sci. USA 2013, 110, 13386–13391. [Google Scholar] [CrossRef]
- Hosseini Balef, S.S.; Piramoon, M.; Hosseinimehr, S.J.; Irannejad, H. In vitro and in silico evaluation of P-glycoprotein inhibition through Tc-99m-methoxyisobutylisonitrile uptake. Chem. Biol. Drug Des. 2019, 93, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Caflisch, A. Molecular dynamics in drug design. Eur. J. Med. Chem. 2015, 91, 4–14. [Google Scholar] [CrossRef]
- Shahraki, O.; Edraki, N.; Khoshneviszadeh, M.; Zargari, F.; Ranjbar, S.; Saso, L.; Firuzi, O.; Miri, R. Novel 5-oxo-hexahydroquinoline derivatives: Design, synthesis, in vitro P-glycoprotein-mediated multidrug resistance reversal profile and molecular dynamics simulation study. Drug Des. Dev. Ther. 2017, 11, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Mukhametov, A.; Raevsky, O.A. On the mechanism of substrate/non-substrate recognition by P-glycoprotein. J. Mol. Graph. Model. 2017, 71, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; O’Mara, M.L. Effect of the Force Field on Molecular Dynamics Simulations of the Multidrug Efflux Protein P-Glycoprotein. J. Chem. Theory Comput. 2021, 17, 6491–6508. [Google Scholar] [CrossRef] [PubMed]
- Barreto-Ojeda, E.; Corradi, V.; Gu, R.-X.; Tieleman, D.P. Coarse-grained molecular dynamics simulations reveal lipid access pathways in P-glycoprotein. J. Gen. Physiol. 2018, 150, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jaimes, K.F.; Aller, S.G. Refined structures of mouse P-glycoprotein. Protein Sci. 2014, 23, 34–46. [Google Scholar] [CrossRef]
- Domicevica, L.; Koldsø, H.; Biggin, P.C. Multiscale molecular dynamics simulations of lipid interactions with P-glycoprotein in a complex membrane. J. Mol. Graph. Model. 2018, 80, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Thangapandian, S.; Kapoor, K.; Tajkhorshid, E. Probing cholesterol binding and translocation in P-glycoprotein. Biochim. Biophys. Acta (BBA)–Biomembr. 2020, 1862, 183090. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, K.; Pant, S.; Tajkhorshid, E. Active participation of membrane lipids in inhibition of multidrug transporter P-glycoprotein. Chem. Sci. 2021, 12, 6293–6306. [Google Scholar] [CrossRef]
- Behmard, E.; Barzegari, E.; Najafipour, S.; Kouhpayeh, A.; Ghasemi, Y.; Asadi-Pooya, A.A. Efflux dynamics of the antiseizure drug, levetiracetam, through the P-glycoprotein channel revealed by advanced comparative molecular simulations. Sci. Rep. 2022, 12, 13674. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.J.; Locher, K.P. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 2007, 581, 935–938. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Sun, Y. Efflux mechanism and pathway of verapamil pumping by human P-glycoprotein. Arch. Biochem. Biophys. 2020, 696, 108675. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Zhang, L.; Liu, F.F.; Sun, Y. Molecular Energetics of Doxorubicin Pumping by Human P-Glycoprotein. J. Chem. Inf. Model. 2019, 59, 3889–3898. [Google Scholar] [CrossRef]
- Zhang, B.; Kang, Z.Z.; Zhang, J.Q.; Kang, Y.; Liang, L.J.; Liu, Y.C.; Wang, Q. Simultaneous binding mechanism of multiple substrates for multidrug resistance transporter P-glycoprotein. Phys. Chem. Chem. Phys. 2021, 23, 4530–4543. [Google Scholar] [CrossRef] [PubMed]
- Bonito, C.A.; Ferreira, R.J.; Ferreira, M.-J.U.; Gillet, J.-P.; Cordeiro, M.; Dos Santos, D.J. Theoretical insights on helix repacking as the origin of P-glycoprotein promiscuity. Sci. Rep. 2020, 10, 9823. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.A.; Abdeljawaad, K.A.A.; Abdelrahman, A.H.M.; Jaragh-Alhadad, L.A.; Oraby, H.F.; Elkaeed, E.B.; Mekhemer, G.A.H.; Gabr, G.A.; Shawky, A.M.; Sidhom, P.A.; et al. Exploring Natural Product Activity and Species Source Candidates for Hunting ABCB1 Transporter Inhibitors: An In Silico Drug Discovery Study. Molecules 2022, 27, 3104. [Google Scholar] [CrossRef] [PubMed]
- Mora Lagares, L.; Pérez-Castillo, Y.; Minovski, N.; Novič, M. Structure–Function Relationships in the Human P-Glycoprotein (ABCB1): Insights from Molecular Dynamics Simulations. Int. J. Mol. Sci. 2021, 23, 362. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Name | Structure | Molecular Weight | Experimental Validation | Ref. |
|---|---|---|---|---|
| Quinoline and 1,2,4-oxadiazole derivative 15 |  | 421.45 | IC50: 8.59 μM | [35] |
| Quinoline 1,3,4-thiadiazole derivative 18 |  | 446.49 | IC50: 2.53 μM | [35] |
| Quinazoline and 1,2,4-oxadiazole derivative 21 |  | 444.41 | IC50: 2.64 μM | [35] |
| Quinoline and thieno [3,2-c]pyridine derivative 22 |  | 424.54 | IC50: 3.64 μM | [35] |
| Quinoline and 1,2,4-oxadiazole derivative 26 |  | 391.47 | IC50: 2.00 μM | [35] |
| Baicalein |  | 270.24 | n.p. 1 | [42] |
| Quercetin-3-glucoside |  | 464.4 | n.p. | [42] |
| 3-(5-{[4-(diphenylamino)phenyl]methylidene}-4-oxo-2-sulfanylidene-1,3-thiazolidin-3-yl)propanoic acid |  | 424.54 | IC50: 42.10 μM | [43] |
| 4-(3,5-bis((E)-3,4-dimethoxystyryl)-1H-pyrazol-1-yl)-Nethylbenzamide |  | 540.24 | IC50 > 50 μM | [44] |
| (4-(3,5-bis((E)-3,4-dimethoxystyryl)-1H-pyrazol-1-yl)phenyl)( pyrrolidin-1-yl) methanone |  | 566.67 | IC50 >50 μM | [44] |
| (2E,4E)-5-(benzo[d][1,3]dioxol-5-yl)-1-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1 H)-yl)penta-2,4-dien-1-one |  | 393.43 | IC50: 2.93 nM | [45] |
| 20-Hydroxyecdysone-2,3,20,22-dicyclohexyl-ketal |  | 640.88 | Decrease P-gp expression in the multi-drug-resistant CEMVbl100 cell | [46] |
| 20-Hydroxyecdysone 2,3,22-tribenzoate |  | 792.95 | Decrease P-gp expression in the multi-drug-resistant CEMVbl100 cell | [46] |
| DL0410 |  | 432.28 | ER 2 (μM): 4.51 | [47] |
| N-(4-methoxyphenyl)-2-methyl-4-(2-nitrophenyl)-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3- carboxamide |  | 433.20 | n.p. | [52] |
| NPC104372 |  | 1012.56 | n.p. | [66] |
| NPC475164 |  | 1120.60 | n.p. | [66] |
| NPC2313 |  | 1038.61 | n.p. | [66] |
| NPC197736 |  | 1006.59 | n.p. | [66] |
| NPC477344 |  | 1008.55 | n.p. | [66] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mora Lagares, L.; Novič, M. Recent Advances on P-Glycoprotein (ABCB1) Transporter Modelling with In Silico Methods. Int. J. Mol. Sci. 2022, 23, 14804. https://doi.org/10.3390/ijms232314804
Mora Lagares L, Novič M. Recent Advances on P-Glycoprotein (ABCB1) Transporter Modelling with In Silico Methods. International Journal of Molecular Sciences. 2022; 23(23):14804. https://doi.org/10.3390/ijms232314804
Chicago/Turabian StyleMora Lagares, Liadys, and Marjana Novič. 2022. "Recent Advances on P-Glycoprotein (ABCB1) Transporter Modelling with In Silico Methods" International Journal of Molecular Sciences 23, no. 23: 14804. https://doi.org/10.3390/ijms232314804
APA StyleMora Lagares, L., & Novič, M. (2022). Recent Advances on P-Glycoprotein (ABCB1) Transporter Modelling with In Silico Methods. International Journal of Molecular Sciences, 23(23), 14804. https://doi.org/10.3390/ijms232314804