

Antiarrhythmic and Inotropic Effects of Selective Na+/Ca2+ Exchanger Inhibition: What Can We Learn from the Pharmacological Studies?
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
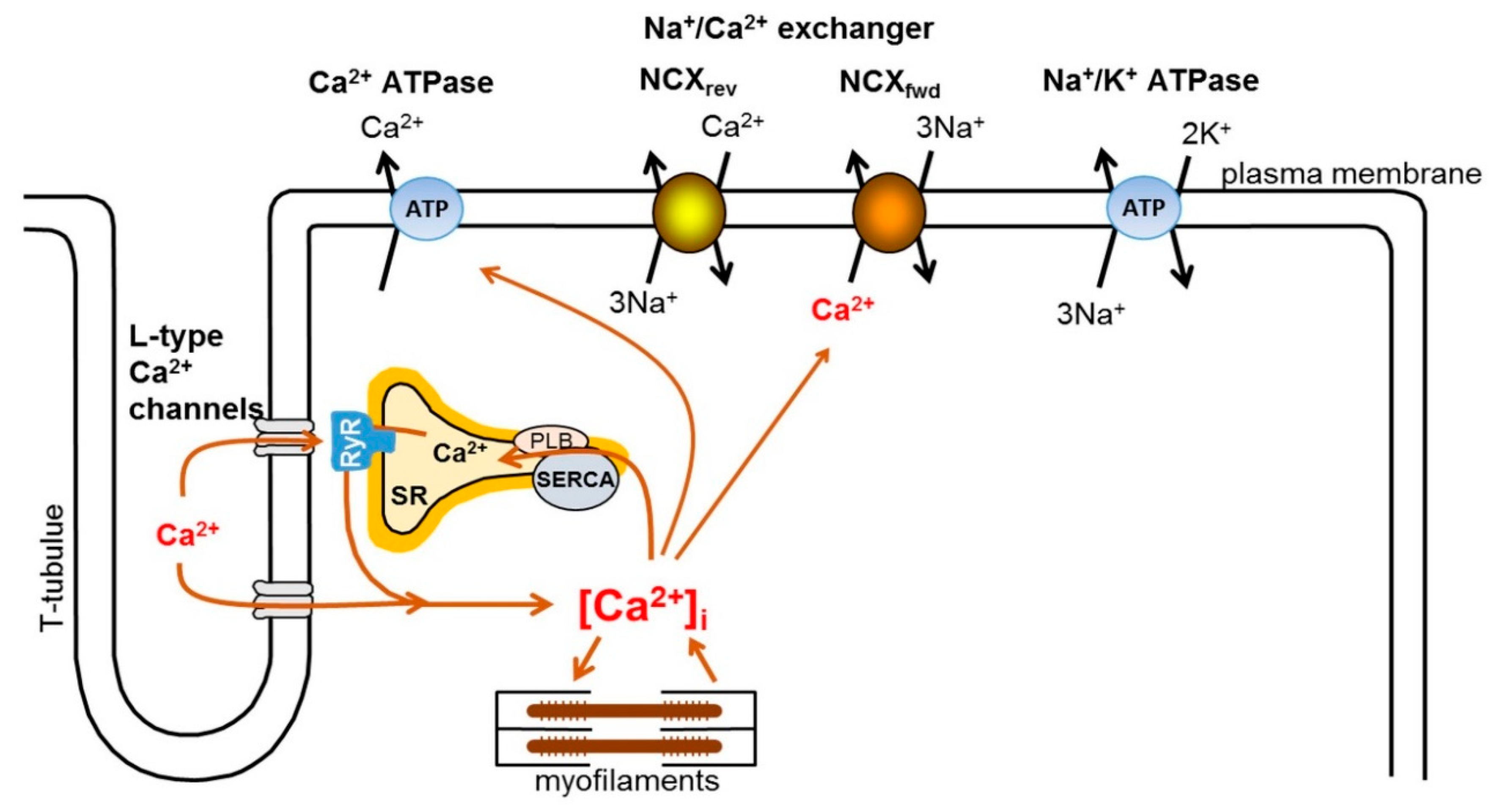
2. Intracellular Ca2+ Handling in the Heart
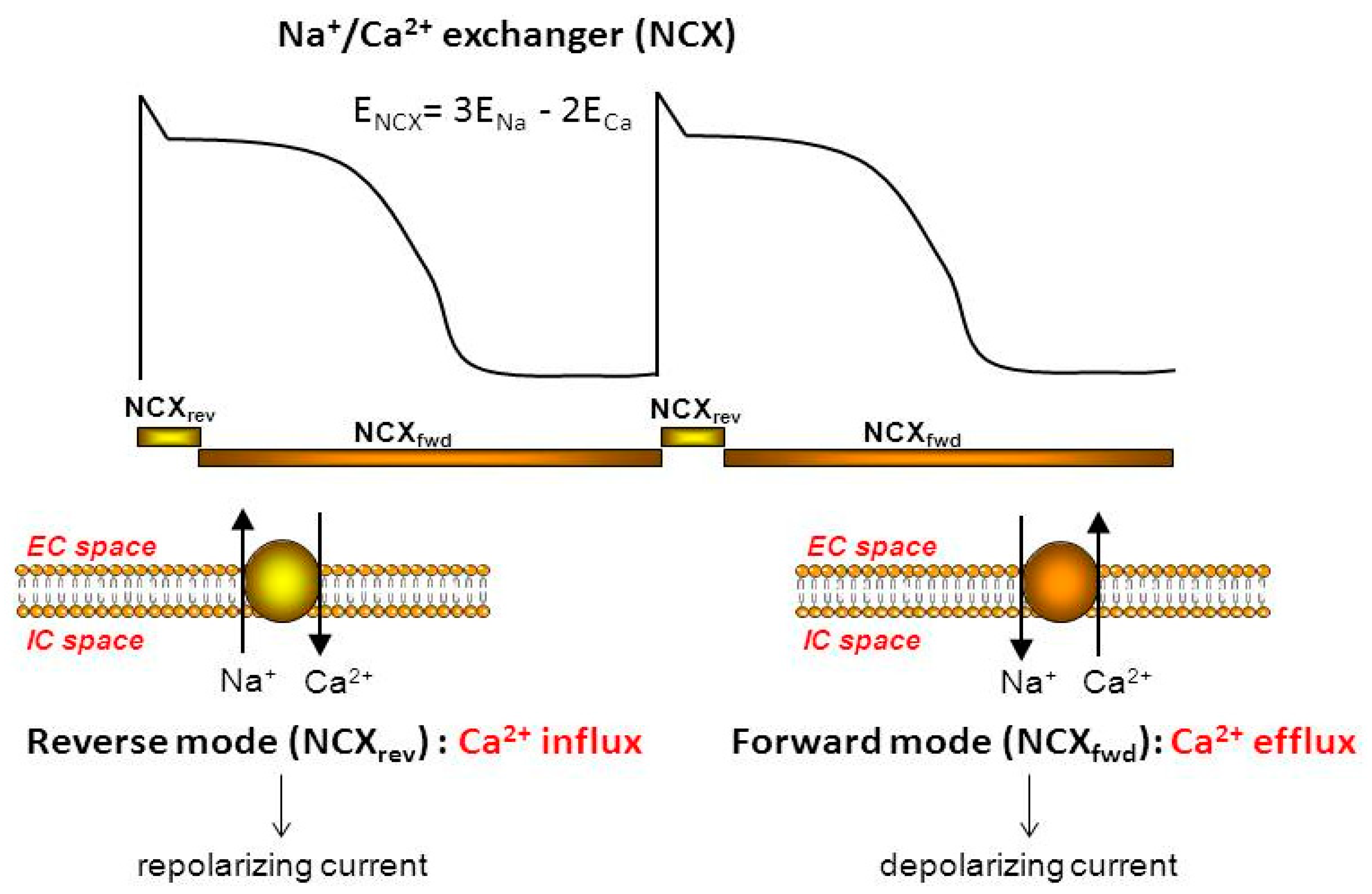
3. Physiology and Pharmacology of the Exchanger
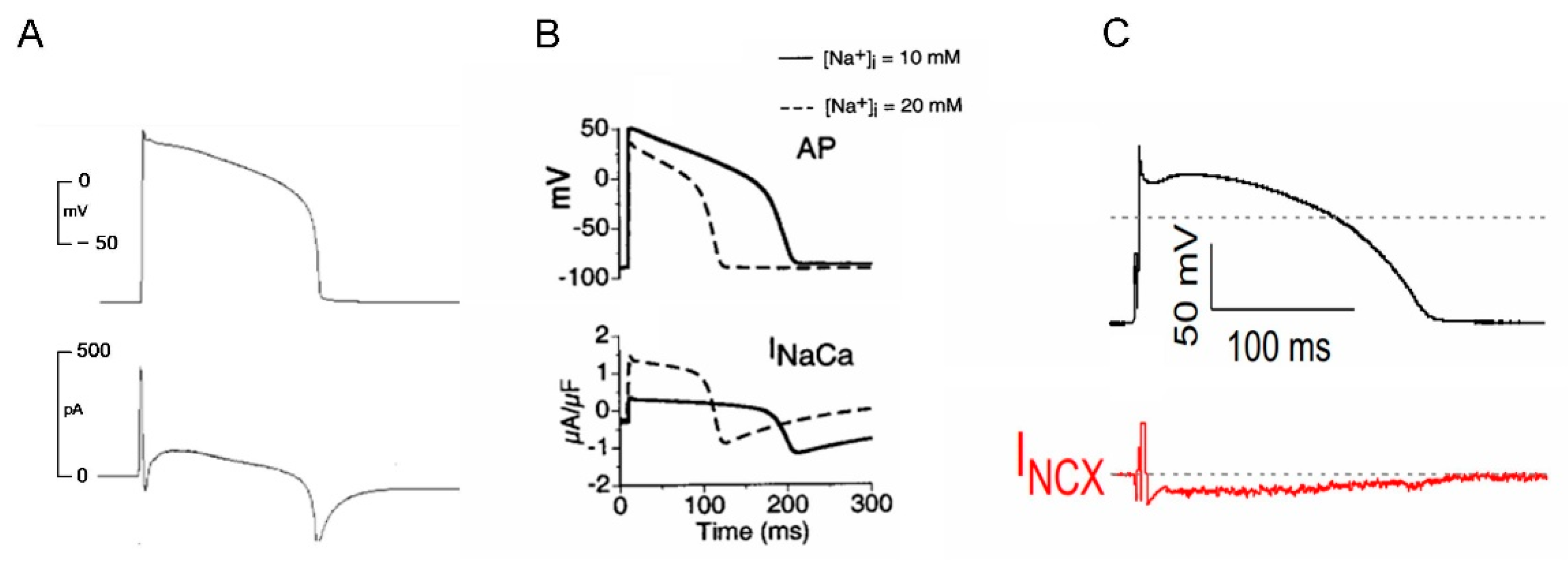
3.1. NCX Current during an Action Potential
3.2. Pharmacology of Novel NCX Inhibitors
3.3. Potential Therapeutic Benefit of Pharmacological NCX Inhibition
4. Effect of Novel NCX Inhibitors on Cellular Arrhythmogenic Mechanisms
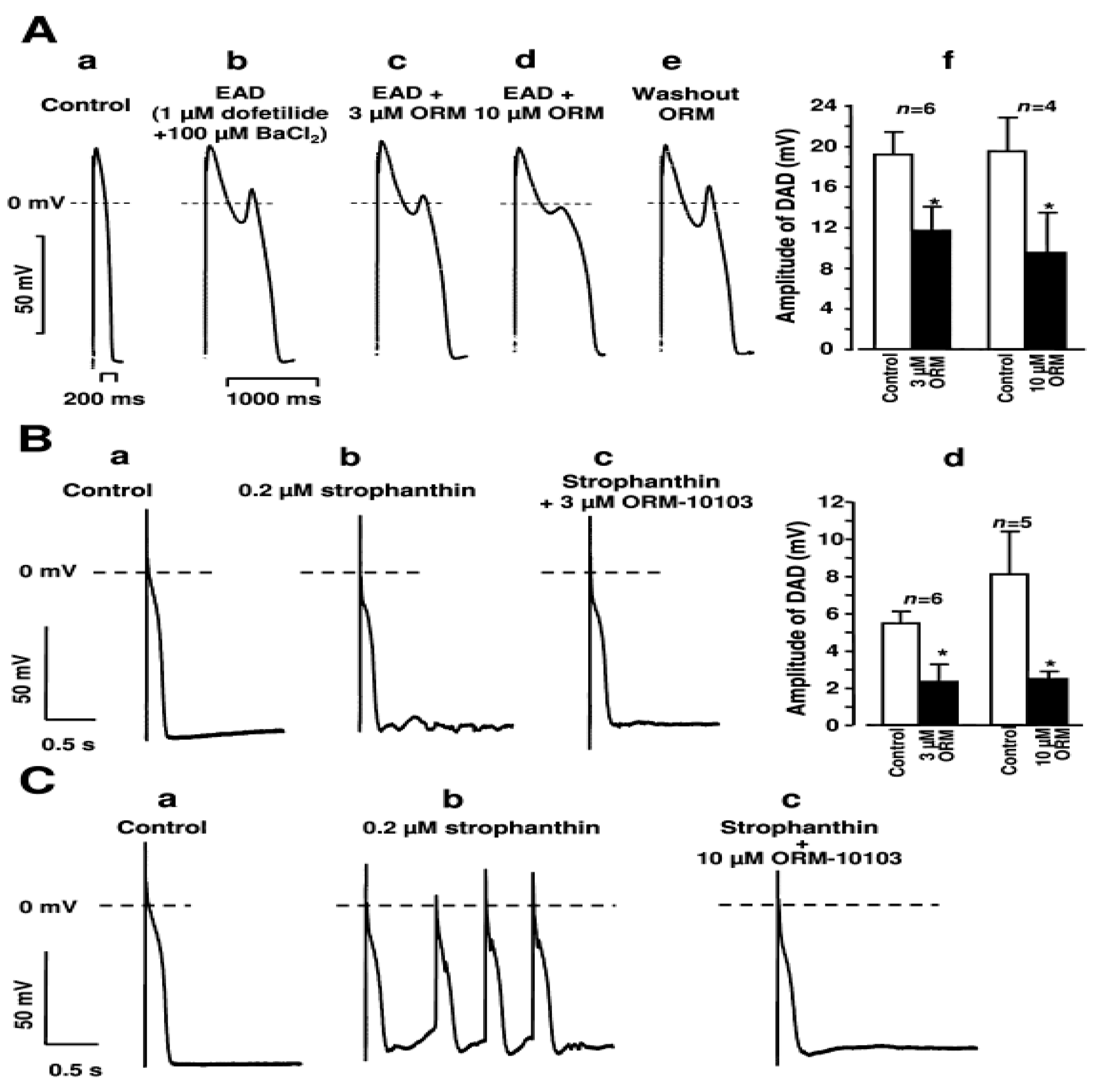
4.1. DAD and Ca2+ Waves
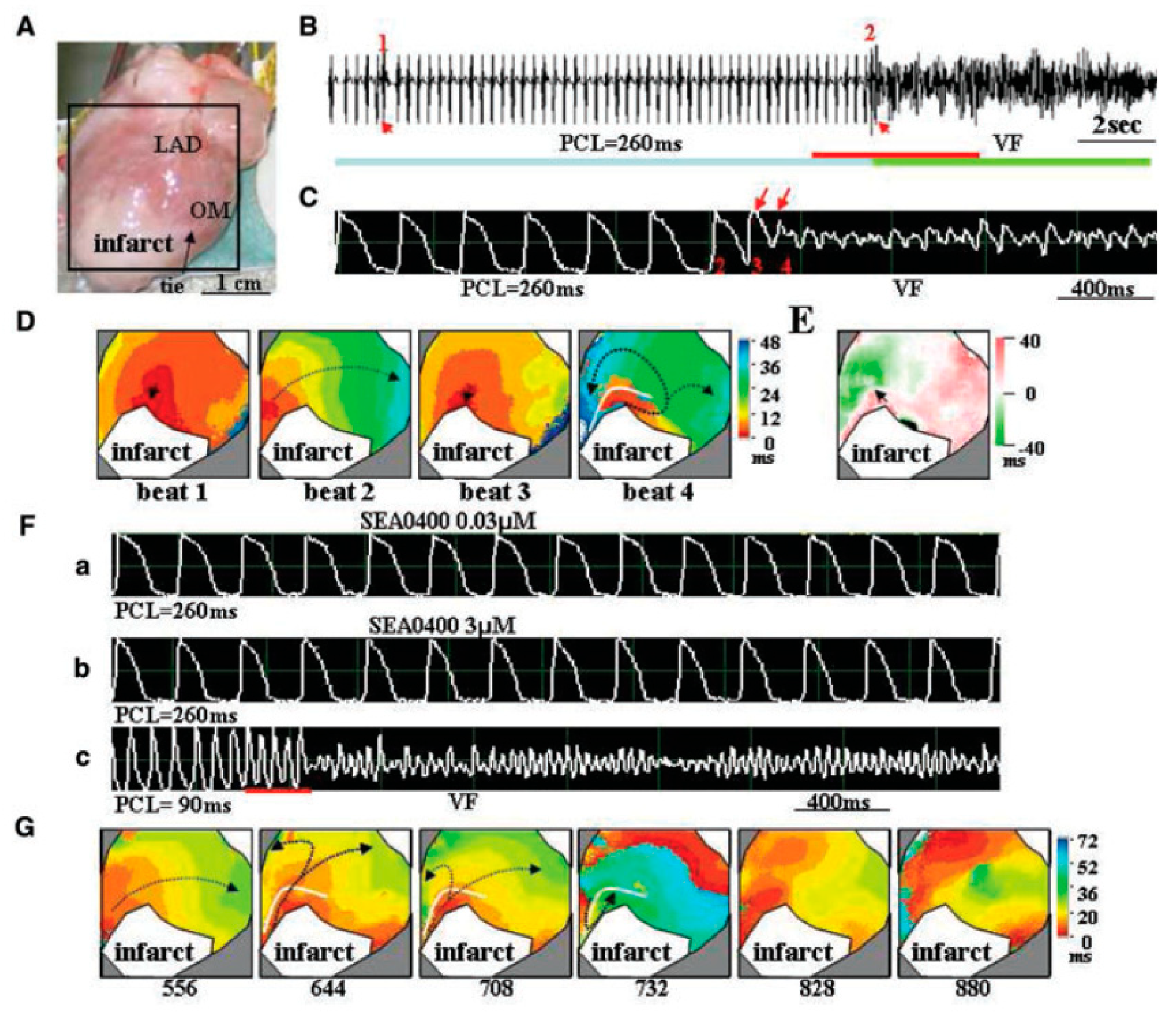
4.2. EAD, LQT and TdP
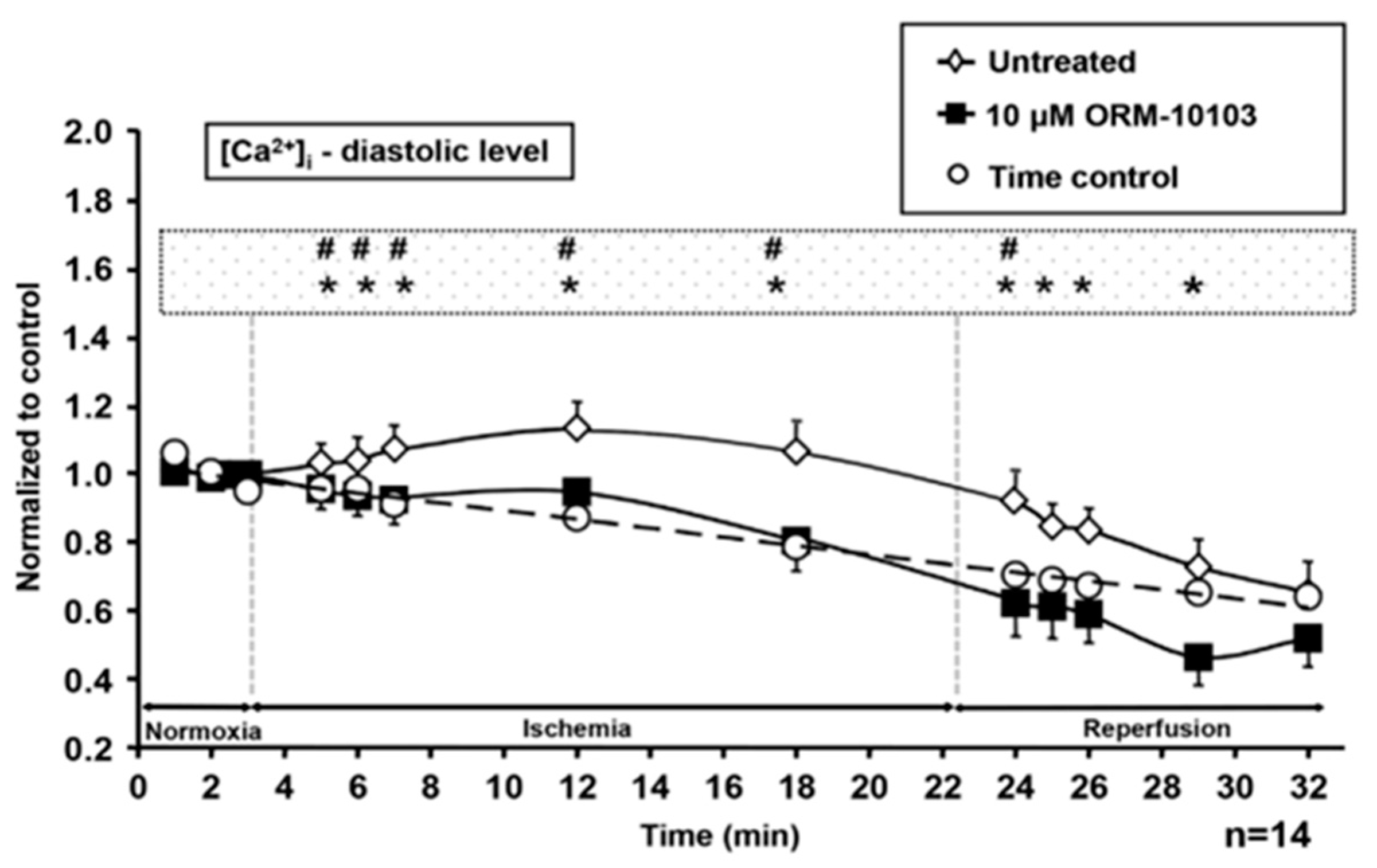
4.3. Ischemia-Reperfusion
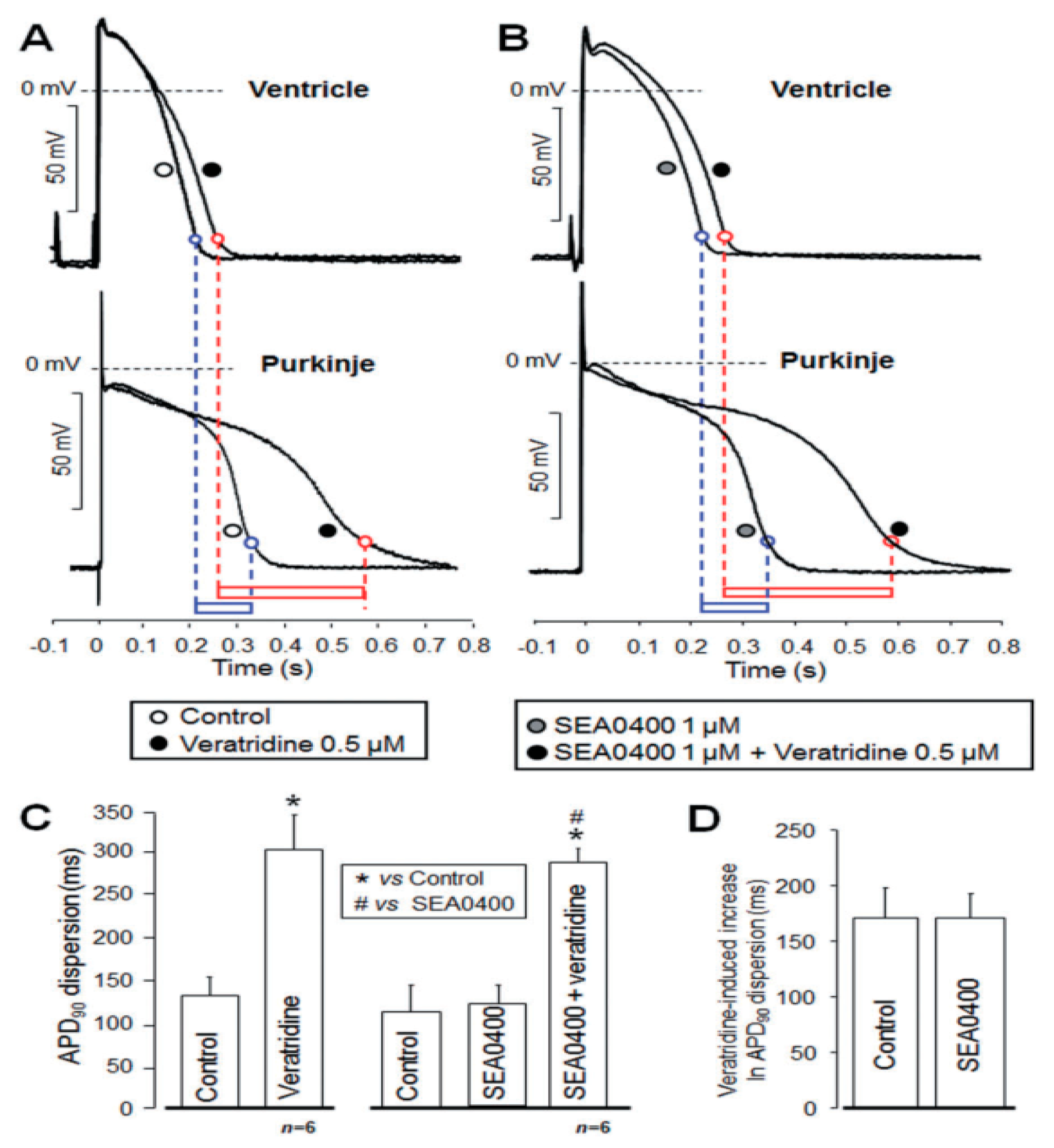
4.4. Action Potential Duration, Refractory Period, and APD-Dispersion
4.5. Action Potential and Ca2+ Transient Alternans
5. Inotropic Effects of Novel NCX Inhibitors
5.1. Selective NCX Inhibition Results in Positive Inotropy in Rat and Mouse and Human Atrium
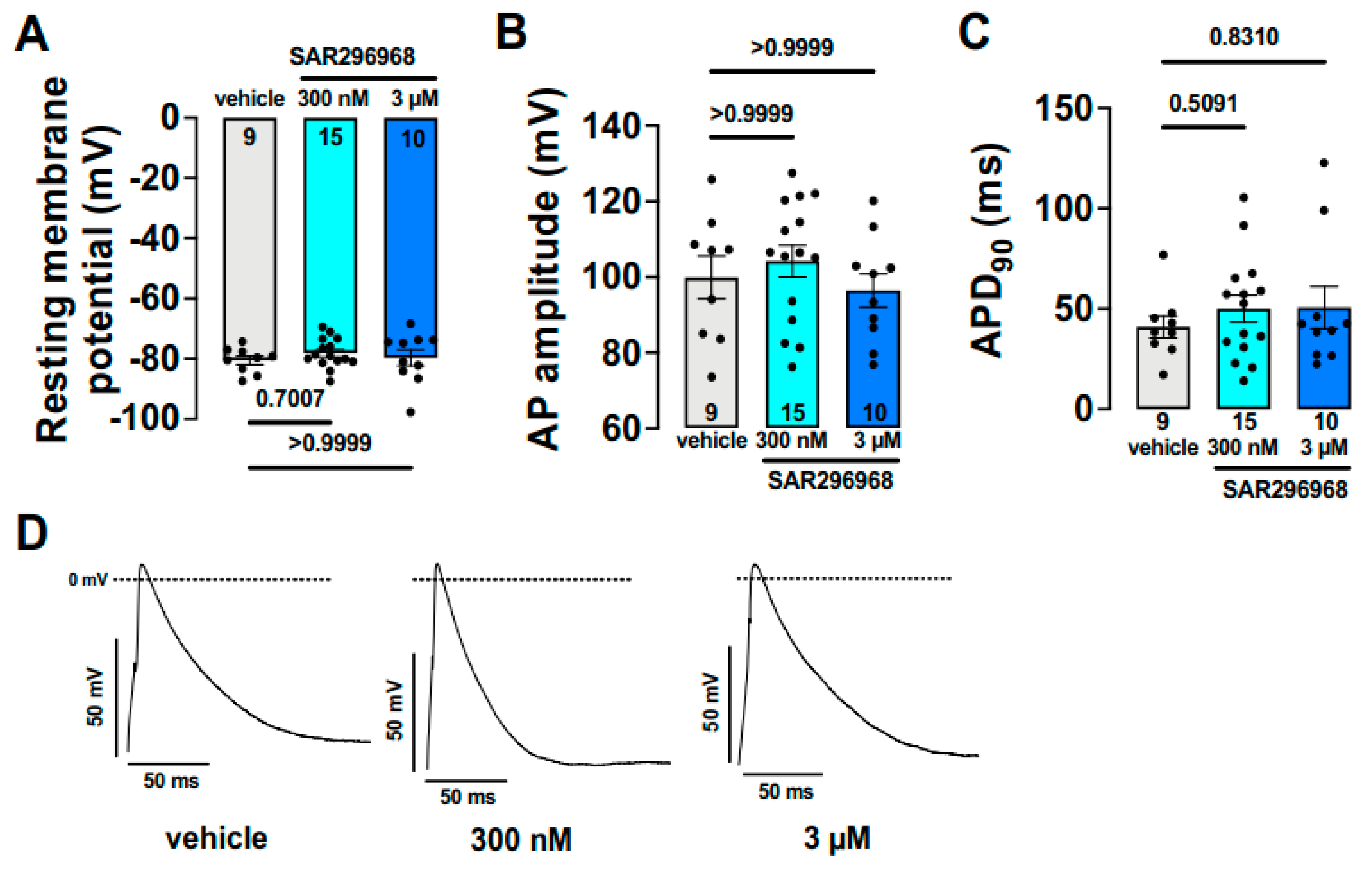
5.2. Selective NCX Inhibition Failed to Exert Positive Inotropy in Guinea Pigs, Rabbits and Dogs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Eisner, D.A.; Choi, H.S.; Díaz, M.E.; O’Neill, S.C.; Trafford, A. Integrative Analysis of Calcium Cycling in Cardiac Muscle. Circ. Res. 2000, 87, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Eisner, D.; Trafford, A.; Dñaz, M.; Overend, C.; O’Neill, S. The control of Ca release from the cardiac sarcoplasmic reticulum: Regulation versus autoregulation. Cardiovasc. Res. 1998, 38, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Parikh, R.R.; Patel, K.R.; Pergolizzi, J.V., Jr.; Breve, F.; Magnusson, P. Effects of Digoxin in Heart Failure (HF) With Reduced Ejection Fraction (EF). Cureus 2022, 14, e22778. [Google Scholar] [CrossRef] [PubMed]
- Hashemi-Shahri, S.H.; Aghajanloo, A.; Ghavami, V.; Arasteh, O.; Mohammadpour, A.H.; Reiner, Ž.; Sahebkar, A. Digoxin and Outcomes in Patients with Heart Failure and Preserved Ejection Fraction [HFpEF] Patients: A Systematic Review and Meta-analysis. Curr. Drug Targets 2022. [Google Scholar] [CrossRef]
- Ding, W.Y.; Boriani, G.; Marin, F.; Blomstrom-Lundqvist, C.; Potpara, T.S.; Fauchier, L.; Lip, G.Y.H.; ESC–EHRA EORP AF Long-Term General Registry Investigators. Outcomes of digoxin vs. beta blocker in atrial fibrillation: Report from ESC-EHRA EORP AF Long-Term General Registry. Eur. Heart J. Cardiovasc. Pharmacother. 2022, 8, 372–382. [Google Scholar] [CrossRef]
- Horváth, B.; Hézső, T.; Kiss, D.; Kistamas, K.; Magyar, J.; Nánási, P.P.; Bányász, T. Late Sodium Current Inhibitors as Potential Antiarrhythmic Agents. Front. Pharmacol. 2020, 11, 413. [Google Scholar] [CrossRef] [PubMed]
- Soliman, D.; Wang, L.; Hamming, K.S.; Yang, W.; Fatehi, M.; Carter, C.C.; Clanachan, A.S.; Light, P.E. Late sodium current inhibition alone with ranolazine is sufficient to reduce ischemia- and cardiac glycoside-induced calcium overload and contractile dysfunction mediated by reverse-mode sodium/calcium exchange. J. Pharmacol. Exp. Ther. 2012, 343, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Udvary, É.; Papp, J.G.; Végh, Á. Cardiovascular effects of the calcium sensitizer, levosimendan, in heart failure induced by rapid pacing in the presence of aortic constriction. Br. J. Pharmacol. 1995, 114, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Virág, L.; Hála, O.; Marton, A.; Varró, A.; Papp, J.G. Cardiac electrophysiological effects of levosimendan, a new calcium sensitizer. Gen. Pharmacol. Vasc. Syst. 1996, 27, 551–556. [Google Scholar] [CrossRef]
- Papp, Z.; Csapó, K.; Pollesello, P.; Haikala, H.; Édes, I. Pharmacological Mechanisms Contributing to the Clinical Efficacy of Levosimendan. Cardiovasc. Drug Rev. 2005, 23, 71–98. [Google Scholar] [CrossRef] [PubMed]
- Horváth, B.; Szentandrássy, N.; Veress, R.; Almássy, J.; Magyar, J.; Bányász, T.; Tóth, A.; Papp, Z.; Nánási, P.P. Frequency-dependent effects of omecamtiv mecarbil on cell shortening of isolated canine ventricular cardiomyocytes. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2017, 390, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, G.; Oláh, A.; Csipo, T.; Kovács, Á.; Pórszász, R.; Veress, R.; Horváth, B.; Nagy, L.; Bódi, B.; Fagyas, M.; et al. Omecamtiv mecarbil evokes diastolic dysfunction and leads to periodic electromechanical alternans. Basic Res. Cardiol. 2021, 116, 24. [Google Scholar] [CrossRef] [PubMed]
- Acsai, K.; Antoons, G.; Livshitz, L.; Rudy, Y.; Sipido, K.R. Microdomain [Ca2+] near ryanodine receptors as reported by L-type Ca2+ and Na+/Ca2+ exchange currents. J. Physiol. 2011, 589, 2569–2583. [Google Scholar] [CrossRef]
- Cannell, M.; Cheng, H.; Lederer, W. Spatial non-uniformities in [Ca2+]i during excitation-contraction coupling in cardiac myocytes. Biophys. J. 1994, 67, 1942–1956. [Google Scholar] [CrossRef]
- Cheng, H.; Lederer, W.J.; Cannell, M.B. Calcium sparks: Elementary events underlying excitation-contraction coupling in heart muscle. Science 1993, 262, 740–744. [Google Scholar] [CrossRef]
- Lukyanenko, V.; Gyorke, S. Ca2+ sparks and Ca2+ waves in saponin-permeabilized rat ventricular myocytes. J. Physiol. 1999, 521 Pt 3, 575–585. [Google Scholar] [CrossRef]
- Luo, D.; Yang, D.; Lan, X.; Li, K.; Li, X.; Chen, J.; Zhang, Y.; Xiao, R.-P.; Han, Q.; Cheng, H. Nuclear Ca2+ sparks and waves mediated by inositol 1,4,5-trisphosphate receptors in neonatal rat cardiomyocytes. Cell Calcium 2008, 43, 165–174. [Google Scholar] [CrossRef]
- Song, L.-S.; Sham, J.S.; Stern, M.D.; Lakatta, E.G.; Cheng, H. Direct measurement of SR release flux by tracking ‘Ca2+ spikes’ in rat cardiac myocytes. J. Physiol. 1998, 512, 677–691. [Google Scholar] [CrossRef]
- Cheng, H.; Lederer, W.J. Calcium sparks. Physiol. Rev. 2008, 88, 1491–1545. [Google Scholar] [CrossRef]
- Franzini-Armstrong, C.; Protasi, F.; Ramesh, V. Shape, Size, and Distribution of Ca2+ Release Units and Couplons in Skeletal and Cardiac Muscles. Biophys. J. 1999, 77, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Lytton, J. Na+/Ca2+ exchangers: Three mammalian gene families control Ca2+ transport. Biochem. J. 2007, 406, 365–382. [Google Scholar] [CrossRef]
- Bers, D.M.; Weber, C.R. Na/Ca Exchange Function in Intact Ventricular Myocytes. Ann. N. Y. Acad. Sci. 2006, 976, 500–512. [Google Scholar] [CrossRef]
- Egan, T.; Noble, D.; Noble, S.J.; Powell, T.; Spindler, A.J.; Twist, V.W. Sodium-calcium exchange during the action potential in guinea-pig ventricular cells. J. Physiol. 1989, 411, 639–661. [Google Scholar] [CrossRef] [PubMed]
- Dubell, W.H.; Boyett, M.R.; Spurgeon, H.A.; Talo, A.; Stern, M.; Lakatta, E. The cytosolic calcium transient modulates the action potential of rat ventricular myocytes. J. Physiol. 1991, 436, 347–369. [Google Scholar] [CrossRef] [PubMed]
- Noble, D.; Noble, S.J.; Bett, G.C.L.; Earm, Y.E.; Ho, W.K.; So, I.K. The Role of Sodium–Calcium Exchange during the Cardiac Action Potential. Ann. N. Y. Acad. Sci. 1991, 639, 334–353. [Google Scholar] [CrossRef]
- Luo, C.H.; Rudy, Y. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ. Res. 1994, 74, 1071–1096. [Google Scholar] [CrossRef]
- Grantham, C.; Cannell, M. Ca2+ Influx During the Cardiac Action Potential in Guinea Pig Ventricular Myocytes. Circ. Res. 1996, 79, 194–200. [Google Scholar] [CrossRef]
- Noble, D.; Varghese, A.; Kohl, P.; Noble, P. Improved guinea-pig ventricular cell model incorporating a diadic space, IKr and IKs, and length- and tension-dependent processes. Can. J. Cardiol. 1998, 14, 123–134. [Google Scholar]
- Faber, G.M.; Rudy, Y. Action Potential and Contractility Changes in [Na+]i Overloaded Cardiac Myocytes: A Simulation Study. Biophys. J. 2000, 78, 2392–2404. [Google Scholar] [CrossRef]
- Weber, C.R.; Piacentino, V., III; Ginsburg, K.S.; Houser, S.R.; Bers, D.M. Na+-Ca2+ exchange current and submembrane [Ca2+] during the cardiac action potential. Circ. Res. 2002, 90, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Armoundas, A.A.; Hobai, I.A.; Tomaselli, G.F.; Winslow, R.L.; O’Rourke, B. Role of Sodium-Calcium Exchanger in Modulating the Action Potential of Ventricular Myocytes from Normal and Failing Hearts. Circ. Res. 2003, 93, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Banyasz, T.; Horvath, B.; Jian, Z.; Izu, L.T.; Chen-Izu, Y. Profile of L-type Ca2+ current and Na+/Ca2+ exchange current during cardiac action potential in ventricular myocytes. Heart Rhythm 2012, 9, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Nagy, N.; Kormos, A.; Kohajda, Z.; Szebeni, A.; Szepesi, J.; Pollesello, P.; Levijoki, J.; Acsai, K.; Virag, L.; Nanasi, P.P.; et al. Selective Na+/Ca2+ exchanger inhibition prevents Ca2+ overload-induced triggered arrhythmias. Br. J. Pharmacol. 2014, 171, 5665–5681. [Google Scholar] [CrossRef]
- Horváth, B.; Kiss, D.; Dienes, C.; Hézső, T.; Kovács, Z.; Szentandrássy, N.; Almássy, J.; Magyar, J.; Bányász, T.; Nánási, P.P. Ion current profiles in canine ventricular myocytes obtained by the “onion peeling” technique. J. Mol. Cell. Cardiol. 2021, 158, 153–162. [Google Scholar] [CrossRef]
- Birinyi, P.; Acsai, K.; Banyasz, T.; Toth, A.; Horvath, B.; Virag, L.; Szentandrassy, N.; Magyar, J.; Varro, A.; Fulop, F.; et al. Effects of SEA0400 and KB-R7943 on Na+/Ca2+ exchange current and L-type Ca2+ current in canine ventricular cardiomyocytes. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2005, 372, 63–70. [Google Scholar] [CrossRef]
- Jost, N.; Nagy, N.; Corici, C.; Kohajda, Z.; Horváth, A.; Acsai, K.; Biliczki, P.; Levijoki, J.; Pollesello, P.; Koskelainen, T.; et al. ORM-10103, a novel specific inhibitor of the Na+/Ca2+ exchanger, decreases early and delayed afterdepolarizations in the canine heart. Br. J. Pharmacol. 2013, 170, 768–778. [Google Scholar] [CrossRef]
- Kohajda, Z.; Farkas-Morvay, N.; Jost, N.; Nagy, N.; Geramipour, A.; Horváth, A.; Varga, R.S.; Hornyik, T.; Corici, C.; Acsai, K.; et al. The Effect of a Novel Highly Selective Inhibitor of the Sodium/Calcium Exchanger (NCX) on Cardiac Arrhythmias in In Vitro and In Vivo Experiments. PLoS ONE 2016, 11, e0166041. [Google Scholar] [CrossRef]
- Eisner, D.; Bode, E.; Venetucci, L.; Trafford, A. Calcium flux balance in the heart. J. Mol. Cell. Cardiol. 2013, 58, 110–117. [Google Scholar] [CrossRef]
- Despa, S.; Bers, D.M. Na+ transport in the normal and failing heart—Remember the balance. J. Mol. Cell. Cardiol. 2013, 61, 2–10. [Google Scholar] [CrossRef]
- Venetucci, L.A.; Trafford, A.; O’Neill, S.C.; Eisner, D.A. Na/Ca Exchange: Regulator of Intracellular Calcium and Source of Arrhythmias in the Heart. Ann. N. Y. Acad. Sci. 2007, 1099, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Antoons, G.; Willems, R.; Sipido, K.R. Alternative strategies in arrhythmia therapy: Evaluation of Na/Ca exchange as an anti-arrhythmic target. Pharmacol. Ther. 2012, 134, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Cranefield, P.F. Action potentials, afterpotentials, and arrhythmias. Circ. Res. 1977, 41, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Wit, A.L. Afterdepolarizations and triggered activity as a mechanism for clinical arrhythmias. Pacing Clin. Electrophysiol. 2018, 41, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Shimada, H.; Namekata, I.; Kawanishi, T.; Iida-Tanaka, N.; Shigenobu, K. Involvement of the Na+/Ca2+ exchanger in ouabain-induced inotropy and arrhythmogenesis in guinea-pig myocardium as revealed by SEA0400. J. Pharmacol. Sci. 2007, 103, 241–246. [Google Scholar] [CrossRef]
- Namekata, I.; Tsuneoka, Y.; Takahara, A.; Shimada, H.; Sugimoto, T.; Takeda, K.; Nagaharu, M.; Shigenobu, K.; Kawanishi, T.; Tanaka, H. Involvement of the Na+/Ca2+ exchanger in the automaticity of guinea-pig pulmonary vein myocardium as revealed by SEA0400. J. Pharmacol. Sci. 2009, 110, 111–116. [Google Scholar] [CrossRef]
- Nagy, Z.A.; Virag, L.; Toth, A.; Biliczki, P.; Acsai, K.; Banyasz, T.; Nanasi, P.; Papp, J.G.; Varro, A. Selective inhibition of sodium-calcium exchanger by SEA-0400 decreases early and delayed after depolarization in canine heart. Br. J. Pharmacol. 2009, 143, 827–831. [Google Scholar] [CrossRef]
- Fujiwara, K.; Tanaka, H.; Mani, H.; Nakagami, T.; Takamatsu, T. Burst emergence of intracellular Ca2+ waves evokes arrhythmogenic oscillatory depolarization via the Na+-Ca2+ exchanger: Simultaneous confocal recording of membrane potential and intracellular Ca2+ in the heart. Circ. Res. 2008, 103, 509–518. [Google Scholar] [CrossRef]
- Oravecz, K.; Kormos, A.; Gruber, A.; Márton, Z.; Kohajda, Z.; Mirzaei, L.; Jost, N.; Levijoki, J.; Pollesello, P.; Koskelainen, T.; et al. Inotropic effect of NCX inhibition depends on the relative activity of the reverse NCX assessed by a novel inhibitor ORM-10962 on canine ventricular myocytes. Eur. J. Pharmacol. 2018, 818, 278–286. [Google Scholar] [CrossRef]
- Gussak, G.; Marszalec, W.; Yoo, S.; Modi, R.; O’Callaghan, C.; Aistrup, G.L.; Cordeiro, J.M.; Goodrow, R.; Kanaporis, G.; Blatter, L.A.; et al. Triggered Ca2+ Waves Induce Depolarization of Maximum Diastolic Potential and Action Potential Prolongation in Dog Atrial Myocytes. Circ. Arrhythmia Electrophysiol. 2020, 13, e008179. [Google Scholar] [CrossRef]
- January, C.T.; Riddle, J.M. Early afterdepolarizations: Mechanism of induction and block. A role for L-type Ca2+ current. Circ. Res. 1989, 64, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Horvath, B.; Hezso, T.; Szentandrassy, N.; Kistamas, K.; Arpadffy-Lovas, T.; Varga, R.; Gazdag, P.; Veress, R.; Dienes, C.; Baranyai, D.; et al. Late sodium current in human, canine and guinea pig ventricular myocardium. J. Mol. Cell. Cardiol. 2020, 139, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Horvath, B.; Banyasz, T.; Jian, Z.; Hegyi, B.; Kistamas, K.; Nanasi, P.P.; Izu, L.T.; Chen-Izu, Y. Dynamics of the late Na+ current during cardiac action potential and its contribution to afterdepolarizations. J. Mol. Cell. Cardiol. 2013, 64, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Zang, Y.; Zheng, D.; Xia, L.; Gong, Y. Role of CaMKII and PKA in Early Afterdepolarization of Human Ventricular Myocardium Cell: A Computational Model Study. Comput. Math. Methods Med. 2016, 2016, 4576313. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Mincholé, A.; Zacur, E.; Quinn, T.A.; Taggart, P.; Rodriguez, B. Early afterdepolarizations promote transmural reentry in ischemic human ventricles with reduced repolarization reserve. Prog. Biophys. Mol. Biol. 2016, 120, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.N.; Garfinkel, A.; Karagueuzian, H.S.; Chen, P.-S.; Qu, Z. Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm 2010, 7, 1891–1899. [Google Scholar] [CrossRef]
- Antzelevitch, C.; Oliva, A. Amplification of spatial dispersion of repolarization underlies sudden cardiac death associated with catecholaminergic polymorphic VT, long QT, short QT and Brugada syndromes. J. Intern. Med. 2006, 259, 48–58. [Google Scholar] [CrossRef]
- Amran, M.S.; Hashimoto, K.; Homma, N. Effects of sodium-calcium exchange inhibitors, KB-R7943 and SEA0400, on aconitine-induced arrhythmias in guinea pigs in vivo, in vitro, and in computer simulation studies. J. Pharmacol. Exp. Ther. 2004, 310, 83–89. [Google Scholar] [CrossRef]
- Bourgonje, V.J.; Vos, M.A.; Ozdemir, S.; Doisne, N.; Acsai, K.; Varro, A.; Sztojkov-Ivanov, A.; Zupko, I.; Rauch, E.; Kattner, L.; et al. Combined Na+/Ca2+ exchanger and L-type calcium channel block as a potential strategy to suppress arrhythmias and maintain ventricular function. Circ. Arrhythmia Electrophysiol. 2013, 6, 371–379. [Google Scholar] [CrossRef]
- Farkas, A.S.; Makra, P.; Csik, N.; Orosz, S.; Shattock, M.J.; Fulop, F.; Forster, T.; Csanady, M.; Papp, J.G.; Varro, A.; et al. The role of the Na+/Ca2+ exchanger, INa and ICaL in the genesis of dofetilide-induced torsades de pointes in isolated, AV-blocked rabbit hearts. Br. J. Pharmacol. 2009, 156, 920–932. [Google Scholar] [CrossRef]
- Milberg, P.; Pott, C.; Fink, M.; Frommeyer, G.; Matsuda, T.; Baba, A.; Osada, N.; Breithardt, G.; Noble, D.; Eckardt, L. Inhibition of the Na+/Ca2+ exchanger suppresses torsades de pointes in an intact heart model of long QT syndrome-2 and long QT syndrome-3. Heart Rhythm 2008, 5, 1444–1452. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wen, H.; Fefelova, N.; Allen, C.; Baba, A.; Matsuda, T.; Xie, L.-H. Revisiting the ionic mechanisms of early afterdepolarizations in cardiomyocytes: Predominant by Ca waves or Ca currents? Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1636–H1644. [Google Scholar] [CrossRef]
- Chang, P.-C.; Lu, Y.-Y.; Lee, H.-L.; Lin, S.-F.; Chu, Y.; Wen, M.-S.; Chou, C.-C. Paradoxical Effects of Sodium–Calcium Exchanger Inhibition on Torsade de Pointes and Early Afterdepolarization in a Heart Failure Rabbit Model. J. Cardiovasc. Pharmacol. 2018, 72, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, E. Cardiac Ionic Currents and Acute Ischemia: From Channels to Arrhythmias. Physiol. Rev. 1999, 79, 917–1017. [Google Scholar] [CrossRef]
- Janse, M.J.; Kléber, A.G. Electrophysiological changes and ventricular arrhythmias in the early phase of regional myocardial ischemia. Circ. Res. 1981, 49, 1069–1081. [Google Scholar] [CrossRef]
- Janse, M.J.; Kleber, A.G.; Capucci, A.; Coronel, R.; Wilms-Schopman, F. Electrophysiological basis for arrhythmias caused by acute ischemia: Role of the subendocardium. J. Mol. Cell. Cardiol. 1986, 18, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.M.; Boyden, P.A. Electrical remodeling in ischemia and infarction. Cardiovasc. Res. 1999, 42, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Imahashi, K.; Kusuoka, H.; Hashimoto, K.; Yoshioka, J.; Yamaguchi, H.; Nishimura, T. Intracellular Sodium Accumulation During Ischemia as the Substrate for Reperfusion Injury. Circ. Res. 1999, 84, 1401–1406. [Google Scholar] [CrossRef] [PubMed]
- Ladilov, Y.; Haffner, S.; Balser-Schafer, C.; Maxeiner, H.; Piper, H.M. Cardioprotective effects of KB-R7943: A novel inhibitor of the reverse mode of Na+/Ca2+ exchanger. Am. J. Physiol. Heart Circ. Physiol. 1999, 276, H1868–H1876. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Cross, H.; Steenbergen, C. Sodium Regulation During Ischemia Versus Reperfusion and Its Role in Injury. Circ. Res. 1999, 84, 1469–1470. [Google Scholar] [CrossRef]
- Cross, H.R.; Lu, L.; Steenbergen, C.; Philipson, K.D.; Murphy, E. Overexpression of the cardiac Na+/Ca2+ exchanger increases susceptibility to ischemia/reperfusion injury in male, but not female, transgenic mice. Circ. Res. 1998, 83, 1215–1223. [Google Scholar] [CrossRef]
- Sugishita, K.; Su, Z.; Li, F.; Philipson, K.D.; Barry, W.H. Gender influences [Ca2+]i during metabolic inhibition in myocytes overexpressing the Na+-Ca2+ exchanger. Circulation 2001, 104, 2101–2106. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, Y.; Zhu, B.-M.; Chen, J.; Kamiya, K.; Miyamoto, S.; Hashimoto, K. Effects of SEA0400, a Na+/Ca2+ exchange inhibitor, on ventricular arrhythmias in the in vivo dogs. Eur. J. Pharmacol. 2005, 506, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.R.; Yang, P.; Remeysen, P.; Saels, A.; Dai, D.Z.; De Clerck, F. Ischemia/reperfusion-induced arrhythmias in anaesthetized rats: A role of Na+ and Ca2+ influx. Eur. J. Pharmacol. 1999, 365, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Zhu, B.-M.; Kamiya, K.; Nagasawa, Y.; Hashimoto, K. KB-R7943, a Na+/Ca2+ Exchange Inhibitor, Does Not Suppress Ischemia/Reperfusion Arrhythmias nor Digitalis Arrhythmias in Dogs. Jpn. J. Pharmacol. 2002, 90, 229–235. [Google Scholar] [CrossRef][Green Version]
- Chou, C.-C.; Chang, P.-C.; Wen, M.-S.; Lee, H.-L.; Chu, Y.; Baba, A.; Matsuda, T.; Yeh, S.-J.; Wu, D. Effects of SEA0400 on Arrhythmogenicity in a Langendorff-Perfused 1-Month Myocardial Infarction Rabbit Model. Pacing Clin. Electrophysiol. 2013, 36, 596–606. [Google Scholar] [CrossRef]
- Takahashi, K.; Takahashi, T.; Suzuki, T.; Onishi, M.; Tanaka, Y.; Hamano-Takahashi, A.; Ota, T.; Kameo, K.; Matsuda, T.; Baba, A. Protective effects of SEA0400, a novel and selective inhibitor of the Na+/Ca2+ exchanger, on myocardial ischemia-reperfusion injuries. Eur. J. Pharmacol. 2003, 458, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Feng, N.C.; Satoh, H.; Urushida, T.; Katoh, H.; Terada, H.; Watanabe, Y.; Hayashi, H. A selective inhibitor of Na+/Ca2+ exchanger, SEA0400, preserves cardiac function and high-energy phosphates against ischemia/reperfusion injury. J. Cardiovasc. Pharmacol. 2006, 47, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Szepesi, J.; Acsai, K.; Sebok, Z.; Prorok, J.; Pollesello, P.; Papp, J.G.; Varro, A.; Toth, A. Comparison of the efficiency of Na+/Ca2+ exchanger or Na+/H+ exchanger inhibition and their combination in reducing coronary reperfusion-induced arrhythmias. J. Physiol. Pharmacol. 2015, 66, 215–226. [Google Scholar] [PubMed]
- Morita, N.; Lee, J.-H.; Bapat, A.; Fishbein, M.C.; Mandel, W.J.; Chen, P.-S.; Weiss, J.N.; Karagueuzian, H.S. Glycolytic inhibition causes spontaneous ventricular fibrillation in aged hearts. Am. J. Physiol. Circ. Physiol. 2011, 301, H180–H191. [Google Scholar] [CrossRef] [PubMed]
- Kormos, A.; Nagy, N.; Acsai, K.; Váczi, K.; Ágoston, S.; Pollesello, P.; Levijoki, J.; Szentandrassy, N.; Papp, J.G.; Varro, A.; et al. Efficacy of selective NCX inhibition by ORM-10103 during simulated ischemia/reperfusion. Eur. J. Pharmacol. 2014, 740, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Souza, A.C.M.; Grabe-Guimarães, A.; Cruz, J.D.S.; Santos-Miranda, A.; Farah, C.; Oliveira, L.T.; Lucas, A.; Aimond, F.; Sicard, P.; Mosqueira, V.C.F.; et al. Mechanisms of artemether toxicity on single cardiomyocytes and protective effect of nanoencapsulation. Br. J. Pharmacol. 2020, 177, 4448–4463. [Google Scholar] [CrossRef]
- Santos-Miranda, A.; Costa, A.D.; Joviano-Santos, J.V.; Rhana, P.; Bruno, A.S.; Rocha, P.; Cau, S.B.; Vieira, L.Q.; Cruz, J.S.; Roman-Campos, D. Inhibition of calcium/calmodulin (Ca2+/CaM)-Calcium/calmodulin-dependent protein kinase II (CaMKII) axis reduces in vitro and ex vivo arrhythmias in experimental Chagas disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21901. [Google Scholar]
- Tadros, G.M.; Zhang, X.-Q.; Song, J.; Carl, L.L.; Rothblum, L.I.; Tian, Q.; Dunn, J.; Lytton, J.; Cheung, J.Y. Effects of Na+/Ca2+ exchanger downregulation on contractility and [Ca2+]i transients in adult rat myocytes. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1616–H1626. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tanaka, H.; Namekata, I.; Takeda, K.; Kazama, A.; Shimizu, Y.; Moriwaki, R.; Hirayama, W.; Sato, A.; Kawanishi, T.; Shigenobu, K. Unique excitation-contraction characteristics of mouse myocardium as revealed by SEA0400, a specific inhibitor of Na+-Ca2+ exchanger. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2005, 371, 526–534. [Google Scholar] [CrossRef]
- Szlovák, J.; Tomek, J.; Zhou, X.; Tóth, N.; Veress, R.; Horváth, B.; Szentandrássy, N.; Levijoki, J.; Papp, J.G.; Herring, N.; et al. Blockade of sodium-calcium exchanger via ORM-10962 attenuates cardiac alternans. J. Mol. Cell. Cardiol. 2020, 153, 111–122. [Google Scholar] [CrossRef]
- Antzelevitch, C. Role of spatial dispersion of repolarization in inherited and acquired sudden cardiac death syndromes. Am. J. Physiol. Circ. Physiol. 2007, 293, H2024–H2038. [Google Scholar] [CrossRef] [PubMed]
- Coronel, R.; Wilms-Schopman, F.J.; Opthof, T.; Janse, M.J. Dispersion of repolarization and arrhythmogenesis. Heart Rhythm 2009, 6, 537–543. [Google Scholar] [CrossRef]
- Milberg, P.; Pott, C.; Frommeyer, G.; Fink, M.; Ruhe, M.; Matsuda, T.; Baba, A.; Klocke, R.; Quang, T.H.; Nikol, S.; et al. Acute inhibition of the Na+/Ca2+ exchanger reduces proarrhythmia in an experimental model of chronic heart failure. Heart Rhythm 2012, 9, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Hegner, P.; Drzymalski, M.; Biedermann, A.; Memmel, B.; Durczok, M.; Wester, M.; Floerchinger, B.; Provaznik, Z.; Schmid, C.; Zausig, Y.; et al. SAR296968, a Novel Selective Na+/Ca2+ Exchanger Inhibitor, Improves Ca2+ Handling and Contractile Function in Human Atrial Cardiomyocytes. Biomedicines 2022, 10, 1932. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.D.; Rosenbaum, D.S. Mechanisms of arrythmogenic cardiac alternans. EP Eur. 2007, 9 Suppl. S6, vi77–vi82. [Google Scholar] [CrossRef] [PubMed]
- Narayan, S.M.; Franz, M.R.; Clopton, P.; Pruvot, E.J.; Krummen, D.E. Repolarization alternans reveals vulnerability to human atrial fibrillation. Circulation 2011, 123, 2922–2930. [Google Scholar] [CrossRef] [PubMed]
- Nolasco, J.B.; Dahlen, R.W. A graphic method for the study of alternation in cardiac action potentials. J. Appl. Physiol. 1968, 25, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Díaz, M.E.; O’Neill, S.C.; Eisner, D.A. Sarcoplasmic Reticulum Calcium Content Fluctuation Is the Key to Cardiac Alternans. Circ. Res. 2004, 94, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.J.; McHarg, J.L.; Gilmour, R.F., Jr. Ionic mechanism of electrical alternans. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H516–H530. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.N.; Blatter, L.A. Cardiac alternans and intracellular calcium cycling. Clin. Exp. Pharmacol. Physiol. 2014, 41, 524–532. [Google Scholar] [CrossRef]
- Wan, X.; Cutler, M.; Song, Z.; Karma, A.; Matsuda, T.; Baba, A.; Rosenbaum, D.S. New experimental evidence for mechanism of arrhythmogenic membrane potential alternans based on balance of electrogenic INCX/ICa currents. Heart Rhythm 2012, 9, 1698–1705. [Google Scholar] [CrossRef]
- Hobai, I.A.; Maack, C.; O’Rourke, B. Partial Inhibition of Sodium/Calcium Exchange Restores Cellular Calcium Handling in Canine Heart Failure. Circ. Res. 2004, 95, 292–299. [Google Scholar] [CrossRef]
- Acsai, K.; Kun, A.; Farkas, A.S.; Fulop, F.; Nagy, N.; Balazs, M.; Szentandrassy, N.; Nanasi, P.P.; Papp, J.G.; Varro, A.; et al. Effect of partial blockade of the Na+/Ca2+-exchanger on Ca2+ handling in isolated rat ventricular myocytes. Eur. J. Pharmacol. 2007, 576, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Grandi, E.; Morotti, S.; Ginsburg, K.S.; Severi, S.; Bers, D.M. Interplay of voltage and Ca-dependent inactivation of L-type Ca current. Prog. Biophys. Mol. Biol. 2010, 103, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Farkas, A.S.; Acsai, K.; Nagy, N.; Toth, A.; Fulop, F.; Seprenyi, G.; Birinyi, P.; Nanasi, P.P.; Forster, T.; Csanady, M.; et al. Na+/Ca2+ exchanger inhibition exerts a positive inotropic effect in the rat heart, but fails to influence the contractility of the rabbit heart. Br. J. Pharmacol. 2009, 154, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Szentandrassy, N.; Birinyi, P.; Szigeti, G.; Farkas, A.; Magyar, J.; Toth, A.; Csernoch, L.; Varro, A.; Nanasi, P.P. SEA0400 fails to alter the magnitude of intracellular Ca2+ transients and contractions in Langendorff-perfused guinea pig heart. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2008, 378, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, S.; Bito, V.; Holemans, P.; Vinet, L.; Mercadier, J.-J.; Varro, A.; Sipido, K.R. Pharmacological Inhibition of Na/Ca Exchange Results in Increased Cellular Ca2+ Load Attributable to the Predominance of Forward Mode Block. Circ. Res. 2008, 102, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Primessnig, U.; Bracic, T.; Levijoki, J.; Otsomaa, L.; Pollesello, P.; Falcke, M.; Pieske, B.; Heinzel, F.R. Long-term effects of Na+/Ca2+ exchanger inhibition with ORM-11035 improves cardiac function and remodelling without lowering blood pressure in a model of heart failure with preserved ejection fraction. Eur. J. Heart Fail. 2019, 21, 1543–1552. [Google Scholar] [CrossRef]
- Birinyi, P.; Toth, A.; Jona, I.; Acsai, K.; Almassy, J.; Nagy, N.; Prorok, J.; Gherasim, I.; Papp, Z.; Hertelendi, Z.; et al. The Na+/Ca2+ exchange blocker SEA0400 fails to enhance cytosolic Ca2+ transient and contractility in canine ventricular cardiomyocytes. Cardiovasc. Res. 2008, 78, 476–484. [Google Scholar] [CrossRef]
- Jin, G.; Manninger, M.; Adelsmayr, G.; Schwarzl, M.; Alogna, A.; Schönleitner, P.; Zweiker, D.; Blaschke, F.; Sherif, M.; Radulovic, S.; et al. Cellular contribution to left and right atrial dysfunction in chronic arterial hypertension in pigs. ESC Heart Fail. 2021, 8, 151–161. [Google Scholar] [CrossRef]
- Otsomaa, L.; Levijoki, J.; Wohlfahrt, G.; Chapman, H.; Koivisto, A.P.; Syrjanen, K.; Koskelainen, T.; Peltokorpi, S.E.; Finckenberg, P.; Heikkila, A.; et al. Discovery and characterization of ORM-11372, a novel inhibitor of the sodium-calcium exchanger with positive inotropic activity. Br. J. Pharmacol. 2020, 177, 5534–5554. [Google Scholar] [CrossRef]
- Pelat, M.; Barbe, F.; Daveu, C.; Ly-Nguyen, L.; Lartigue, T.; Marque, S.; Tavares, G.; Ballet, V.; Guillon, J.-M.; Steinmeyer, K.; et al. SAR340835, a Novel Selective Na+/Ca2+ Exchanger Inhibitor, Improves Cardiac Function and Restores Sympathovagal Balance in Heart Failure. J. Pharmacol. Exp. Ther. 2021, 377, 293–304. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagy, N.; Tóth, N.; Nánási, P.P. Antiarrhythmic and Inotropic Effects of Selective Na+/Ca2+ Exchanger Inhibition: What Can We Learn from the Pharmacological Studies? Int. J. Mol. Sci. 2022, 23, 14651. https://doi.org/10.3390/ijms232314651
Nagy N, Tóth N, Nánási PP. Antiarrhythmic and Inotropic Effects of Selective Na+/Ca2+ Exchanger Inhibition: What Can We Learn from the Pharmacological Studies? International Journal of Molecular Sciences. 2022; 23(23):14651. https://doi.org/10.3390/ijms232314651
Chicago/Turabian StyleNagy, Norbert, Noémi Tóth, and Péter P. Nánási. 2022. "Antiarrhythmic and Inotropic Effects of Selective Na+/Ca2+ Exchanger Inhibition: What Can We Learn from the Pharmacological Studies?" International Journal of Molecular Sciences 23, no. 23: 14651. https://doi.org/10.3390/ijms232314651
APA StyleNagy, N., Tóth, N., & Nánási, P. P. (2022). Antiarrhythmic and Inotropic Effects of Selective Na+/Ca2+ Exchanger Inhibition: What Can We Learn from the Pharmacological Studies? International Journal of Molecular Sciences, 23(23), 14651. https://doi.org/10.3390/ijms232314651