
The Downstaging Concept in Treatment-Resistant Depression: Spotlight on Ketamine


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Clinical Definitions
2.1.1. Treatment-Resistant Depression
2.1.2. Difficult-to-Treat Depression
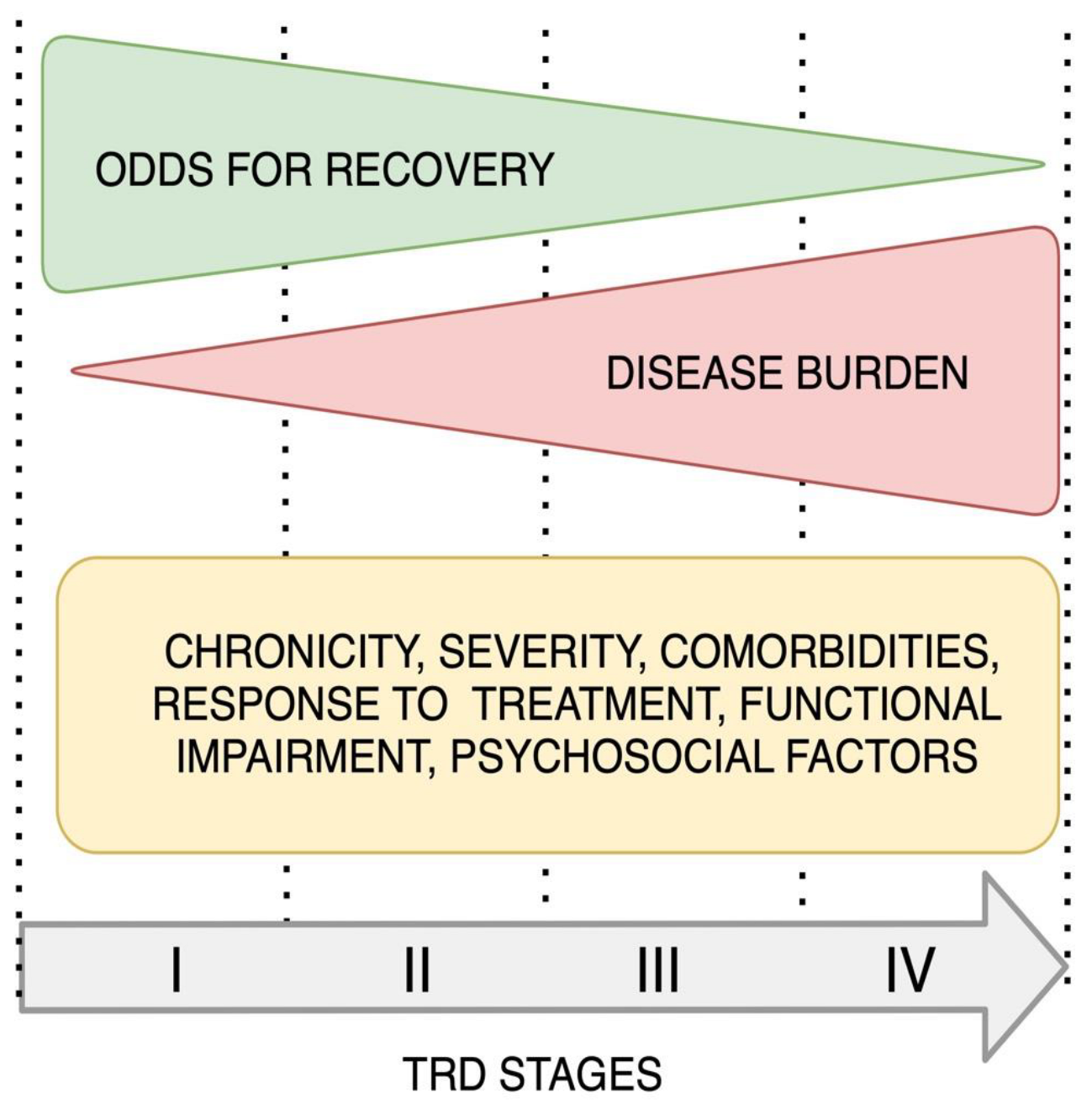
2.1.3. Staging
2.2. Neuroplasticity
2.2.1. Adult Neuroplasticity
2.2.2. Neuroplastic Changes in Treatment-Resistant Depression
2.3. Ketamine in Treatment-Resistant Depression
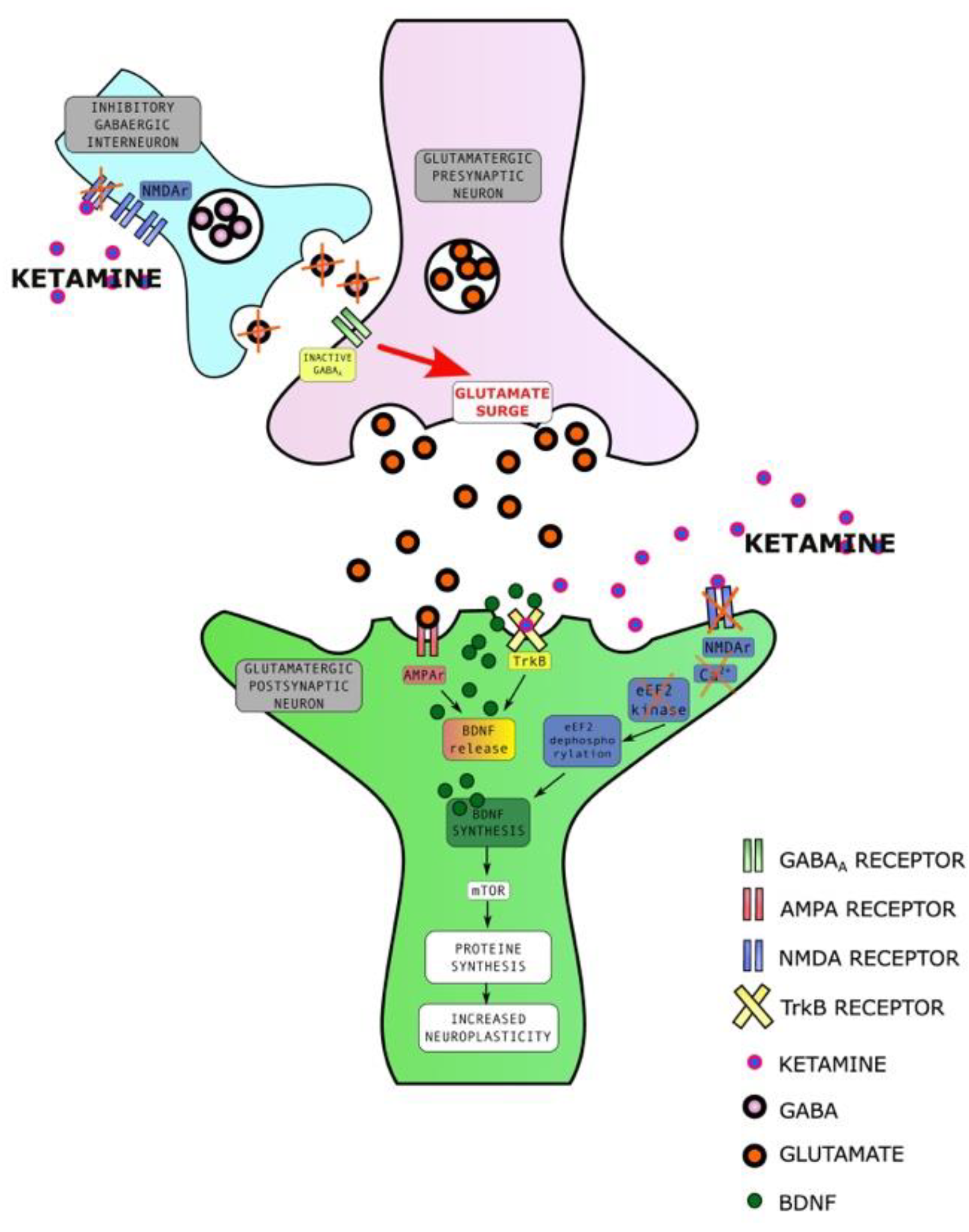
2.3.1. Ketamine Enhances Synaptic Plasticity
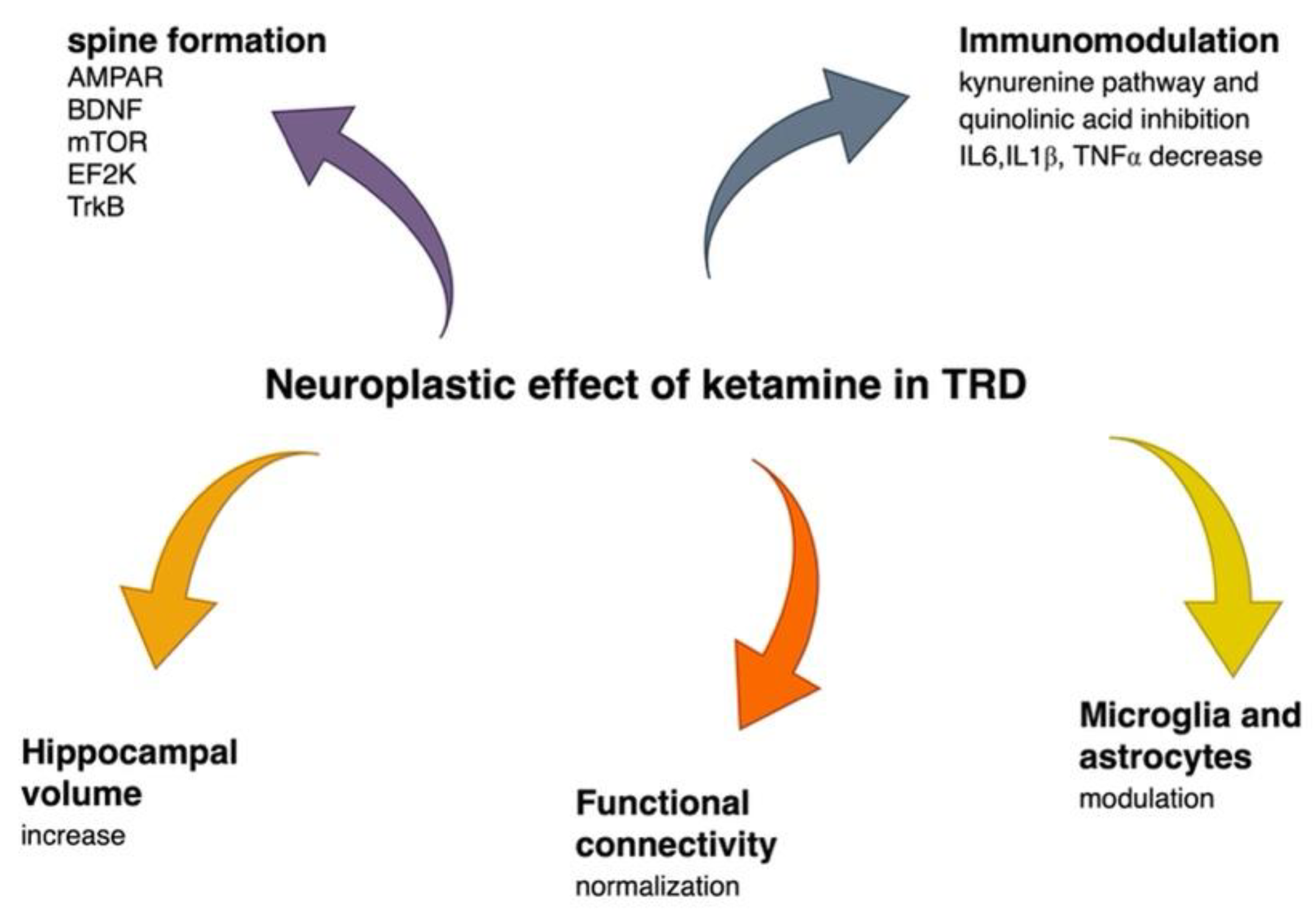
2.3.2. Ketamine Affects Brain Volume
2.3.3. Ketamine Alters Functional Connectivity
2.3.4. Ketamine Has an Immunomodulatory Effect
2.3.5. Ketamine Enhances Resilience
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olesen, J.; Gustavsson, A.; Svensson, M.; Wittchen, H.U.; Jönsson, B.; CDBE2010 Study Group; European Brain Council. The economic cost of brain disorders in Europe. Eur. J. Neurol. 2012, 19, 155–162. [Google Scholar] [CrossRef]
- Crown, W.H.; Finkelstein, S.; Berndt, E.R.; Ling, D.; Poret, A.W.; Rush, A.J.; Russell, J.M. The impact of treatment-resistant depression on health care utilization and costs. J. Clin. Psychiatry 2002, 63, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Gibson, T.B.; Jing, Y.; Smith, C.G.; Kim, E.; Bagalman, J.E.; Burton, W.N.; Tran, Q.V.; Pikalov, A.; Goetzel, R.Z. The cost burden of treatment resistance in patients with depression. Am. J. Manag. Care 2010, 16, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, D.; Hancock, K.J.; Kisely, S. The gap in life expectancy from preventable physical illness in psychiatric patients in Western Australia: Retrospective analysis of population-based registers. BMJ 2013, 21, f2539. [Google Scholar] [CrossRef] [PubMed]
- Heerlein, K.; Young, A.H.; Otte, C.; Frodl, T.; Degraeve, G.; Hagedoorn, W.; Oliveira-Maia, A.J.; Perez Sola, V.; Rathod, S.; Rosso, G.; et al. Real-world evidence from a European cohort study of patients with treatment-resistant depression: Baseline patient characteristics. J. Affect. Disord. 2021, 15, 115–122. [Google Scholar] [CrossRef]
- Heerlein, K.; De Giorgi, S.; Degraeve, G.; Frodl, T.; Hagedoorn, W.; Oliveira-Maia, A.J.; Otte, C.; Perez Sola, V.; Rathod, S.; Rosso, G.; et al. Real-world evidence from a European cohort study of patients with treatment-resistant depression: Healthcare resource utilization. J. Affect. Disord. 2022, 1, 442–450. [Google Scholar] [CrossRef]
- Rush, A.J.; Trivedi, M.H.; Wisniewski, S.R.; Nierenberg, A.A.; Stewart, J.W.; Warden, D.; Niederehe, G.; Thase, M.E.; Lavori, P.W.; Lebowitz, B.D.; et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: A STAR*D report. Am. J. Psychiatry 2006, 163, 1905–1917. [Google Scholar] [CrossRef]
- Pigott, H.E.; Leventhal, A.M.; Alter, G.S.; Boren, J.J. Efficacy and effectiveness of antidepressants: Current status of research. Psychother. Psychosom. 2010, 79, 267–279. [Google Scholar] [CrossRef]
- Heerlein, K.; Perugi, G.; Otte, C.; Frodl, T.; Degraeve, G.; Hagedoorn, W.; Oliveira-Maia, A.J.; Perez Sola, V.; Rathod, S.; Rosso, G.; et al. Real-world evidence from a European cohort study of patients with treatment-resistant depression: Treatment patterns and clinical outcomes. J. Affect. Disord. 2021, 1, 334. [Google Scholar] [CrossRef]
- Kautzky, A.; Dold, M.; Bartova, L.; Spies, M.; Kranz, G.S.; Souery, D.; Montgomery, S.; Mendlewicz, J.; Zohar, J.; Fabbri, C.; et al. Clinical factors predicting treatment-resistant depression: Affirmative results from the European multicenter study. Acta Psychiatr Scand. 2019, 139, 78–88. [Google Scholar] [CrossRef]
- Brown, S.; Rittenbach, K.; Cheung, S.; McKean, G.; MacMaster, F.P.; Clement, F. Current and Common Definitions of Treatment-Resistant Depression: Findings from a Systematic Review and Qualitative Interviews. Can. J. Psychiatry 2019, 64, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Kasper, S.; Frazer, A. Editorial for Treatment-Resistant Depression (TRD). Int. J. Neuropsychopharmacol. 2019, 1, 83–84. [Google Scholar] [CrossRef]
- Rybak, Y.E.; Lai, K.S.P.; Ramasubbu, R.; Vila-Rodriguez, F.; Blumberger, D.M.; Chan, P.; Delva, N.; Giacobbe, P.; Gosselin, C.; Kennedy, S.H.; et al. Treatment-resistant major depressive disorder: Canadian expert consensus on definition and assessment. Depress. Anxiety 2021, 38, 456–467. [Google Scholar] [CrossRef]
- Demyttenaere, K.; Van Duppen, Z. The Impact of (the Concept of) Treatment-Resistant Depression: An Opinion Review. Int. J. Neuropsychopharmacol. 2019, 1, 85–92. [Google Scholar] [CrossRef]
- Scott, F.; Hampsey, E.; Gnanapragasam, S.; Carter, B.; Marwood, L.; Taylor, R.W.; Emre, C.; Korotkova, L.; Martín-Dombrowski, J.; Cleare, A.J.; et al. Systematic review and meta-analysis of augmentation and combination treatments for early-stage treatment-resistant depression. J. Psychopharmacol. 2022, 21, 2698811221104058. [Google Scholar] [CrossRef]
- Gaynes, B.N.; Rush, A.J.; Trivedi, M.H.; Wisniewski, S.R.; Spencer, D.; Fava, M. The STAR*D study: Treating depression in the real world. Cleve Clin. J. Med. 2008, 75, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Kasper, S.; Cubała, W.J.; Fagiolini, A.; Ramos-Quiroga, J.A.; Souery, D.; Young, A.H. Practical recommendations for the management of treatment-resistant depression with esketamine nasal spray therapy: Basic science, evidence-based knowledge and expert guidance. World J. Biol. Psychiatry 2021, 22, 468–482. [Google Scholar] [CrossRef]
- Rush, A.J.; Aaronson, S.T.; Demyttenaere, K. Difficult-to-treat depression: A clinical and research roadmap for when remission is elusive. Aust. N. Z. J. Psychiatry 2019, 53, 109–118. [Google Scholar] [CrossRef]
- McAllister-Williams, R.H.; Arango, C.; Blier, P.; Demyttenaere, K.; Falkai, P.; Gorwood, P.; Hopwood, M.; Javed, A.; Kasper, S.; Malhi, G.S.; et al. The identification, assessment and management of difficult-to-treat depression: An international consensus statement. J. Affect. Disord. 2020, 15, 264–282. [Google Scholar] [CrossRef]
- Ladwig, K.H.; Baghai, T.C.; Doyle, F.; Hamer, M.; Herrmann-Lingen, C.; Kunschitz, E.; Lemogne, C.; Beresnevaite, M.; Compare, A.; von Känel, R.; et al. Mental health-related risk factors and interventions in patients with heart failure: A position paper endorsed by the European Association of Preventive Cardiology (EAPC). Eur. J. Prev. Cardiol. 2022, 25, 1124–1141. [Google Scholar] [CrossRef]
- Rush, A.J.; Sackeim, H.A.; Conway, C.R.; Bunker, M.T.; Hollon, S.D.; Demyttenaere, K.; Young, A.H.; Aaronson, S.T.; Dibué, M.; Thase, M.E.; et al. Clinical research challenges posed by difficult-to-treat depression. Psychol. Med. 2022, 52, 419–432. [Google Scholar] [CrossRef]
- Thase, M.E.; Rush, A.J. When at first you don’t succeed: Sequential strategies for antidepressant nonresponders. J. Clin. Psychiatry 1997, 58 (Suppl. S13), 23–29. [Google Scholar]
- Salloum, N.C.; Papakostas, G.I. Staging Treatment Intensity and Defining Resistant Depression: Historical Overview and Future Directions. J. Clin. Psychiatry 2019, 4, 18r12250. [Google Scholar] [CrossRef] [PubMed]
- Fekadu, A.; Wooderson, S.; Donaldson, C.; Markopoulou, K.; Masterson, B.; Poon, L.; Cleare, A.J. The Maudsley staging method is a multidimensional tool to quantify treatment resistance in depression. J. Clin. Psychiatry 2009, 70, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Van Belkum, S.M.; Geugies, H.; Lysen, T.S.; Cleare, A.J.; Peeters, F.P.M.L.; Penninx, B.W.J.H.; Schoevers, R.A.; Ruhe, H.G. Validity of the Maudsley Staging Method in Predicting Treatment-Resistant Depression Outcome Using the Netherlands Study of Depression and Anxiety. J. Clin. Psychiatry 2018, 79, 17m11475. [Google Scholar] [CrossRef]
- National Institute for Clinical Excellence. Depression: The Treatment and Management of Depression in Adults (Update). National Institute for Health and Clinical Excellence NICE Guideline. Available online: www.nice.org.uk/guidance/ng222 (accessed on 29 June 2022).
- Day, E.; Shah, R.; Taylor, R.W.; Marwood, L.; Nortey, K.; Harvey, J.; McAllister-Williams, R.H.; Geddes, J.R.; Barrera, A.; Young, A.H.; et al. A retrospective examination of care pathways in individuals with treatment-resistant depression. BJPsych Open 2021, 14, e101. [Google Scholar] [CrossRef]
- Albert, P.R. Adult neuroplasticity: A new “cure” for major depression? J. Psychiatry Neurosci. 2019, 44, 147–150. [Google Scholar] [CrossRef]
- La Rosa, C.; Parolisi, R.; Bonfanti, L. Brain Structural Plasticity: From Adult Neurogenesis to Immature Neurons. Front. Neurosci. 2020, 14, 75. [Google Scholar] [CrossRef] [PubMed]
- Castrén, E.; Monteggia, L.M. Brain-Derived Neurotrophic Factor Signaling in Depression and Antidepressant Action. Biol. Psychiatry 2021, 90, 128–136. [Google Scholar] [CrossRef]
- Duman, R.S.; Deyama, S.; Fogaça, M.V. Role of BDNF in the pathophysiology and treatment of depression: Activity-dependent effects distinguish rapid-acting antidepressants. Eur. J. Neurosci. 2021, 53, 126–139. [Google Scholar] [CrossRef]
- De Menezes Galvão, A.C.; Almeida, R.N.; de Sousa, G.M., Jr.; Leocadio-Miguel, M.A.; Palhano-Fontes, F.; de Araujo, D.B.; Lobão-Soares, B.; Maia-de-Oliveira, J.P.; Nunes, E.A.; Hallak, J.E.C.; et al. Pathophysiology of Major Depression by Clinical Stages. Front. Psychol. 2021, 12, 641779. [Google Scholar] [CrossRef]
- Autry, A.E.; Adachi, M.; Nosyreva, E.; Na, E.S.; Los, M.F.; Cheng, P.F.; Kavalali, E.T.; Monteggia, L.M. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 2011, 475, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Frodl, T.; Jäger, M.; Smajstrlova, I.; Born, C.; Bottlender, R.; Palladino, T.; Reiser, M.; Möller, H.J.; Meisenzahl, E.M. Effect of hippocampal and amygdala volumes on clinical outcomes in major depression: A 3-year prospective magnetic resonance imaging study. J. Psychiatry Neurosci. 2008, 33, 423–430. [Google Scholar] [PubMed]
- Fu, C.H.; Steiner, H.; Costafreda, S.G. Predictive neural biomarkers of clinical response in depression: A meta-analysis of functional and structural neuroimaging studies of pharmacological and psychological therapies. Neurobiol. Dis. 2013, 52, 75–83. [Google Scholar] [CrossRef]
- Sandu, A.L.; Artiges, E.; Galinowski, A.; Gallarda, T.; Bellivier, F.; Lemaitre, H.; Granger, B.; Ringuenet, D.; Tzavara, E.T.; Martinot, J.L.; et al. Amygdala and regional volumes in treatment-resistant versus nontreatment-resistant depression patients. Depress. Anxiety 2017, 34, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lee, B.; Liu, R.J.; Banasr, M.; Dwyer, J.M.; Iwata, M.; Li, X.Y.; Aghajanian, G.; Duman, R.S. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010, 329, 959–964. [Google Scholar] [CrossRef]
- Runia, N.; Yücel, D.E.; Lok, A.; de Jong, K.; Denys, D.A.J.P.; van Wingen, G.A.; Bergfeld, I.O. The neurobiology of treatment-resistant depression: A systematic review of neuroimaging studies. Neurosci. Biobehav. Rev. 2022, 132, 433–448. [Google Scholar] [CrossRef]
- Andrews-Hanna, J.R.; Reidler, J.S.; Huang, C.; Buckner, R.L. Evidence for the default network’s role in spontaneous cognition. J. Neurophysiol. 2010, 104, 322–335. [Google Scholar] [CrossRef]
- Liu, J.; Fan, Y.; Zheng, L.-L.; Liu, B.; Ju, Y.; Wang, M.; Dong, Q.; Lu, X.; Sun, J.; Zhang, L.; et al. The neuroprogressive nature of major depressive disorder: Evidence from an intrinsic connectome analysis. Transl. Psychiatry 2021, 11, 102. [Google Scholar] [CrossRef]
- Drysdale, A.T.; Grosenick, L.; Downar, J.; Dunlop, K.; Mansouri, F.; Meng, Y.; Fetcho, R.N.; Zebley, B.; Oathes, D.J.; Etkin, A.; et al. Resting-state connectivity biomarkers define neurophysiological subtypes of depression. Nat. Med. 2017, 23, 28–38, Erratum in Nat. Med. 2017, 23, 264. [Google Scholar] [CrossRef]
- Fettes, P.W.; Moayedi, M.; Dunlop, K.; Mansouri, F.; Vila-Rodriguez, F.; Giacobbe, P.; Davis, K.D.; Lam, R.W.; Kennedy, S.H.; Daskalakis, Z.J.; et al. Abnormal Functional Connectivity of Frontopolar Subregions in Treatment-Nonresponsive Major Depressive Disorder. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2018, 3, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, E.; Wilson, A.A.; Mizrahi, R.; Rusjan, P.M.; Miler, L.; Rajkowska, G.; Suridjan, I.; Kennedy, J.L.; Rekkas, P.V.; Houle, S.; et al. Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry 2015, 72, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Cakmak, J.D.; Liu, L.; Poirier, S.E.; Schaefer, B.; Poolacherla, R.; Burhan, A.M.; Sabesan, P.; St Lawrence, K.; Théberge, J.; Hicks, J.W.; et al. The functional and structural associations of aberrant microglial activity in major depressive disorder. J. Psychiatry Neurosci. 2022, 47, E197–E208. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; O’Connor, J.C.; Lawson, M.A.; Kelley, K.W. Inflammation-associated depression: From serotonin to kynurenine. Psychoneuroendocrinology 2011, 36, 426–436. [Google Scholar] [CrossRef]
- Felger, J.C.; Lotrich, F.E. Inflammatory cytokines in depression: Neurobiological mechanisms and therapeutic implications. Neuroscience 2013, 246, 199–229. [Google Scholar] [CrossRef] [PubMed]
- Strawbridge, R.; Arnone, D.; Danese, A.; Papadopoulos, A.; Herane Vives, A.; Cleare, A.J. Inflammation and clinical response to treatment in depression: A meta-analysis. Eur. Neuropsychopharmacol. 2015, 25, 1532–1543. [Google Scholar] [CrossRef]
- Chamberlain, S.R.; Cavanagh, J.; de Boer, P.; Mondelli, V.; Jones, D.N.C.; Drevets, W.C.; Cowen, P.J.; Harrison, N.A.; Pointon, L.; Pariante, C.M.; et al. Treatment-resistant depression and peripheral C-reactive protein. Br. J. Psychiatry 2019, 214, 11–19. [Google Scholar] [CrossRef]
- Yang, C.; Wardenaar, K.J.; Bosker, F.J.; Li, J.; Schoevers, R.A. Inflammatory markers and treatment outcome in treatment-resistant depression: A systematic review. J. Affect. Disord. 2019, 257, 640–649. [Google Scholar] [CrossRef]
- Liu, J.J.; Wei, Y.B.; Strawbridge, R.; Bao, Y.; Chang, S.; Shi, L.; Que, J.; Gadad, B.S.; Trivedi, M.H.; Kelsoe, J.R.; et al. Peripheral cytokine levels and response to antidepressant treatment in depression: A systematic review and meta-analysis. Mol. Psychiatry 2020, 25, 339–350. [Google Scholar] [CrossRef]
- Rush, A.J.; Fava, M.; Wisniewski, S.R.; Lavori, P.W.; Trivedi, M.H.; Sackeim, H.A.; Thase, M.E.; Nierenberg, A.A.; Quitkin, F.M.; Kashner, T.M.; et al. Sequenced treatment alternatives to relieve depression (STAR*D): Rationale and design. Control Clin. Trials 2004, 25, 119–142. [Google Scholar] [CrossRef]
- McIntyre, R.S.; Filteau, M.J.; Martin, L.; Patry, S.; Carvalho, A.; Cha, D.S.; Barakat, M.; Miguelez, M. Treatment-resistant depression: Definitions, review of the evidence, and algorithmic approach. J. Affect. Disord. 2014, 156, 1–7. [Google Scholar] [CrossRef]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Zarate, C.A., Jr.; Singh, J.B.; Carlson, P.J.; Brutsche, N.E.; Ameli, R.; Luckenbaugh, D.A.; Charney, D.S.; Manji, H.K. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry. 2006, 63, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Price, R.B.; Nock, M.K.; Charney, D.S.; Mathew, S.J. Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biol. Psychiatry 2009, 66, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Daly, E.J.; Singh, J.B.; Fedgchin, M.; Cooper, K.; Lim, P.; Shelton, R.C.; Thase, M.E.; Winokur, A.; Van Nueten, L.; Manji, H.; et al. Efficacy and Safety of Intranasal Esketamine Adjunctive to Oral Antidepressant Therapy in Treatment-Resistant Depression: A Randomized Clinical Trial. JAMA Psychiatry 2018, 75, 139–148. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration (FDA). FDA Approves New Nasal Spray Medication for Treatment-Resistant Depression; Available Only at a Certified Doctor’s Office or Clinic. 2019. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-nasal-spray-medication-treatmentresistant-depression-available-only-certified (accessed on 31 May 2019).
- Lepack, A.E.; Bang, E.; Lee, B.; Dwyer, J.M.; Duman, R.S. Fast-acting antidepressants rapidly stimulate ERK signaling and BDNF release in primary neuronal cultures. Neuropharmacology 2016, 111, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Papp, M.; Cubala, W.J.; Swiecicki, L.; Newman-Tancredi, A.; Willner, P. Perspectives for therapy of treatment-resistant depression. Br. J. Pharmacol. 2022, 179, 4181–4200. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.Y.; Hawken, E.; Vazquez, G.H. The Mechanisms Behind Rapid Antidepressant Effects of Ketamine: A Systematic Review with a Focus on Molecular Neuroplasticity. Front. Psychiatry 2022, 13, 860882. [Google Scholar] [CrossRef] [PubMed]
- Harward, S.C.; Hedrick, N.G.; Hall, C.E.; Parra-Bueno, P.; Milner, T.A.; Pan, E.; Laviv, T.; Hempstead, B.L.; Yasuda, R.; McNamara, J.O. Autocrine BDNF-TrkB signalling within a single dendritic spine. Nature 2016, 538, 99–103. [Google Scholar] [CrossRef]
- Nestler, E.J.; Barrot, M.; DiLeone, R.J.; Eisch, A.J.; Gold, S.J.; Monteggia, L.M. Neurobiology of depression. Neuron 2002, 34, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Li, C.T.; Lin, C.P.; Chou, K.H.; Chen, I.Y.; Hsieh, J.C.; Wu, C.L.; Lin, W.C.; Su, T.P. Structural and cognitive deficits in remitting and non-remitting recurrent depression: A voxel-based morphometric study. Neuroimage 2010, 50, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krystal, J.H. Synaptic plasticity and depression: New insights from stress and rapid-acting antidepressants. Nat. Med. 2016, 22, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Moda-Sava, R.N.; Murdock, M.H.; Parekh, P.K.; Fetcho, R.N.; Huang, B.S.; Huynh, T.N.; Witztum, J.; Shaver, D.C.; Rosenthal, D.L.; Always, E.J.; et al. Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science 2019, 364, eaat8078. [Google Scholar] [CrossRef] [PubMed]
- Price, J.B.; Yates, C.G.; Morath, B.A.; Van De Wakker, S.K.; Yates, N.J.; Butters, K.; Frye, M.A.; McGee, S.L.; Tye, S.J. Lithium augmentation of ketamine increases insulin signaling and antidepressant-like active stress coping in a rodent model of treatment-resistant depression. Transl. Psychiatry 2021, 11, 598. [Google Scholar] [CrossRef]
- Kavalali, E.T.; Monteggia, L.M. Targeting Homeostatic Synaptic Plasticity for Treatment of Mood Disorders. Neuron 2020, 106, 715–726. [Google Scholar] [CrossRef]
- Deyama, S.; Duman, R.S. Neurotrophic mechanisms underlying the rapid and sustained antidepressant actions of ketamine. Pharmacol. Biochem. Behav. 2020, 188, 172837. [Google Scholar] [CrossRef]
- Höflich, A.; Kraus, C.; Pfeiffer, R.M.; Seiger, R.; Rujescu, D.; Zarate, C.A., Jr.; Kasper, S.; Winkler, D.; Lanzenberger, R. Translating the immediate effects of S-Ketamine using hippocampal subfield analysis in healthy subjects-results of a randomized controlled trial. Transl. Psychiatry 2021, 11, 200. [Google Scholar] [CrossRef]
- Zhou, Y.L.; Wu, F.C.; Wang, C.Y.; Zheng, W.; Lan, X.F.; Deng, X.R.; Ning, Y.P. Relationship between hippocampal volume and inflammatory markers following six infusions of ketamine in major depressive disorder. J. Affect. Disord. 2020, 276, 608–615. [Google Scholar] [CrossRef]
- Abdallah, C.G.; Jackowski, A.; Salas, R.; Gupta, S.; Sato, J.R.; Mao, X.; Coplan, J.D.; Shungu, D.C.; Mathew, S.J. The Nucleus Accumbens and Ketamine Treatment in Major Depressive Disorder. Neuropsychopharmacology 2017, 42, 1739–1746. [Google Scholar] [CrossRef]
- Niciu, M.J.; Iadarola, N.D.; Banerjee, D.; Luckenbaugh, D.A.; Park, M.; Lener, M.; Park, L.; Ionescu, D.F.; Ballard, E.D.; Brutsche, N.E.; et al. The antidepressant efficacy of subanesthetic-dose ketamine does not correlate with baseline subcortical volumes in a replication sample with major depressive disorder. J. Psychopharmacol. 2017, 31, 1570–1577. [Google Scholar] [CrossRef]
- Nakamura, T.; Tomita, M.; Horikawa, N.; Ishibashi, M.; Uematsu, K.; Hiraki, T.; Abe, T.; Uchimura, N. Functional connectivity between the amygdala and subgenual cingulate gyrus predicts the antidepressant effects of ketamine in patients with treatment-resistant depression. Neuropsychopharmacol. Rep. 2021, 41, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chen, X.; Hu, Y.; Zhou, Y.; Wang, C.; Zheng, W.; Liu, W.; Lan, X.; Ning, Y.; Zhang, B. Functional connectivity between the habenula and default mode network and its association with the antidepressant effect of ketamine. Depress. Anxiety 2022, 39, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yao, L.; Wang, P.; Liu, Z. Immunoregulation and antidepressant effect of ketamine. Transl. Neurosci. 2021, 12, 218–236. [Google Scholar] [CrossRef]
- Al Jurdi, R.K.; Swann, A.; Mathew, S.J. Psychopharmacological Agents and Suicide Risk Reduction: Ketamine and Other Approaches. Curr. Psychiatry Rep. 2015, 17, 81. [Google Scholar] [CrossRef]
- Verdonk, F.; Petit, A.C.; Abdel-Ahad, P.; Vinckier, F.; Jouvion, G.; de Maricourt, P.; De Medeiros, G.F.; Danckaert, A.; Van Steenwinckel, J.; Blatzer, M.; et al. Microglial production of quinolinic acid as a target and a biomarker of the antidepressant effect of ketamine. Brain Behav. Immun. 2019, 81, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xie, L.; Gao, C.; Zhai, L.; Zhang, N.; Guo, L. Astrocytes activation contributes to the antidepressant-like effect of ketamine but not scopolamine. Pharmacol. Biochem. Behav. 2018, 170, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Maffioletti, E.; Bocchio-Chiavetto, L.; Perusi, G.; Carvalho Silva, R.; Sacco, C.; Bazzanella, R.; Zampieri, E.; Bortolomasi, M.; Gennarelli, M.; Minelli, A. Inflammation-related microRNAs are involved in stressful life events exposure and in trauma-focused psychotherapy in treatment-resistant depressed patients. Eur. J. Psychotraumatol. 2021, 12, 1987655. [Google Scholar] [CrossRef]
- Krzystyniak, A.; Baczynska, E.; Magnowska, M.; Antoniuk, S.; Roszkowska, M.; Zareba-Koziol, M.; Das, N.; Basu, S.; Pikula, M.; Wlodarczyk, J. Prophylactic Ketamine Treatment Promotes Resilience to Chronic Stress and Accelerates Recovery: Correlation with Changes in Synaptic Plasticity in the CA3 Subregion of the Hippocampus. Int. J. Mol. Sci. 2019, 20, 1726. [Google Scholar] [CrossRef]
- Evers, A.G.; Murrough, J.W.; Charney, D.S.; Costi, S. Ketamine as a prophylactic resilience-enhancing agent. Front. Psychiatry 2022, 13, 833259. [Google Scholar] [CrossRef]
- Sella, T.; Weiss, A.; Mittendorf, E.A.; King, T.A.; Pilewskie, M.; Giuliano, A.E.; Metzger-Filho, O. Neoadjuvant Endocrine Therapy in Clinical Practice: A Review. JAMA Oncol. 2021, 7, 1700–1708. [Google Scholar] [CrossRef]
- Leidner, R.; Crittenden, M.; Young, K.; Xiao, H.; Wu, Y.; Couey, M.A.; Patel, A.A.; Cheng, A.C.; Watters, A.L.; Bifulco, C.; et al. Neoadjuvant immunoradiotherapy results in high rate of complete pathological response and clinical to pathological downstaging in locally advanced head and neck squamous cell carcinoma. J. Immunother. Cancer 2021, 9, e002485. [Google Scholar] [CrossRef] [PubMed]
- Levenson, G.; Voron, T.; Paye, F.; Balladur, P.; Debove, C.; Chafai, N.; De Dios, A.G.; Lefevre, J.H.; Parc, Y. Tumor downstaging after neoadjuvant chemotherapy determines survival after surgery for gastric adenocarcinoma. Surgery 2021, 170, 1711–1717. [Google Scholar] [CrossRef] [PubMed]
- Caraballo, C.; Desai, N.R.; Mulder, H.; Alhanti, B.; Wilson, F.P.; Fiuzat, M.; Felker, G.M.; Piña, I.L.; O’Connor, C.M.; Lindenfeld, J.; et al. Clinical Implications of the New York Heart Association Classification. J. Am. Heart Assoc. 2019, 8, e014240. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, C.; Studer, R.; Rajput, T.; Jindal, R.; Agrawal, R.; Corda, S.; Senni, M. Real-world effectiveness and safety of sacubitril/valsartan in heart failure: A systematic review. Int. J. Cardiol. 2021, 331, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Levinta, A.; Meshkat, S.; McIntyre, R.S.; Ho, C.; Lui, L.M.W.; Lee, Y.; Mansur, R.B.; Teopiz, K.M.; Rodrigues, N.B.; Di Vincenzo, J.D.; et al. The association between stage of treatment-resistant depression and clinical utility of ketamine/esketamine: A systematic review. J. Affect. Disord. 2022, 318, 139–149. [Google Scholar] [CrossRef] [PubMed]
- McAllister-Williams, R.H.; Christmas, D.M.B.; Cleare, A.J.; Currie, A.; Gledhill, J.; Insole, L.; Malizia, A.L.; McGeever, M.; Morriss, R.; Robinson, L.J.; et al. Multiple-therapy-resistant major depressive disorder: A clinically important concept. Br. J. Psychiatry 2018, 212, 274–278. [Google Scholar] [CrossRef]
- Lascelles, K.; Marzano, L.; Brand, F.; Trueman, H.; McShane, R.; Hawton, K. Ketamine treatment for individuals with treatment-resistant depression: Longitudinal qualitative interview study of patient experiences. BJPsych Open 2020, 7, e9. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilkowska, A.; Cubała, W.J. The Downstaging Concept in Treatment-Resistant Depression: Spotlight on Ketamine. Int. J. Mol. Sci. 2022, 23, 14605. https://doi.org/10.3390/ijms232314605
Wilkowska A, Cubała WJ. The Downstaging Concept in Treatment-Resistant Depression: Spotlight on Ketamine. International Journal of Molecular Sciences. 2022; 23(23):14605. https://doi.org/10.3390/ijms232314605
Chicago/Turabian StyleWilkowska, Alina, and Wiesław Jerzy Cubała. 2022. "The Downstaging Concept in Treatment-Resistant Depression: Spotlight on Ketamine" International Journal of Molecular Sciences 23, no. 23: 14605. https://doi.org/10.3390/ijms232314605
APA StyleWilkowska, A., & Cubała, W. J. (2022). The Downstaging Concept in Treatment-Resistant Depression: Spotlight on Ketamine. International Journal of Molecular Sciences, 23(23), 14605. https://doi.org/10.3390/ijms232314605