
Recombinant Humanized IgG1 Antibody Promotes Reverse Cholesterol Transport through FcRn-ERK1/2-PPARα Pathway in Hepatocytes

Abstract
:1. Introduction
2. Results
2.1. The Antibody Inhibits the Development of Hyperlipidemia in ApoE−/− Mice
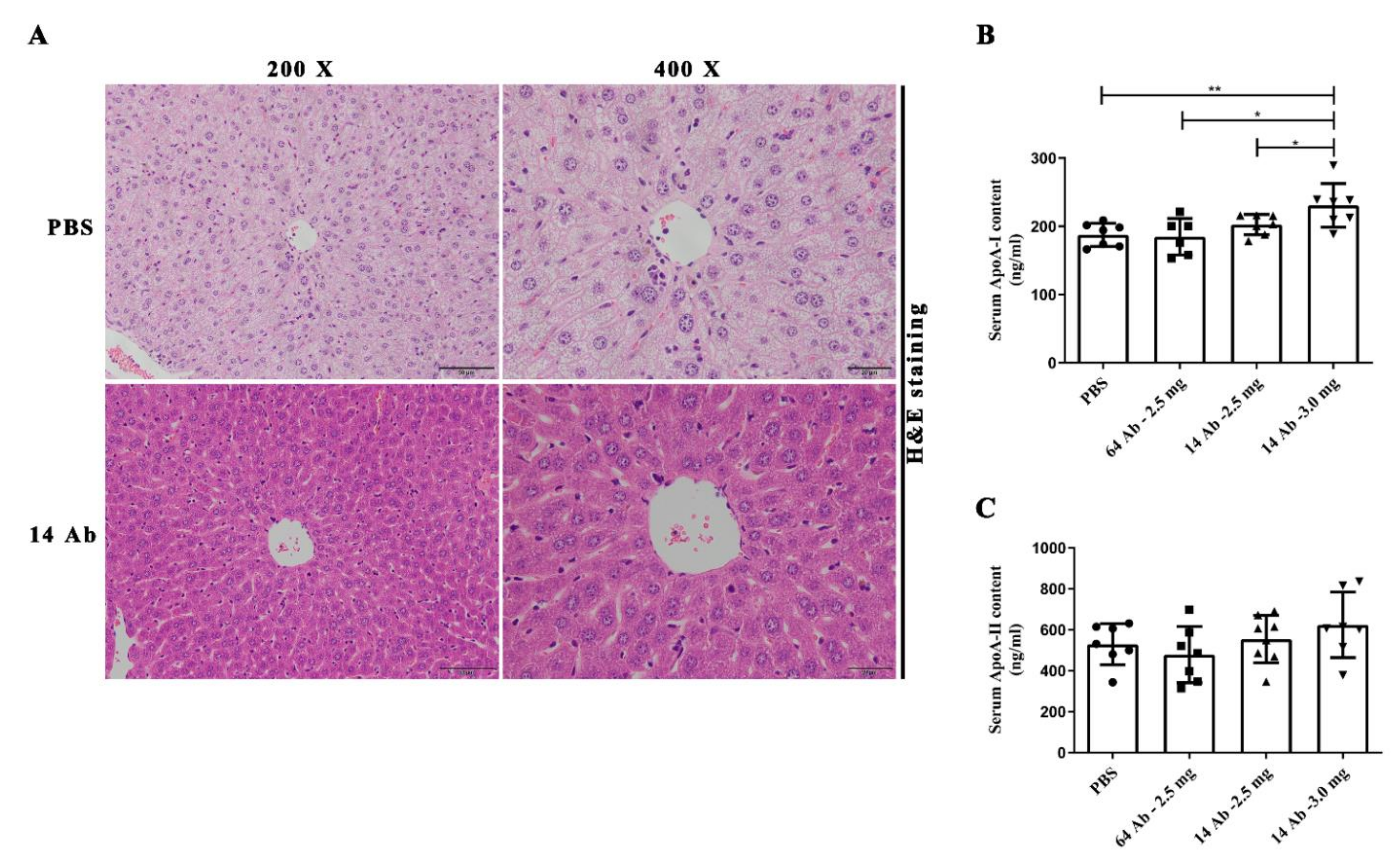
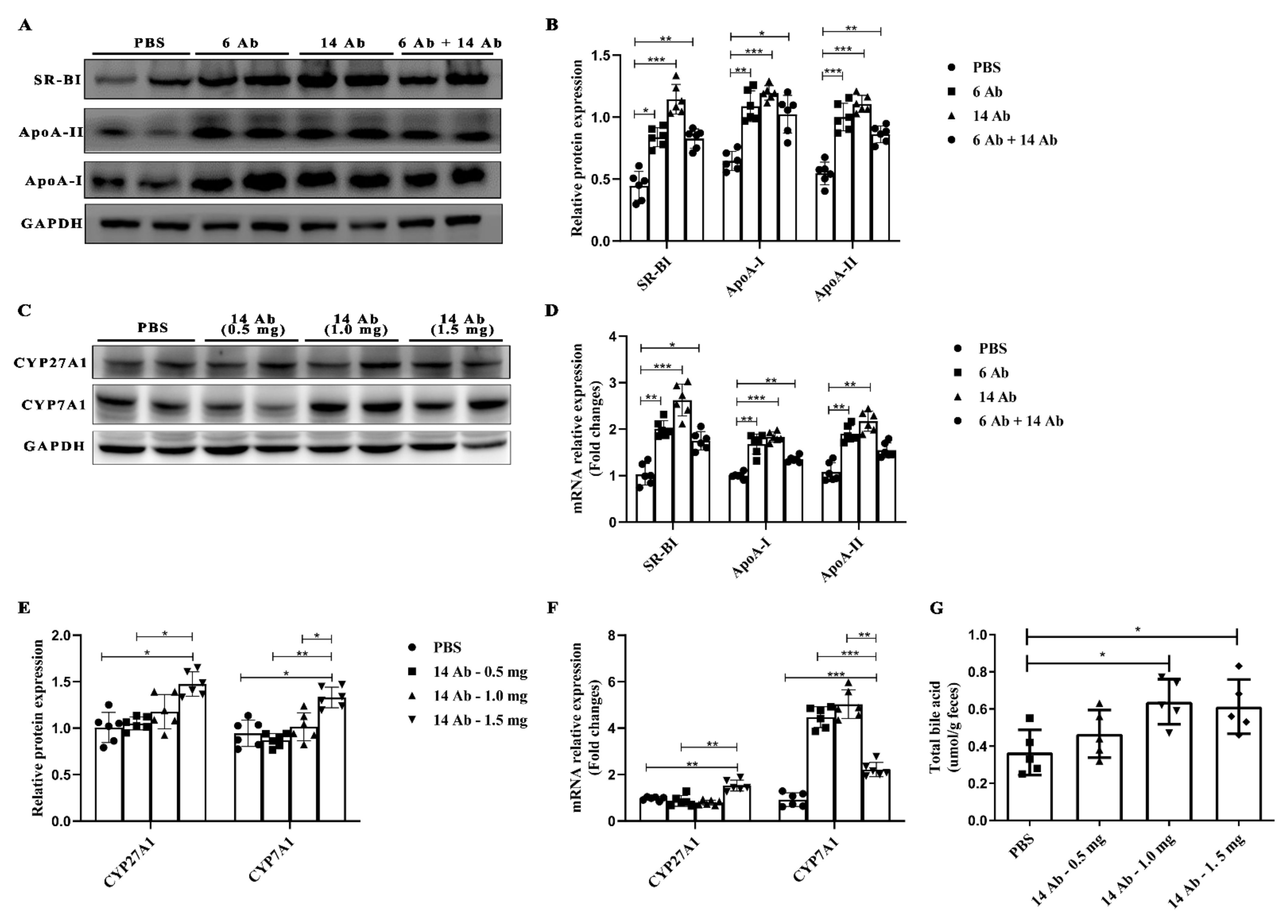
2.2. The Antibody Promotes Liver Reverse Cholesterol Transport in ApoE−/− Mice
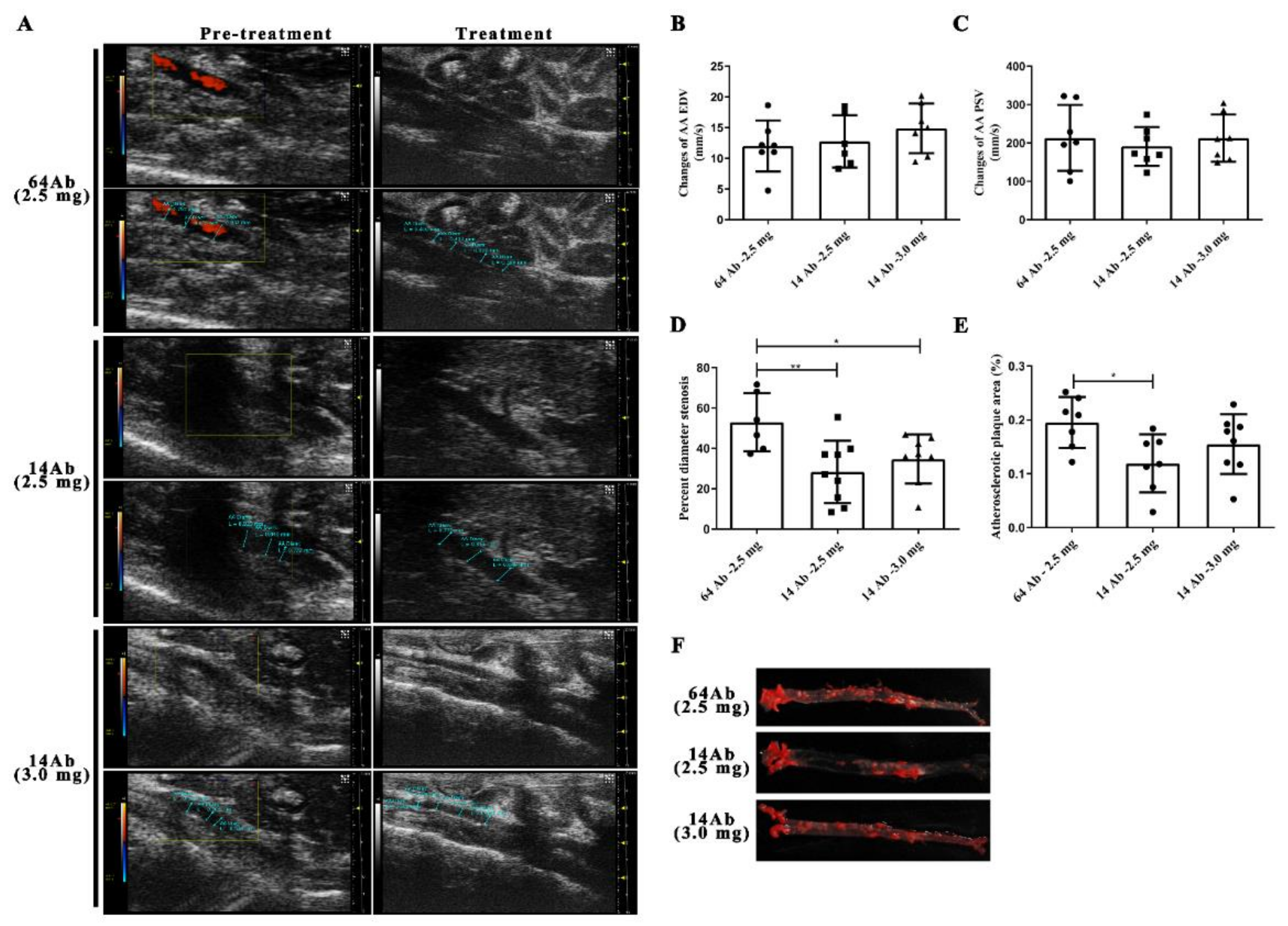
2.3. The Antibody Improves HFD-Induced Aorta Atherosclerosis
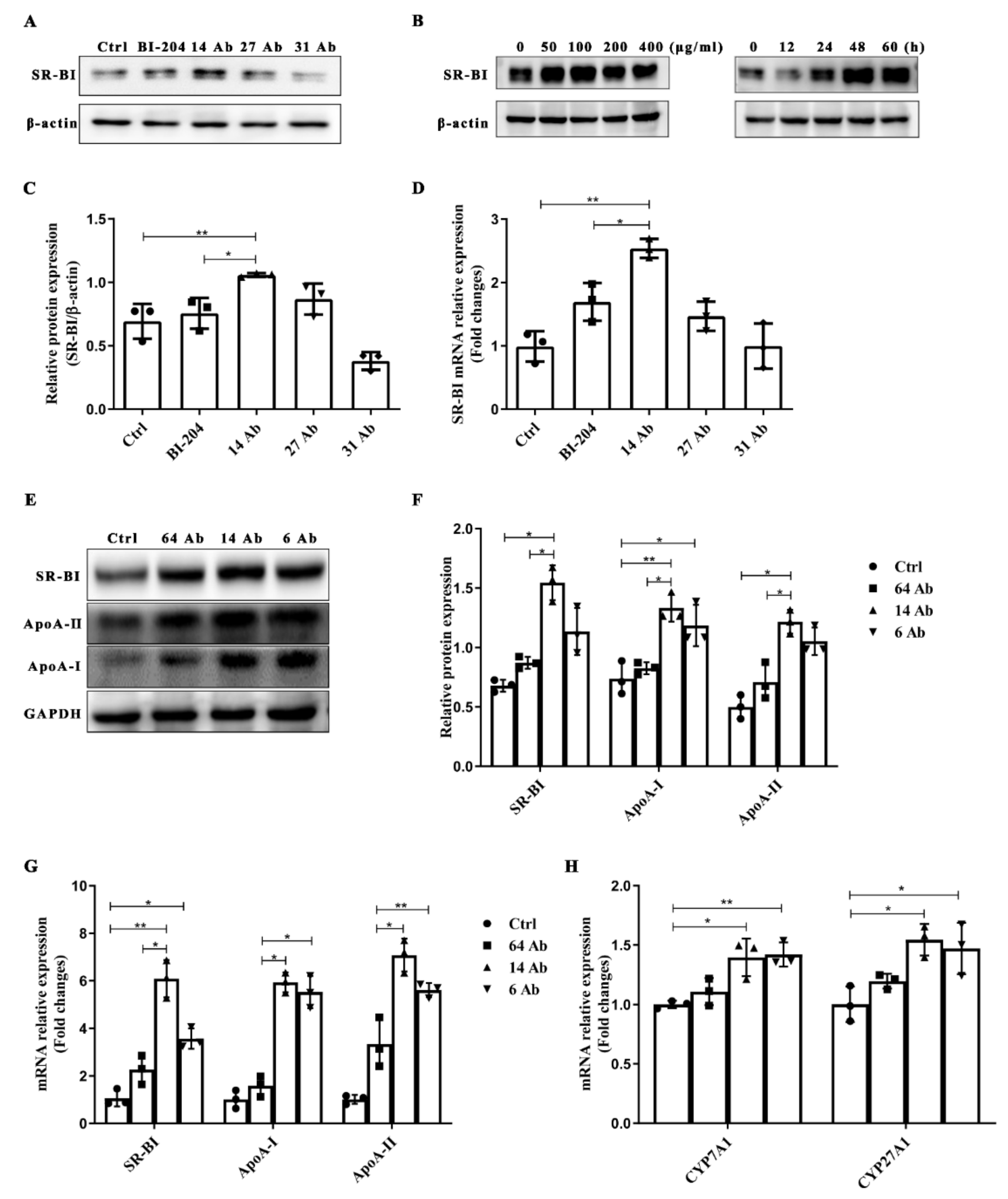
2.4. The Antibody Upregulates the Expression of SR-BI, ApoA-II, and ApoA-I in HepG2 Cells
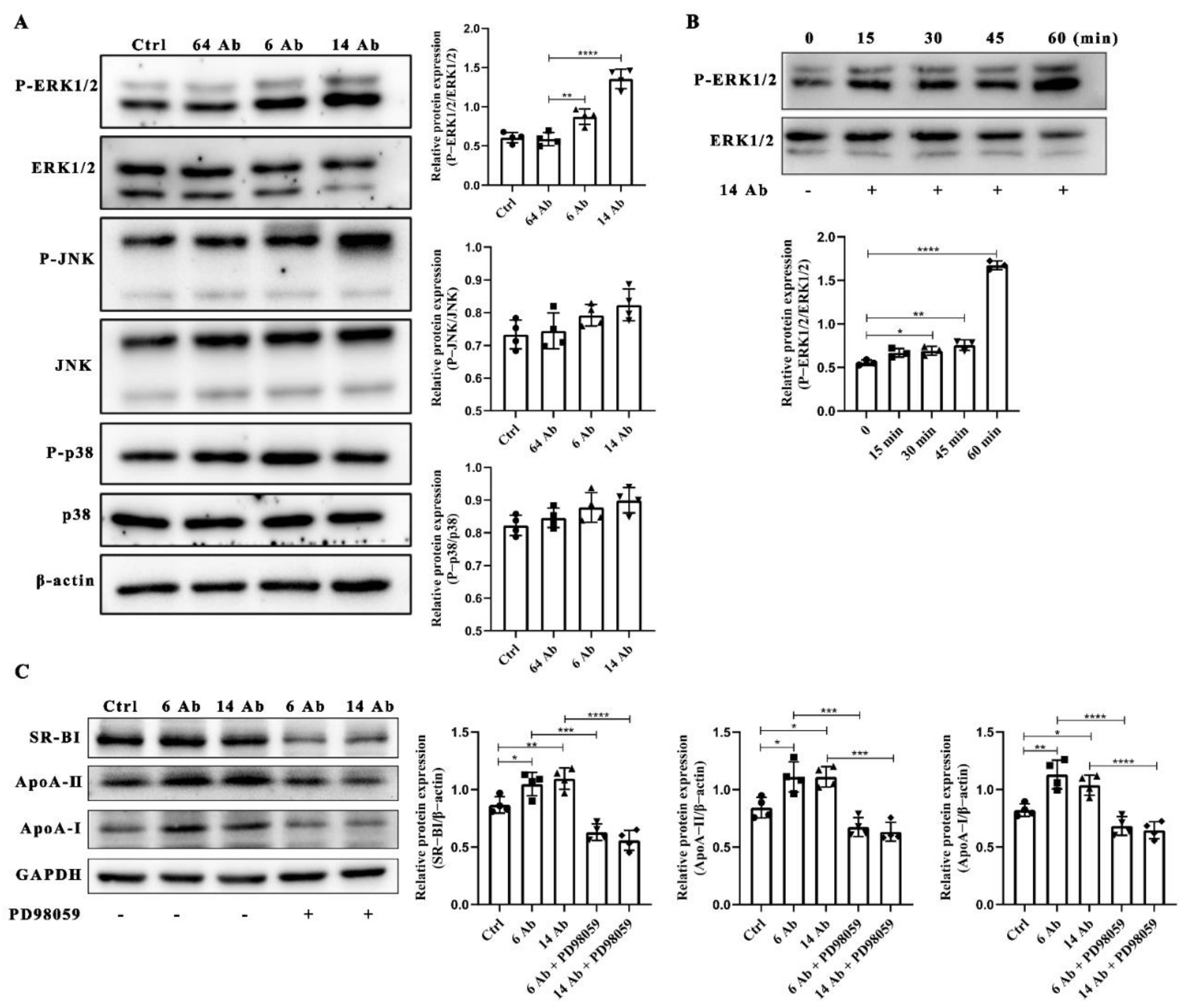
2.5. The Antibody Upregulates the Expression of ApoA-I, ApoA-II and SR-BI Is ERK1/2 Dependent
2.6. The Antibody Upregulates SR-BI, ApoA-I, and ApoA-II Protein Expression through FcRn
2.7. The Antibody Upregulates the Expression of SR-BI Is Regulated by PPARα
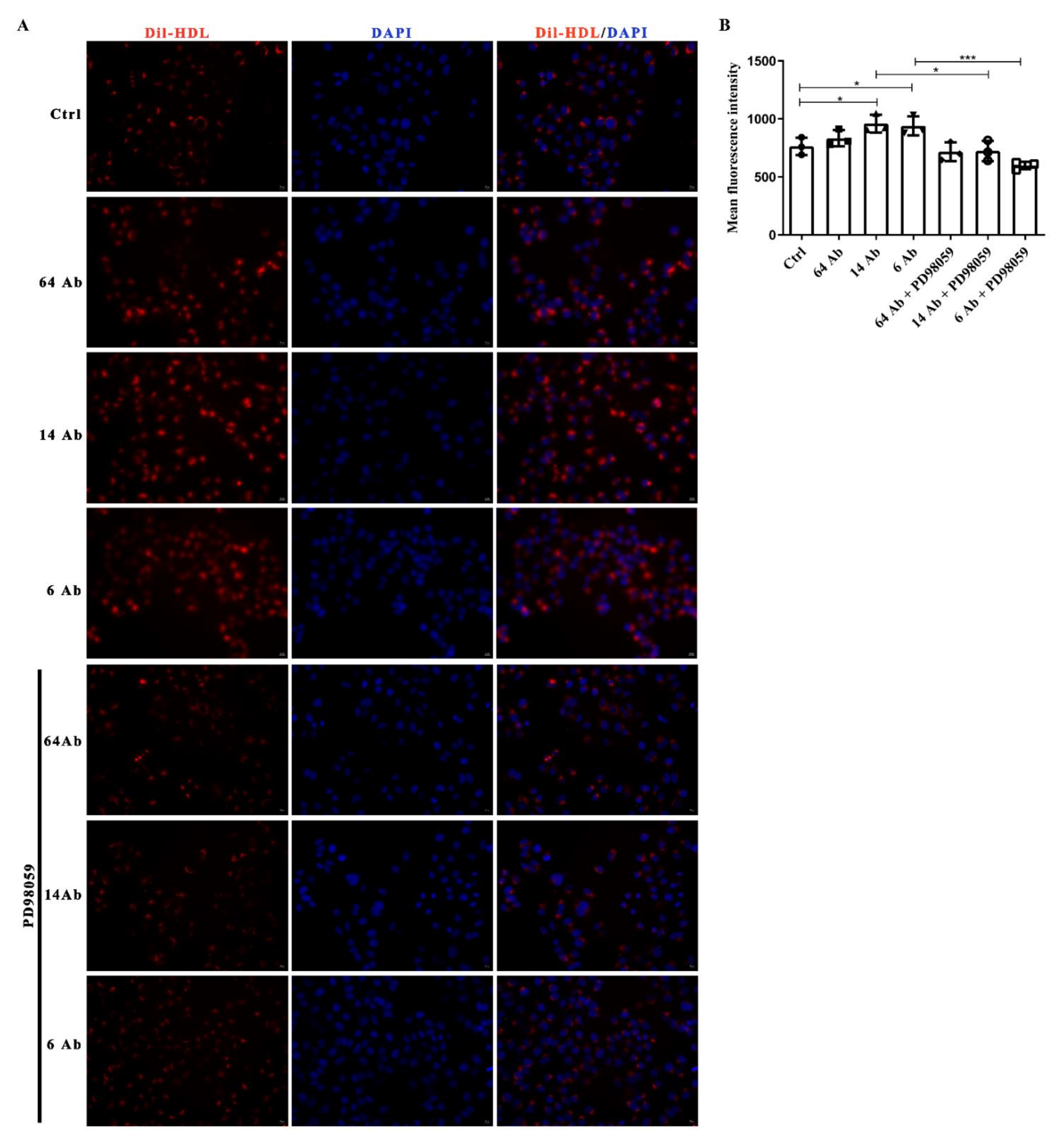
2.8. The Antibody Promotes Dil-HDL Uptake Is ERK1/2 Dependent
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. RNA Isolation and RT-PCR Analysis
4.4. Western Blot Analysis
4.5. Cell Transfection
4.6. Luciferase Reporter Gene Assays
4.7. Dil-HDL Uptake Assay
4.8. Mice and Treatments
4.9. Ultrasound Imaging Analysis of Abdominal Aorta
4.10. Oil Red O Staining of Plaque
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Hunter, P.M.; Hegele, R.A. Functional foods and dietary supplements for the management of dyslipidaemia. Nat. Rev. Endocrinol. 2017, 13, 278–288. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, Z.Y. Roles of Spicy Foods and Their Bioactive Compounds in Management of Hypercholesterolemia. J. Agric. Food Chem. 2018, 66, 8662–8671. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, E.C.; Peotta, V.A.; Gava, A.L.; Pereira, T.M.; Meyrelles, S.S. Cardiac and vascular phenotypes in the apolipoprotein E-deficient mouse. J. Biomed. Sci. 2012, 19, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouimet, M.; Barrett, T.J.; Fisher, E.A. HDL and Reverse Cholesterol Transport. Circ. Res. 2019, 124, 1505–1518. [Google Scholar] [CrossRef] [PubMed]
- Acton, S.; Rigotti, A.; Landschulz, K.T.; Xu, S.; Hobbs, H.H.; Krieger, M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996, 271, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Saddar, S.; Carriere, V.; Lee, W.R.; Tanigaki, K.; Yuhanna, I.S.; Parathath, S.; Morel, E.; Warrier, M.; Sawyer, J.K.; Gerard, R.D.; et al. Scavenger Receptor Class B Type I Is a Plasma Membrane Cholesterol Sensor. Circ. Res. 2013, 112, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, S.; Lin, R.; Duan, R.; Li, Z.; Tang, D.; Liu, X.; Liu, Y.; Zhao, M. Recombinant humanized IgG1 maintain liver triglyceride homeostasis through Arylacetamide deacetylase in ApoE(−/−) mice. Int. Immunopharmacol. 2022, 108, 108741. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.; Mulay, V.; Darabi, M.; Chan, K.C.; Heeren, J.; Pol, A.; Lambert, G.; Rye, K.A.; Enrich, C.; Grewal, T. Ras/mitogen-activated protein kinase (MAPK) signaling modulates protein stability and cell surface expression of scavenger receptor SR-BI. J. Biol. Chem. 2011, 286, 23077–23092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, D.; Zou, J.; Su, D.; Mai, H.; Zhang, S.; Li, P.; Zheng, X. Curcumin prevents high-fat diet-induced hepatic steatosis in ApoE(−/−) mice by improving intestinal barrier function and reducing endotoxin and liver TLR4/NF-κB inflammation. Nutr. Metab. 2019, 16, 79. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.K.; Choe, M.K.; Kim, H.J.; Kim, Y.S.; Binas, B.; Kim, H.J. An ApoB100-mimetic vaccine prevents obesity and liver steatosis in ApoE−/− mice. Pharmacol. Rep. PR 2017, 69, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Seidman, J.S.; Zhao, P.; Troutman, T.D.; Spann, N.J.; Que, X.; Zhou, F.; Liao, Z.; Pasillas, M.; Yang, X.; et al. Neutralization of Oxidized Phospholipids Ameliorates Non-alcoholic Steatohepatitis. Cell Metab. 2020, 31, 189–206.e8. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Bile acids: Regulation of synthesis. J. Lipid. Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzolo, D.; Buckley, K.; Kong, B.; Zhan, L.; Shen, J.; Stofan, M.; Brinker, A.; Goedken, M.; Buckley, B.; Guo, G.L. Bile Acid Homeostasis in a Cholesterol 7α-Hydroxylase and Sterol 27-Hydroxylase Double Knockout Mouse Model. Hepatology 2019, 70, 389–402. [Google Scholar] [CrossRef]
- Liu, X.; Su, J.; Zhou, H.; Zeng, Z.; Li, Z.; Xiao, Z.; Zhao, M. Collagen VI antibody reduces atherossclerosis by activating monocyte/macrophage polarization in ApoE−/− mice. Int. Immunopharmacol. 2022, 111, 109100. [Google Scholar] [CrossRef] [PubMed]
- Chinetti, G.; Gbaguidi, F.G.; Griglio, S.; Mallat, Z.; Antonucci, M.; Poulain, P.; Chapman, J.; Fruchart, J.C.; Tedgui, A.; Najib-Fruchart, J.; et al. CLA-1/SR-BI is expressed in atherosclerotic lesion macrophages and regulated by activators of peroxisome proliferator-activated receptors. Circulation 2000, 101, 2411–2417. [Google Scholar] [CrossRef] [Green Version]
- Duez, H.; Lefebvre, B.; Poulain, P.; Torra, I.P.; Percevault, F.; Luc, G.; Peters, J.M.; Gonzalez, F.J.; Gineste, R.; Helleboid, S.; et al. Regulation of human apoA-I by gemfibrozil and fenofibrate through selective peroxisome proliferator-activated receptor alpha modulation. Arter. Thromb. Vasc. Biol. 2005, 25, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Guo, Q.; Lu, M.; Li, Y. An experimental study on amelioration of dyslipidemia-induced atherosclesis by Clematichinenoside through regulating Peroxisome proliferator-activated receptor-α mediated apolipoprotein A-I, A-II and C-III. Eur. J. Pharmacol. 2015, 761, 362–374. [Google Scholar] [CrossRef]
- Barish, G.D.; Evans, R.M. PPARs and LXRs: Atherosclerosis goes nuclear. Trends Endocrinol. Metab. 2004, 15, 158–165. [Google Scholar] [CrossRef] [PubMed]
- El-Tantawy, W.H.; Temraz, A. Natural products for controlling hyperlipidemia: Review. Arch. Physiol. Biochem. 2019, 125, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Hirata, H.; Uto-Kondo, H.; Ogura, M.; Ayaori, M.; Shiotani, K.; Ota, A.; Tsuchiya, Y.; Ikewaki, K. Xanthohumol, a hop-derived prenylated flavonoid, promotes macrophage reverse cholesterol transport. J. Nutr. Biochem. 2017, 47, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Després, J.P.; Fullerton, H.J.; et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 2016, 133, e38–e360. [Google Scholar] [CrossRef] [PubMed]
- Stone, N.J.; Robinson, J.G.; Lichtenstein, A.H.; Bairey Merz, C.N.; Blum, C.B.; Eckel, R.H.; Goldberg, A.C.; Gordon, D.; Levy, D.; Lloyd-Jones, D.M.; et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2014, 63, 2889–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellner-Weibel, G.; de La Llera-Moya, M.; Connelly, M.A.; Stoudt, G.; Christian, A.E.; Haynes, M.P.; Williams, D.L.; Rothblat, G.H. Expression of scavenger receptor BI in COS-7 cells alters cholesterol content and distribution. Biochemistry 2000, 39, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Yancey, P.G.; de la Llera-Moya, M.; Swarnakar, S.; Monzo, P.; Klein, S.M.; Connelly, M.A.; Johnson, W.J.; Williams, D.L.; Rothblat, G.H. High density lipoprotein phospholipid composition is a major determinant of the bi-directional flux and net movement of cellular free cholesterol mediated by scavenger receptor BI. J. Biol. Chem. 2000, 275, 36596–36604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linton, M.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. The Role of Lipids and Lipoproteins in Atherosclerosis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Thacker, S.G.; Rousset, X.; Esmail, S.; Zarzour, A.; Jin, X.; Collins, H.L.; Sampson, M.; Stonik, J.; Demosky, S.; Malide, D.A.; et al. Increased plasma cholesterol esterification by LCAT reduces diet-induced atherosclerosis in SR-BI knockout mice. J. Lipid Res. 2015, 56, 1282–1295. [Google Scholar] [CrossRef] [Green Version]
- Malerød, L.; Sporstøl, M.; Juvet, L.K.; Mousavi, A.; Gjøen, T.; Berg, T. Hepatic scavenger receptor class B, type I is stimulated by peroxisome proliferator-activated receptor gamma and hepatocyte nuclear factor 4alpha. Biochem. Biophys. Res. Commun. 2003, 305, 557–565. [Google Scholar] [CrossRef]
- Kämmerer, I.; Ringseis, R.; Biemann, R.; Wen, G.; Eder, K. 13-hydroxy linoleic acid increases expression of the cholesterol transporters ABCA1, ABCG1 and SR-BI and stimulates apoA-I-dependent cholesterol efflux in RAW264.7 macrophages. Lipids Health Dis. 2011, 10, 222. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Bradley, M.N.; Beaven, S.W.; Tontonoz, P. Phosphorylation of the liver X receptors. FEBS Lett. 2006, 580, 4835–4841. [Google Scholar] [CrossRef] [Green Version]
- Blumberg, R.S.; Koss, T.; Story, C.M.; Barisani, D.; Polischuk, J.; Lipin, A.; Pablo, L.; Green, R.; Simister, N.E. A major histocompatibility complex class I-related Fc receptor for IgG on rat hepatocytes. J. Clin. Investig. 1995, 95, 2397–2402. [Google Scholar] [CrossRef] [Green Version]
- Ren, K.; Mo, Z.C.; Liu, X.; Tang, Z.L.; Jiang, Y.; Peng, X.S.; Zhang, Q.H.; Shi, J.F.; Yi, G.H. TGF-beta Down-regulates Apolipoprotein M Expression through the TAK-1-JNK-c-Jun Pathway in HepG2 Cells. Lipids 2017, 52, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; An, H.; Yu, Y.; Zhang, M.; Qi, R.; Cao, X. Ras participates in CpG oligodeoxynucleotide signaling through association with toll-like receptor 9 and promotion of interleukin-1 receptor-associated kinase/tumor necrosis factor receptor-associated factor 6 complex formation in macrophages. J. Biol. Chem. 2003, 278, 36334–36340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, K.; Zhu, X.; Zheng, Z.; Mo, Z.C.; Peng, X.S.; Zeng, Y.Z.; Ou, H.X.; Zhang, Q.H.; Qi, H.Z.; Zhao, G.J.; et al. MicroRNA-24 aggravates atherosclerosis by inhibiting selective lipid uptake from HDL cholesterol via the post-transcriptional repression of scavenger receptor class B type I. Atherosclerosis 2018, 270, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Schiopu, A.; Frendeus, B.; Jansson, B.; Soderberg, I.; Ljungcrantz, I.; Araya, Z.; Shah, P.K.; Carlsson, R.; Nilsson, J.; Fredrikson, G.N. Recombinant antibodies to an oxidized low-density lipoprotein epitope induce rapid regression of atherosclerosis in apobec-1(-/-)/low-density lipoprotein receptor(-/-) mice. J. Am. Coll. Cardiol. 2007, 50, 2313–2318. [Google Scholar] [CrossRef] [PubMed]
- Sawada, H.; Chen, J.Z.; Wright, B.C.; Moorleghen, J.J.; Lu, H.S.; Daugherty, A. Ultrasound Imaging of the Thoracic and Abdominal Aorta in Mice to Determine Aneurysm Dimensions. J. Vis. Exp. JoVE 2019, 145, e59013. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Total Cholesterol (mmol/g) | Triglyceride (mmol/g) | HDL-C (mmol/g) | LDL-C (mmol/g) |
|---|---|---|---|---|
| PBS | 0.053 ± 0.013 | 0.273 ± 0.131 | 0.341 ± 0.153 | 0.017 ± 0.005 |
| 6 Ab | 0.050 ± 0.014 | 0.180 ± 0.025 * | 0.552 ± 0.153 | 0.015 ± 0.003 |
| 14 Ab | 0.048 ± 0.005 | 0.128 ± 0.017 ** | 0.811 ± 0.365 ** | 0.010 ± 0.007 * |
| 6 Ab + 14 Ab | 0.044 ± 0.007 | 0.153 ± 0.080 * | 0.636 ± 0.208 * | 0.011 ± 0.005 * |
| Group | Pre-Treatment | Treatment | ||||
|---|---|---|---|---|---|---|
| AA Diam (mm) | AA PSV (mm/s) | AA EDV (mm/s) | AA Diam (mm) | AA PSV (mm/s) | AA EDV (mm/s) | |
| 64 Ab-2.5 mg (n = 7) | 0.809 ± 0.051 | 470.723 ± 41.270 | 40.841 ± 4.996 | 0.381 ± 0.055 ** | 237.897 ± 26.342 ** | 23.651 ± 1.483 ** |
| 14 Ab-2.5 mg (n = 9) | 0.744 ± 0.021 | 416.076 ± 20.283 | 33.270 ± 6.050 | 0.536 ± 0.044 **# | 253.507 ± 35.949 * | 20.381 ± 3.205 |
| 14 Ab-3.0 mg (n = 8) | 0.778 ± 0.043 | 457.699 ± 30.357 | 35.246 ± 3.727 | 0.498 ± 0.019 *** | 247.004 ± 26.367 *** | 27.266 ± 6.459 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Zhang, Q.; Liu, X.; Zhao, M. Recombinant Humanized IgG1 Antibody Promotes Reverse Cholesterol Transport through FcRn-ERK1/2-PPARα Pathway in Hepatocytes. Int. J. Mol. Sci. 2022, 23, 14607. https://doi.org/10.3390/ijms232314607
Li Z, Zhang Q, Liu X, Zhao M. Recombinant Humanized IgG1 Antibody Promotes Reverse Cholesterol Transport through FcRn-ERK1/2-PPARα Pathway in Hepatocytes. International Journal of Molecular Sciences. 2022; 23(23):14607. https://doi.org/10.3390/ijms232314607
Chicago/Turabian StyleLi, Zhonghao, Qi Zhang, Xianyan Liu, and Ming Zhao. 2022. "Recombinant Humanized IgG1 Antibody Promotes Reverse Cholesterol Transport through FcRn-ERK1/2-PPARα Pathway in Hepatocytes" International Journal of Molecular Sciences 23, no. 23: 14607. https://doi.org/10.3390/ijms232314607
APA StyleLi, Z., Zhang, Q., Liu, X., & Zhao, M. (2022). Recombinant Humanized IgG1 Antibody Promotes Reverse Cholesterol Transport through FcRn-ERK1/2-PPARα Pathway in Hepatocytes. International Journal of Molecular Sciences, 23(23), 14607. https://doi.org/10.3390/ijms232314607