
Treatment of HFpEF beyond the SGLT2-Is: Does the Addition of GLP-1 RA Improve Cardiometabolic Risk and Outcomes in Diabetic Patients?

 , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
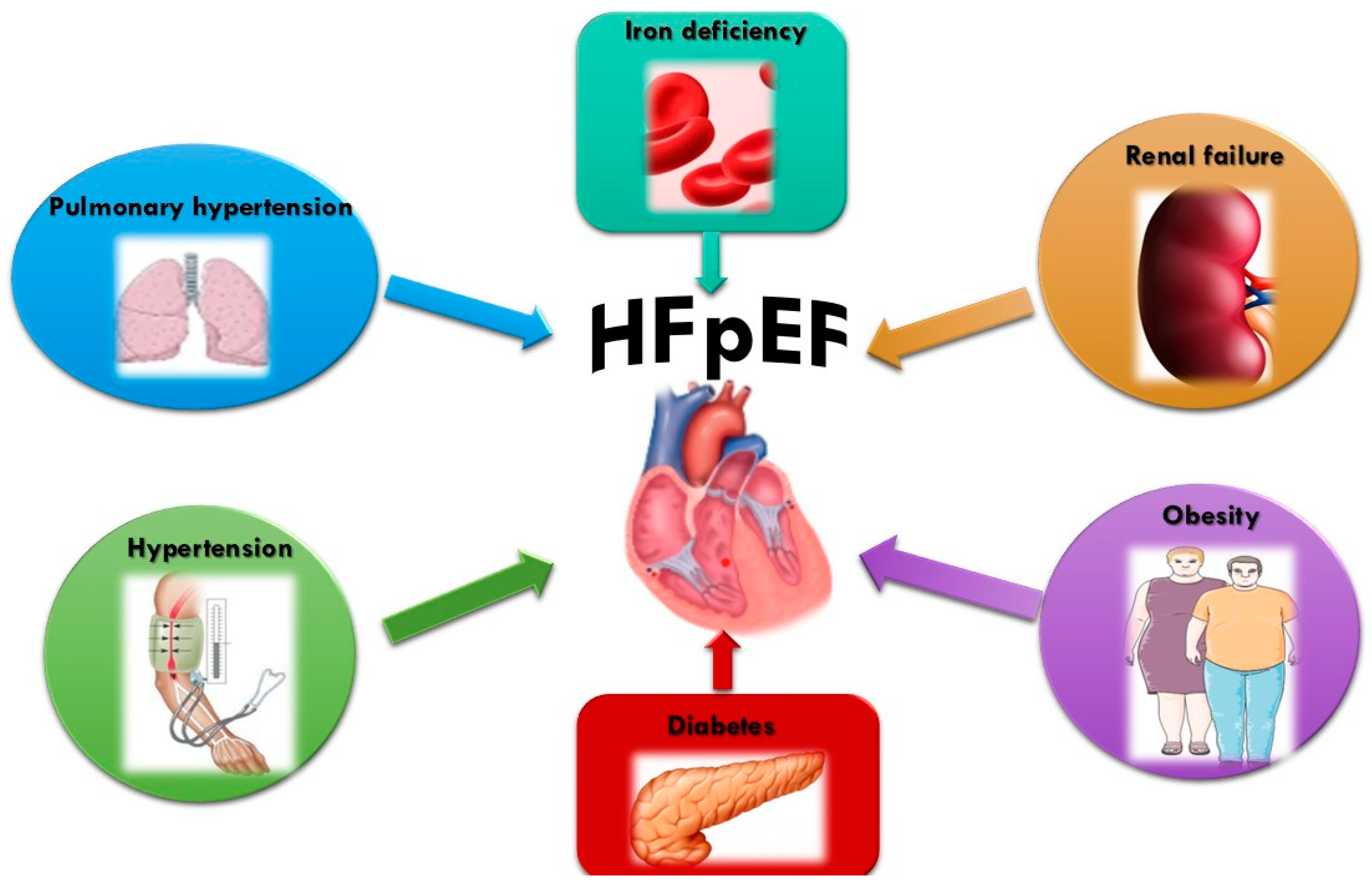
2. Phenotyping Aspects and the Burden of Cardio-Nephro-Metabolic Risk in HFpEF
3. Cardiovascular Effects of GLP-1 RAs in T2DM
4. The SGLT2-Is Effects on Cardio-Renal Axis
Sodium-glucose cotransporter 2 belongs to a family of glucose transporter proteins localized in the first segment of the proximal renal tubule and responsible for the majority of filtered glucose and sodium reabsorption. SGLT2-Is are a novel class of drugs for the treatment of T2DM and HF [80,81,82].Maximal glucose tubular transport (TmG) in healthy adult men is approximately 375 mg/min, which corresponds to 300 mg of glucose per day [83,84,85]. Because glucose filtration rate is lower than TmG under normoglycemic and hypoglycemic conditions, filtered glucose is completely reabsorbed in proximal convoluted tubules by SGLT2 receptor; conversely, glycosuria occurs when glycemia exceeds the threshold of 180 mg/day.
CV beneficial effects of SGLT2-Is include positive hemodynamic effects with a reduction in preload and afterload and improved cardiac contractility, related not only to the increased diuresis and natriuresis, but also to nephron remodeling, endothelial function improvement and, through glycosuria, also loss of body weight and adipose tissue with reduced insulin resistance [87,97,98]. A systematic review of preclinical studies showed that SGLT2-Is attenuated vascular dysfunction through a combination of mechanisms that appear to act independently of glucose-lowering benefits [99]. SGLT2-Is reduce blood pressure also in the absence of an increased heart rate, suggesting that these agents may lead to a reduction in sympathetic nervous system (SNS) activity [100,101]. As HF progresses, a continual decline in mitochondrial oxidative metabolism occurs; SGLT2-Is were shown to increase circulating ketone levels, secondary to mobilizing adipose tissue fatty acids, which are then used by the liver for ketogenesis [102]. These ketones have been proposed to improve cardiac energetics and cardiac efficiency [103,104].
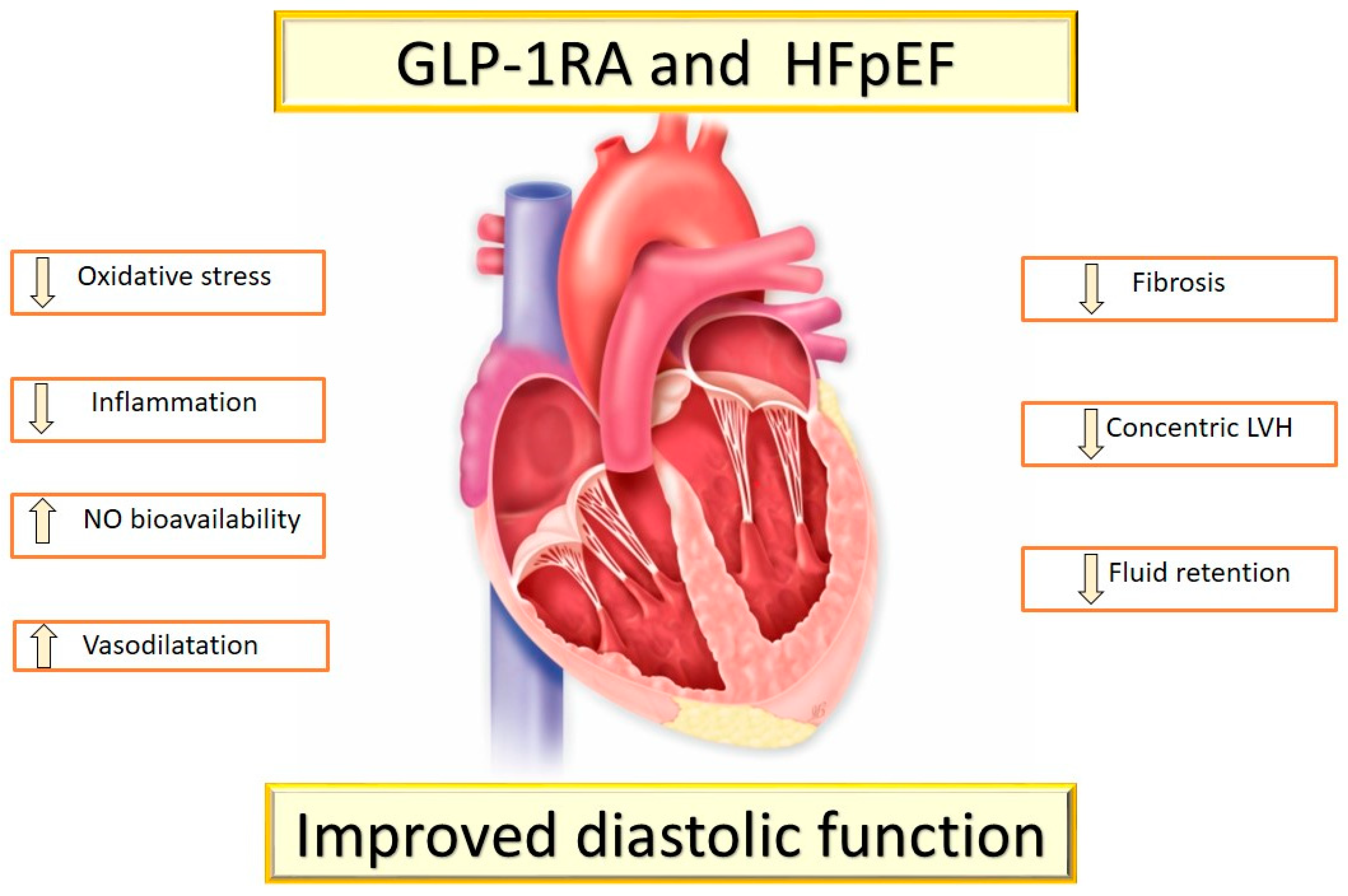
5. The Rationale for the Use of GLP-1RAs and SGLT2-Is in T2DM Patients with HFpEF
6. Discussion
7. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Borlaug, B.A.; Paulus, W.J. Heart failure with preserved and reduced ejection fraction: Different risk profiles for different diseases. Eur. Heart J. 2013, 34, 1393–1395. [Google Scholar] [CrossRef] [Green Version]
- Cleland, J.G.; Pellicori, P.; Dierckx, R. Clinical Trials in Patients with Heart Failure and Preserved Left Ventricular Ejection Fraction. Heart Fail. Clin. 2014, 10, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Lekavich, C.L.; Barksdale, D.J.; Neelon, V.J.; Wu, J.-R. Heart failure preserved ejection fraction (HFpEF): An integrated and strategic review. Heart Fail. Rev. 2015, 20, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Zhao, X.; Hammill, B.G.; Hernandez, A.F.; Fonarow, G.C.; Felker, G.M.; Yancy, C.W.; Heidenreich, P.A.; Ezekowitz, J.A.; DeVore, A.D. Trends in Noncardiovascular Comorbidities Among Patients Hospitalized for Heart Failure: In- sights from the Get with The Guidelines-Heart Failure Registry. Circ. Heart Fail. 2018, 11, e004646. [Google Scholar] [CrossRef] [PubMed]
- Cas, A.D.; Khan, S.S.; Butler, J.; Mentz, R.J.; Bonow, R.O.; Avogaro, A.; Tschoepe, D.; Doehner, W.; Greene, S.J.; Senni, M.; et al. Impact of Diabetes on Epidemiology, Treatment, and Outcomes of Patients with Heart Failure. JACC Heart Fail. 2015, 3, 136–145. [Google Scholar] [CrossRef]
- Lam, C.S.P.; Chandramouli, C.; Ahooja, V.; Verma, S. SGLT-2 Inhibitors in Heart Failure: Current Management, Unmet Needs, and Therapeutic Prospects. J. Am. Heart Assoc. 2019, 8, e013389. [Google Scholar] [CrossRef]
- Withaar, C.; Meems, L.M.G.; Markousis-Mavrogenis, G.; Boogerd, C.J.; Silljé, H.H.W.; Schouten, E.M.; Dokter, M.M.; Voors, A.A.; Westenbrink, B.D.; Lam, C.S.P.; et al. The effects of liraglutide and dapagliflozin on cardiac function and structure in a multi-hit mouse model of heart failure with preserved ejection fraction. Cardiovasc. Res. 2021, 117, 2108–2124. [Google Scholar] [CrossRef]
- Verma, S.; Rawat, S.; Ho, K.L.; Wagg, C.S.; Zhang, L.; Teoh, H.; Dyck, J.E.; Uddin, G.M.; Oudit, G.Y.; Mayoux, E.; et al. Empagliflozin Increases Cardiac Energy Production in Diabetes: Novel translational insights into the heart failure bene- fits of SGLT2 inhibitors. JACC Basic Transl. Sci. 2018, 3, 575–587. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [Green Version]
- Johansson, I.; Dahlström, U.; Edner, M.; Näsman, P.; Rydén, L.; Norhammar, A. Prognostic Implications of Type 2 Diabetes Mellitus in Ischemic and Nonischemic Heart Failure. J. Am. Coll. Cardiol. 2016, 68, 1404–1416. [Google Scholar] [CrossRef]
- Pocock, S.J.; Ariti, C.A.; Mcmurray, J.; Maggioni, A.P.; Køber, L.; Squire, I.B.; Swedberg, K.; Dobson, J.; Poppe, K.K.; Whalley, G.; et al. Predicting survival in heart failure: A risk score based on 39 372 patients from 30 studies. Eur. Heart J. 2012, 34, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Dauriz, M.; Laroche, C.; Temporelli, P.L.; Hassanein, M.; Seferovic, P.M.; Drozdz, J.; Ferrari, R.; Anker, S.; Coats, A.; et al. In-hospital and 1-year mortality associated with diabetes in patients with acute heart failure: Results from theESC-HFAHeart Failure Long-Term Registry. Eur. J. Heart Fail. 2016, 19, 54–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunlay, S.M.; Givertz, M.M.; Aguilar, D.; Allen, L.A.; Chan, M.; Desai, A.S.; Deswal, A.; Dickson, V.V.; Kosiborod, M.N.; Lekavich, C.L.; et al. Type 2 diabetes mellitus and heart failure: A scientific statement from the American Heart Association and the Heart Failure Society of America: This statement does not represent an update of the 2017 ACC/AHA/HFSA heart failure guideline update. Circulation 2019, 140, e294–e324. [Google Scholar] [CrossRef] [PubMed]
- Kenny, H.C.; Abel, E.D. Heart Failure in Type 2 Diabetes Mellitus. Circ. Res. 2019, 124, 121–141. [Google Scholar] [CrossRef]
- Pop-Busui, R.; Cleary, P.A.; Braffett, B.H.; Martin, C.L.; Herman, W.H.; Low, P.A.; Lima, J.A.; Bluemke, D.A.; DCCT/EDIC Research Group. Association between cardiovascular autonomic neuropathy and left ventricular dysfunction: DCCT/EDIC study (Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications). J. Am. Coll. Cardiol. 2013, 61, 447–454. [Google Scholar] [CrossRef] [Green Version]
- Rahman, H.; Ryan, M.; Lumley, M.; Modi, B.; McConkey, H.; Ellis, H.; Scannell, C.; Clapp, B.; Marber, M.; Webb, A.; et al. Coronary Microvascular Dysfunction Is Associated with Myocardial Ischemia and Abnormal Coronary Perfusion during Exercise. Circulation 2019, 140, 1805–1816. [Google Scholar] [CrossRef]
- Pop-Busui, R.; Kirkwood, I.; Schmid, H.; Marinescu, V.; Schroeder, J.; Larkin, D.; Yamada, E.; Raffel, D.M.; Stevens, M.J. Sympathetic dysfunction in type 1 diabetes: Association with impaired myocardial blood "ow reserve and diastolic dysfunction. J. Am. Coll. Cardiol. 2004, 44, 2368–2374. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.S.; Fonarow, G.C.; McGuire, D.K.; Hernandez, A.F.; Vaduganathan, M.; Rosenstock, J.; Handelsman, Y.; Verma, S.; Anker, S.D.; McMurray, J.J.; et al. Glucagon-Like Peptide 1 Receptor Agonists and Heart Failure: The need for further evidence generation and practice guidelines optimization. Circulation 2020, 142, 1205–1218. [Google Scholar] [CrossRef]
- Volpe, M.; Gallo, G. Cardiometabolic phenotype of heart failure with preserved ejection fraction as a target of sodium-glucose co-transporter 2 inhibitors and glucagon-like peptide receptor agonists. Cardiovasc. Res. 2021, 117, 1992–1994. [Google Scholar] [CrossRef]
- Zile, M.; Gaasch, W.H.; Carroll, J.D.; Feldman, M.D.; Aurigemma, G.P.; Schaer, G.L.; Ghali, J.K.; Liebson, P.R. Heart Failure with a Normal Ejection Fraction: Is measurement of diastolic function necessary to make the diagnosis of diastolic heart failure? Circulation 2001, 104, 779–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ennezat, P.-V.; Le Jemtel, T.; Logeart, D.; Maréchaux, S. Heart failure with preserved ejection fraction: A systemic disorder? Rev. Med. Interne 2012, 33, 370–380. [Google Scholar] [CrossRef]
- Samson, R.; Jaiswal, A.; Ennezat, P.V.; Cassidy, M.; Le Jemtel, T.H. Clinical Phenotypes in Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. 2016, 5, e002477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owan, T.E.; Hodge, D.O.; Herges, R.M.; Jacobsen, S.J.; Roger, V.L.; Redfield, M.M. Trends in Prevalence and Outcome of Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2006, 355, 251–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, R.S.; Tu, J.V.; Lee, D.S.; Austin, P.C.; Fang, J.; Haouzi, A.; Gong, Y.; Liu, P.P. Outcome of Heart Failure with Preserved Ejection Fraction in a Population-Based Study. N. Engl. J. Med. 2006, 355, 260–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitzman, D.W.; Gardin, J.M.; Gottdiener, J.S.; Arnold, A.; Boineau, R.; Aurigemma, G.; Marino, E.K.; Lyles, M.; Cushman, M.; Enright, P.L.; et al. Importance of heart failure with preserved systolic function in patients ≥65 years of age. Am. J. Cardiol. 2001, 87, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Clark, A.L.; Cleland, J.G. Diastolic heart failure: A difficult problem in the elderly. Am. J. Geriatr. Cardiol. 2004, 13, 16–21. [Google Scholar] [CrossRef]
- Epstein, F.H.; Wei, J.Y. Age and the Cardiovascular System. New Engl. J. Med. 1992, 327, 1735–1739. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, P. Hemodynamic Aging as the Consequence of Structural Changes Associated with Early Vascular Aging (EVA). Aging Dis. 2014, 5, 109–113. [Google Scholar] [CrossRef]
- Hart, C.Y.; Meyer, D.M.; Tazelaar, H.D.; Grande, J.P.; Burnett, J.C., Jr.; Housmans, P.R.; Redfield, M.M. Load Versus Humoral Activation in the Genesis of Early Hypertensive Heart Disease. Circulation 2001, 104, 215–220. [Google Scholar] [CrossRef]
- Ndumele, C.E.; Coresh, J.; Lazo, M.; Hoogeveen, R.; Blumenthal, R.S.; Folsom, A.R.; Selvin, E.; Ballantyne, C.M.; Nambi, V. Obesity, Subclinical Myocardial Injury, and Incident Heart Failure. JACC Heart Fail. 2014, 2, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.Y.; O’Moore-Sullivan, T.; Leano, R.; Byrne, N.; Beller, E.; Marwick, T.H. Alterations of Left Ventricular Myocardial Characteristics Associated with Obesity. Circulation 2004, 110, 3081–3087. [Google Scholar] [CrossRef] [Green Version]
- Wildman, R.P.; Mackey, R.H.; Bostom, A.; Thompson, T.; Sutton-Tyrrell, K. Measures of Obesity Are Associated with Vascular Stiffness in Young and Older Adults. Hypertension 2003, 42, 468–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, M.E.; Belohlavek, M.; Romero-Corral, A.; Gami, A.S.; Gilman, G.; Svatikova, A.; Amin, R.S.; Lopez-Jimenez, F.; Khandheria, B.K.; Somers, V.K. Comparison of Cardiac Structural and Functional Changes in Obese Otherwise Healthy Adults with Versus without Obstructive Sleep Apnea. Am. J. Cardiol. 2007, 99, 1298–1302. [Google Scholar] [CrossRef]
- Fung, J.W.; Li, T.S.; Choy, D.K.; Yip, G.W.; Ko, F.W.; Sanderson, J.E.; Hui, D.S. Severe Obstructive Sleep Apnea Is Associated with Left Ventricular Diastolic Dysfunction. Chest 2002, 121, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Obokata, M.; Reddy, Y.; Pislaru, S.; Melenovsky, V.; Borlaug, B.A. Evidence Supporting the Existence of a Distinct Obese Phenotype of Heart Failure with Preserved Ejection Fraction. Circulation 2017, 136, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.D.; Litwin, S.E.; Sweeney, G. Cardiac Remodeling in Obesity. Physiol. Rev. 2008, 88, 389–419. [Google Scholar] [CrossRef] [PubMed]
- Alpert, M.A.; Karthikeyan, K.; Abdullah, O.; Ghadban, R. Obesity and Cardiac Remodeling in Adults: Mechanisms and Clinical Implications. Prog. Cardiovasc. Dis. 2018, 61, 114–123. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Khan, M.S.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Rocca, H.P.B.; Choi, D.; Chopra, V.; et al. Baseline characteristics of patients with heart failure with preserved ejection fraction in the EMPEROR-Preserved trial. Eur. J. Heart Fail. 2020, 22, 2383–2392. [Google Scholar] [CrossRef]
- van de Wouw, J.; Broekhuizen, M.; Sorop, O.; Joles, J.A.; Verhaar, M.C.; Duncker, D.J.; Danser, A.H.J.; Merkus, D. Chronic Kidney Disease as a Risk Factor for Heart Failure with Preserved Ejection Fraction: A Focus on Microcirculatory Factors and Therapeutic Targets. Front. Physiol. 2019, 10, 1108. [Google Scholar] [CrossRef]
- Brouwers, F.P.; De Boer, R.A.; Van Der Harst, P.; Voors, A.A.; Gansevoort, R.T.; Bakker, S.J.; Hillege, H.L.; Van Veldhuisen, D.J.; van Gilst, W. Incidence and epidemiology of new onset heart failure with preserved vs. reduced ejection fraction in a community-based cohort: 11-year follow-up of PREVEND. Eur. Heart J. 2013, 34, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- ter Maaten, J.M.; Damman, K.; Verhaar, M.C.; Paulus, W.J.; Duncker, D.J.; Cheng, C.; Heerebeek, L.; Hillege, H.L.; Lam, C.S.; Navis, G.; et al. Connecting heart failure with preserved ejection fraction and renal dysfunction: The role of endothelial dysfunction and inflammation. Eur. J. Heart Fail. 2016, 18, 588–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obokata, M.; Reddy, Y.N.V.; Melenovsky, V.; Pislaru, S.; Borlaug, B.A. Deterioration in right ventricular structure and function over time in patients with heart failure and preserved ejection fraction. Eur. Heart J. 2019, 40, 689–697. [Google Scholar] [CrossRef] [Green Version]
- Raftery, E.B.; Banks, D.C.; Oram, S. Occlusive disease of the coronary arteries presenting as primary congestive cardiomyopathy. Lancet 1969, 2, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.J.; Wu, E.; Rafael, A.; Chen, E.-L.; Parker, M.A.; Simonetti, O.; Klocke, F.J.; Bonow, R.O.; Judd, R.M. The Use of Contrast-Enhanced Magnetic Resonance Imaging to Identify Reversible Myocardial Dysfunction. N. Engl. J. Med. 2000, 343, 1445–1453. [Google Scholar] [CrossRef]
- Ohara, T.; Little, W.C. Evolving focus on diastolic dysfunction in patients with coronary artery disease. Curr. Opin. Cardiol. 2010, 25, 613–621. [Google Scholar] [CrossRef]
- Wijns, W.; Serruys, P.W.; Slager, C.J.; Grimm, J.; Krayenbuehl, H.P.; Hugenholtz, P.G.; Hess, O.M. Effect of coronary occlusion during percutaneous transluminal angioplasty in humans on left ventricular chamber stiffness and regional diastolic pressure-radius relations. J. Am. Coll. Cardiol. 1986, 7, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Klapholz, M.; Maurer, M.; Lowe, A.M.; Messineo, F.; Meisner, J.S.; Mitchell, J.; Kalman, J.; Phillips, R.A.; Steingart, R.; Brown, E.J.; et al. Hospitalization for heart failure in the presence of a normal left ventricular ejection fraction: Results of the New York heart failure registry. J. Am. Coll. Cardiol. 2004, 43, 1432–1438. [Google Scholar] [CrossRef] [Green Version]
- Gerber, Y.; Weston, S.A.; Berardi, C.; McNallan, S.M.; Jiang, R.; Redfield, M.M.; Roger, V.L. Contemporary Trends in Heart Failure with Reduced and Preserved Ejection Fraction After Myocardial Infarction: A Community Study. Am. J. Epidemiology 2013, 178, 1272–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgendy, I.Y.; Pepine, C.J. Heart Failure with Preserved Ejection Fraction: Is Ischemia Due to Coronary Microvascular Dysfunction a Mechanistic Factor? Am. J. Med. 2019, 132, 692–697. [Google Scholar] [CrossRef]
- Shah, S.J.; Katz, D.; Selvaraj, S.; Burke, M.A.; Yancy, C.W.; Gheorghiade, M.; Bonow, R.O.; Huang, C.-C.; Deo, R.C. Phenomapping for Novel Classification of Heart Failure with Preserved Ejection Fraction. Circulation 2015, 131, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.E.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holman, R.R.; Bethel, M.A.; Mentz, R.J.; Thompson, V.P.; Lokhnygina, Y.; Buse, J.B.; Chan, J.C.; Choi, J.; Gustavson, S.M.; Iqbal, N.; et al. Effects of Once-Weekly Exenatide on Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 1228–1239. [Google Scholar] [CrossRef]
- Hernandez, A.F.; Green, J.B.; Janmohamed, S.; D’Agostino, R.B.; Granger, C.B.; Jones, N.P.; Leiter, L.A.; Rosenberg, A.E.; Sigmon, K.N.; Somerville, M.C.; et al. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): A double-blind, randomised placebo-controlled trial. Lancet 2018, 392, 1519–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerstein, H.C.; Colhoun, H.M.; Dagenais, G.R.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Riesmeyer, J.S.; Riddle, M.C.; Rydén, L.; et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): A double-blind, randomised placebo-controlled trial. Lancet 2019, 394, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, D.E.; Hansen, T.B.; et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2020, 41, 255–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the primary prevention of cardiovascular disease: A report of the american college of cardiology/American heart association task force on clinical practice guidelines. Circulation 2019, 140, e596–e646. [Google Scholar] [CrossRef]
- Ray, K.K.; Seshasai, S.R.K.; Wijesuriya, S.; Sivakumaran, R.; Nethercott, S.; Preiss, D.; Erqou, S.; Sattar, N. Effect of intensive control of glucose on cardiovascular outcomes and death in patients with diabetes mellitus: A meta-analysis of randomised controlled trials. Lancet 2009, 373, 1765–1772. [Google Scholar] [CrossRef]
- Dalsgaard, N.B.; Brønden, A.; Vilsbøll, T.; Knop, F.K. Cardiovascular safety and benefits of GLP-1 receptor agonists. Expert Opin. Drug Saf. 2017, 16, 351–363. [Google Scholar] [CrossRef]
- Saraiva, F.K.; Sposito, A.C. Cardiovascular effects of Glucagon-like peptide 1 (GLP-1) receptor agonists. Cardiovasc. Diabetol. 2014, 13, 142. [Google Scholar] [CrossRef] [Green Version]
- Drucker, D.J. The Cardiovascular Biology of Glucagon-like Peptide-1. Cell Metab. 2016, 24, 15–30. [Google Scholar] [CrossRef]
- Alharby, H.; Abdelati, T.; Rizk, M.; Youssef, E.; Gaber, N.; Moghazy, K.; Yafei, S. Association of fasting glucagon-like peptide-1 with oxidative stress and subclinical atherosclerosis in type 2 diabetes. Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 1077–1080. [Google Scholar] [CrossRef]
- Hirano, T.; Mori, Y. Anti-atherogenic and anti-inflammatory properties of glucagon-like peptide-1, glucose-dependent insulinotropic polypepide, and dipeptidyl peptidase-4 inhibitors in experimental animals. J. Diabetes Investig. 2016, 7, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Nagashima, M.; Watanabe, T.; Terasaki, M.; Tomoyasu, M.; Nohtomi, K.; Kim-Kaneyama, J.; Miyazaki, A.; Hirano, T. Native incretins prevent the development of atherosclerotic lesions in apolipoprotein E knockout mice. Diabetologia 2011, 54, 2649–2659. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Feng, P.; Zhang, X.; Li, D.; Wang, R.; Ji, C.; Li, G.; Hölscher, C. The diabetes drug semaglutide reduces infarct size, inflammation, and apoptosis, and normalizes neurogenesis in a rat model of stroke. Neuropharmacology 2019, 158, 107748. [Google Scholar] [CrossRef]
- Burgmaier, M.; Liberman, A.; Möllmann, J.; Kahles, F.; Reith, S.; Lebherz, C.; Marx, N.; Lehrke, M. Glucagon-like peptide-1 (GLP-1) and its split products GLP-1(9-37) and GLP-1(28-37) stabilize atherosclerotic lesions in apoe−/− mice. Atherosclerosis 2013, 231, 427–435. [Google Scholar] [CrossRef]
- Sudo, M.; Li, Y.; Hiro, T.; Takayama, T.; Mitsumata, M.; Shiomi, M.; Sugitani, M.; Matsumoto, T.; Hao, H.; Hirayama, A. Inhibition of plaque progression and promotion of plaque stability by glucagon-like peptide-1 receptor agonist: Serial in vivo findings from iMap-IVUS in Watanabe heritable hyperlipidemic rabbits. Atherosclerosis 2017, 265, 283–291. [Google Scholar] [CrossRef]
- Dorecka, M.; Siemianowicz, K.; Francuz, T.; Garczorz, W.; Chyra, A.; Klych, A.; Romaniuk, W. Exendin-4 and GLP-1 decreases induced expression of ICAM-1, VCAM-1 and RAGE in human retinal pigment epithelial cells. Pharmacol. Rep. 2013, 65, 884–890. [Google Scholar] [CrossRef]
- Chang, W.; Zhu, F.; Zheng, H.; Zhou, Z.; Miao, P.; Zhao, L.; Mao, Z. Glucagon-like peptide-1 receptor agonist dulaglutide prevents ox-LDL-induced adhesion of monocytes to human endothelial cells: An implication in the treatment of atherosclerosis. Mol. Immunol. 2019, 116, 73–79. [Google Scholar] [CrossRef]
- Vinué, A.; Navarro, J.; Herrero-Cervera, A.; García-Cubas, M.; Andrés-Blasco, I.; Martínez-Hervás, S.; Real, J.T.; Ascaso, J.F.; González-Navarro, H. The GLP-1 analogue lixisenatide decreases atherosclerosis in insulin-resistant mice by modulating macrophage phenotype. Diabetologia 2017, 60, 1801–1812. [Google Scholar] [CrossRef]
- Bruen, R.; Curley, S.; Kajani, S.; Lynch, G.; O’Reilly, M.E.; Dillon, E.T.; Brennan, E.; Barry, M.; Sheehan, S.; McGillicuddy, F.C.; et al. Liraglutide Attenuates Preestablished Atherosclerosis in Apolipoprotein E-Deficient Mice via Regulation of Immune Cell Phenotypes and Proinflammatory Mediators. J. Pharmacol. Exp. Ther. 2019, 370, 447–458. [Google Scholar] [CrossRef]
- Barale, C.; Buracco, S.; Cavalot, F.; Frascaroli, C.; Guerrasio, A.; Russo, I. Glucagon-like peptide 1-related peptides increase nitric oxide effects to reduce platelet activation. Thromb. Haemost. 2017, 117, 1115–1128. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Mehta, J.L.; Chen, M. Glucagon-like Peptide-1 Receptor Agonist Liraglutide Inhibits Endothelin-1 in Endothelial Cell by Repressing Nuclear Factor-Kappa B Activation. Cardiovasc. Drugs Ther. 2013, 27, 371–380. [Google Scholar] [CrossRef]
- Jojima, T.; Uchida, K.; Akimoto, K.; Tomotsune, T.; Yanagi, K.; Iijima, T.; Suzuki, K.; Kasai, K.; Aso, Y. Liraglutide, a GLP-1 receptor agonist, inhibits vascular smooth muscle cell proliferation by enhancing AMP-activated protein kinase and cell cycle regulation, and delays atherosclerosis in ApoE deficient mice. Atherosclerosis 2017, 261, 44–51. [Google Scholar] [CrossRef]
- Rizzo, M.; Rizvi, A.A.; Patti, A.M.; Nikolic, D.; Giglio, R.V.; Castellino, G.; Li Volti, G.; Caprio, M.; Montalto, G.; Provenzano, V.; et al. Liraglutide improves metabolic parameters and carotid intima-media thickness in diabetic patients with the metabolic syndrome: An 18-month prospective study. Cardiovasc. Diabetol. 2016, 15, 162. [Google Scholar] [CrossRef] [Green Version]
- Rakipovski, G.; Rolin, B.; Nøhr, J.; Klewe, I.; Frederiksen, K.S.; Augustin, R.; Hecksher-Sørensen, J.; Ingvorsen, C.; Polex-Wolf, J.; Knudsen, L.B. The GLP-1 Analogs Liraglutide and Semaglutide Reduce Atherosclerosis in ApoE−/− and LDLr−/− Mice by a Mechanism That Includes Inflammatory Pathways. JACC Basic Transl. Sci. 2018, 3, 844–857. [Google Scholar] [CrossRef] [PubMed]
- Anholm, C.; Kumarathurai, P.; Pedersen, L.R.; Samkani, A.; Walzem, R.L.; Nielsen, O.W.; Kristiansen, O.P.; Fenger, M.; Madsbad, S.; Sajadieh, A.; et al. Liraglutide in combination with metformin may improve the atherogenic lipid profile and decrease C-reactive protein level in statin treated obese patients with coronary artery disease and newly diagnosed type 2 diabetes: A randomized trial. Atherosclerosis 2019, 288, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Iacobellis, G.; Mohseni, M.; Bianco, S.D.; Banga, P.K. Liraglutide causes large and rapid epicardial fat reduction. Obesity 2017, 25, 311–316. [Google Scholar] [CrossRef] [Green Version]
- Salvatore, T.; Carbonara, O.; Cozzolino, D.; Torella, R.; Nasti, R.; Lascar, N.; Sasso, F.C. Kidney in Diabetes: From Organ Damage Target to Therapeutic Target. Curr. Drug Metab. 2011, 12, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Tsampasian, V.; Baral, R.; Chattopadhyay, R.; Debski, M.; Joshi, S.S.; Reinhold, J.; Dweck, M.R.; Garg, P.; Vassiliou, V.S. The Role of SGLT2 Inhibitors in Heart Failure: A Systematic Review and Meta-Analysis. Cardiol. Res. Pract. 2021, 2021, 9927533. [Google Scholar] [CrossRef] [PubMed]
- Muscoli, S.; Barillà, F.; Tajmir, R.; Meloni, M.; Della Morte, D.; Bellia, A.; Di Daniele, N.; Lauro, D.; Andreadi, A. The new role of SGLT2 Inhibitors in the Management of Heart Failure; current evidence and future perspective. Pharmaceutics 2022, 14, 1730. [Google Scholar] [CrossRef] [PubMed]
- Ruhnau, B.; Faber, O.; Borch-Johnsen, K.; Thorsteinsson, B. Renal threshold for glucose in non-insulin-dependent diabetic patients. Diabetes Res. Clin. Pract. 1997, 36, 27–33. [Google Scholar] [CrossRef]
- Alsahli, M.; Gerich, J.E. Renal glucose metabolism in normal physiological conditions and in diabetes. Diabetes Res. Clin. Pract. 2017, 133, 1–9. [Google Scholar] [CrossRef]
- Liu, J.; Lee, T.; DeFronzo, R.A. Why Do SGLT2 Inhibitors Inhibit Only 30–50% of Renal Glucose Reabsorption in Humans? Diabetes 2012, 61, 2199–2204. [Google Scholar] [CrossRef] [Green Version]
- Rahmoune, H.; Thompson, P.W.; Ward, J.M.; Smith, C.D.; Hong, G.; Brown, J. Glucose Transporters in Human Renal Proximal Tubular Cells Isolated from the Urine of Patients with Non–Insulin-Dependent Diabetes. Diabetes 2005, 54, 3427–3434. [Google Scholar] [CrossRef] [Green Version]
- Zelniker, T.A.; Braunwald, E. Cardiac and Renal Effects of Sodium-Glucose Co-Transporter 2 Inhibitors in Diabetes. J. Am. Coll. Cardiol. 2018, 72, 1845–1855. [Google Scholar] [CrossRef] [PubMed]
- Boyer, C.C. The vascular pattern of the renal glomerulus as revealed by plastic reconstruction from serial sections. Anat. Rec. 1956, 125, 433–441. [Google Scholar] [CrossRef]
- Prandi, F.R.; Barone, L.; Lecis, D.; Belli, M.; Sergi, D.; Milite, M.; Lerakis, S.; Romeo, F.; Barillà, F. Biomolecular Mechanisms of Cardiorenal Protection with Sodium-Glucose Co-Transporter 2 Inhibitors. Biomolecules 2022, 12, 1349. [Google Scholar] [CrossRef]
- Pruijm, M.; Milani, B.; Pivin, E.; Podhajska, A.; Vogt, B.; Stuber, M.; Burnier, M. Reduced cortical oxygenation predicts a progressive decline of renal function in patients with chronic kidney disease. Kidney Int. 2018, 93, 932–940. [Google Scholar] [CrossRef]
- Mulder, S.; Heerspink, H.J.L.; Darshi, M.; Kim, J.J.; Laverman, G.D.; Sharma, K.; Pena, M.J. Effects of dapagliflozin on urinary metabolites in people with type 2 diabetes. Diabetes Obes. Metab. 2019, 21, 2422–2428. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, S.H.; Kang, J.M.; Heo, J.H.; Kim, D.-J.; Park, S.H.; Sung, M.; Kim, J.; Oh, J.; Yang, D.H.; et al. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am. J. Physiol Renal. Physiol. 2019, 317, F767–F780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Ju, F.; Li, J.; Liu, T.; Zuo, Y.; Abbott, G.W.; Hu, Z. Empagliflozin protects against renal ischemia/reperfusion injury in mice. Sci. Rep. 2022, 12, 19323. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-P.; Zhang, Z.-Y.; Wu, H.-W.; Fang, L.-J.; Hu, B.; Tang, C.; Zhang, Y.-Q.; Yin, L.; Tang, D.-E.; Zheng, Z.-H.; et al. SGLT2 inhibitors improve kidney function and morphology by regulating renal metabolic reprogramming in mice with diabetic kidney disease. J. Transl. Med. 2022, 20, 420. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Takei, M.; Shiraishi, Y.; Suzuki, Y. Increased Hematocrit During Sodium-Glucose Cotransporter 2 Inhibitor Therapy Indicates Recovery of Tubulointerstitial Function in Diabetic Kidneys. J. Clin. Med. Res. 2016, 8, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Mazer, C.D.; Hare, G.M.; Connelly, P.W.; Gilbert, R.E.; Shehata, N.; Quan, A.; Teoh, H.; Leiter, L.A.; Zinman, B.; Jüni, P.; et al. Effect of Empagliflozin on Erythropoietin Levels, Iron Stores, and Red Blood Cell Morphology in Patients with Type 2 Diabetes Mellitus and Coronary Artery Disease. Circulation 2020, 141, 704–707. [Google Scholar] [CrossRef]
- Verma, S. Potential Mechanisms of Sodium-Glucose Co-Transporter 2 Inhibitor-Related Cardiovascular Benefits. Am. J. Med. 2019, 124, S36–S44. [Google Scholar] [CrossRef] [Green Version]
- Kahl, S.; Gancheva, S.; Straßburger, K.; Herder, C.; Machann, J.; Katsuyama, H.; Kabisch, S.; Henkel, E.; Kopf, S.; Lagerpusch, M.; et al. Empagliflozin Effectively Lowers Liver Fat Content in Well-Controlled Type 2 Diabetes: A Randomized, Double-Blind, Phase 4, Placebo-Controlled Trial. Diabetes Care 2020, 43, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Alshnbari, A.S.; Millar, S.A.; O’Sullivan, S.E.; Idris, I. Effect of Sodium-Glucose Cotransporter-2 Inhibitors on Endothelial Function: A Systematic Review of Preclinical Studies. Diabetes Ther. 2020, 11, 1947–1963. [Google Scholar] [CrossRef]
- Chiba, Y.; Yamada, T.; Tsukita, S.; Takahashi, K.; Munakata, Y.; Shirai, Y.; Kodama, S.; Asai, Y.; Sugisawa, T.; Uno, K.; et al. Dapagliflozin, a Sodium-Glucose Co-Transporter 2 Inhibitor, Acutely Reduces Energy Expenditure in BAT via Neural Signals in Mice. PLoS ONE 2016, 11, e0150756. [Google Scholar] [CrossRef]
- Matthews, V.B.; Elliot, R.H.; Rudnicka, C.; Hricova, J.; Herat, L.; Schlaich, M. Role of the sympathetic nervous system in regulation of the sodium glucose cotransporter 2. J. Hypertens. 2017, 35, 2059–2068. [Google Scholar] [CrossRef]
- Ferrannini, E.; Baldi, S.; Frascerra, S.; Astiarraga, B.; Heise, T.; Bizzotto, R.; Mari, A.; Pieber, T.R.; Muscelli, E. Shift to fatty substrates utilization in response to sodium-glucose co-transporter-2 inhibition in nondiabetic subjects and type 2 diabetic patients. Diabetes 2016, 65, 1190–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudaliar, S.; Alloju, S.; Henry, R.R. Can a Shift in Fuel Energetics Explain the Beneficial Cardiorenal Outcomes in the EMPA-REG OUTCOME Study? A Unifying Hypothesis. Diabetes Care 2016, 39, 1115–1122. [Google Scholar] [CrossRef] [Green Version]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME trial: A “thrifty substrate” hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, S.D.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; Shah, S.J.; Lindholm, D.; et al. Dapagliflozin in heart failure with preserved and mildly reduced ejection fraction: Rationale and design of the DELIVER trial. Eur. J. Heart Fail. 2021, 23, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Vaduganathan, M.; Docherty, K.F.; Claggett, B.L.; Jhund, P.S.; de Boer, R.A.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; et al. SGLT-2 inhibitors in patients with heart failure: A comprehensive meta-analysis of five randomised controlled trials. Lancet 2022, 400, 757–767. [Google Scholar] [CrossRef]
- Neuen, B.L.; Young, T.; Heerspink, H.J.L.; Neal, B.; Perkovic, V.; Billot, L.; Mahaffey, K.W.; Charytan, D.M.; Wheeler, D.C.; Arnott, C.; et al. SGLT2 inhibitors for the prevention of kidney failure in patients with type 2 diabetes: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2019, 7, 845–854. [Google Scholar] [CrossRef]
- Steiner, S. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. Z. Gefassmed. 2016, 13, 17–18. [Google Scholar]
- Mahaffey, K.W.; Neal, B.; Perkovic, V.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Fabbrini, E.; Sun, T.; Li, Q.; et al. Canagliflozin for Primary and Secondary Prevention of Cardiovascular Events: Results from the CANVAS Program (Canagliflozin Cardiovascular Assessment Study). Circulation 2018, 137, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Cannon, C.P.; Pratley, R.; Dagogo-Jack, S.; Mancuso, J.; Huyck, S.; Masiukiewicz, U.; Charbonnel, B.; Frederich, R.; Gallo, S.; Cosentino, F.; et al. Cardiovascular Outcomes with Ertugliflozin in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 1425–1435. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.-F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef]
- Brown, A.J.M.; Gandy, S.; McCrimmon, R.; Houston, J.G.; Struthers, A.D.; Lang, C.C. A randomized controlled trial of dapagliflozin on left ventricular hypertrophy in people with type two diabetes: The DAPA-LVH trial. Eur. Heart J. 2020, 41, 3421–3432. [Google Scholar] [CrossRef] [PubMed]
- Vasilakou, D.; Karagiannis, T.; Athanasiadou, E.; Mainou, M.; Liakos, A.; Bekiari, E.; Sarigianni, M.; Matthews, D.R.; Tsapas, A. Sodium–Glucose Cotransporter 2 Inhibitors for Type 2 Diabetes: A systematic review and meta-analysis. Ann. Intern. Med. 2013, 159, 262–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.; McMurray, J.J.V. SGLT2 inhibitors and mechanisms of cardiovascular benefit: A state-of-the-art review. Diabetologia 2018, 61, 2108–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inzucchi, S.E.; Zinman, B.; Fitchett, D.; Wanner, C.; Ferrannini, E.; Schumacher, M.; Schmoor, C.; Ohneberg, K.; Johansen, O.E.; George, J.T.; et al. How does empagliflozin reduce cardiovascular mortality? Insights from a mediation analysis of the EMPA-REG OUTCOME trial. Diabetes Care 2018, 41, 356–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajeev, S.P.; Cuthbertson, D.; Wilding, J.P.H. Energy balance and metabolic changes with sodium-glucose co-transporter 2 inhibition. Diabetes Obes. Metab. 2016, 18, 125–134. [Google Scholar] [CrossRef]
- Ferrannini, E.; Muscelli, E.; Frascerra, S.; Baldi, S.; Mari, A.; Heise, T.; Broedl, U.C.; Woerle, H.-J. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J. Clin. Investig. 2014, 124, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; Garg, A.; Yan, A.T.; Gupta, A.K.; Al-Omran, M.; Sabongui, A.; Teoh, H.; Mazer, C.D.; Connelly, K.A. Effect of Empagliflozin on Left Ventricular Mass and Diastolic Function in Individuals with Diabetes: An Important Clue to the EMPA-REG OUTCOME Trial? Diabetes Care 2016, 39, e212–e213. [Google Scholar] [CrossRef] [Green Version]
- Solini, A.; Giannini, L.; Seghieri, M.; Vitolo, E.; Taddei, S.; Ghiadoni, L.; Bruno, R.M. Dapagliflozin acutely improves endothelial dysfunction, reduces aortic stiffness and renal resistive index in type 2 diabetic patients: A pilot study. Cardiovasc. Diabetol. 2017, 16, 138. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.R.; Lee, Y.-H.; Lee, S.-G.; Kang, E.S.; Cha, B.-S.; Lee, B.-W. The renal tubular damage marker urinary N-acetyl-β-d-glucosaminidase may be more closely associated with early detection of atherosclerosis than the glomerular damage marker albuminuria in patients with type 2 diabetes. Cardiovasc. Diabetol. 2017, 16, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusaka, H.; Koibuchi, N.; Hasegawa, Y.; Ogawa, H.; Kim-Mitsuyama, S. Empagliflozin lessened cardiac injury and reduced visceral adipocyte hypertrophy in prediabetic rats with metabolic syndrome. Cardiovasc. Diabetol. 2016, 15, 157. [Google Scholar] [CrossRef] [PubMed]
- Bizino, M.B.; Jazet, I.M.; Westenberg, J.J.M.; Van Eyk, H.J.; Paiman, E.H.M.; Smit, J.W.A.; Lamb, H.J. Effect of liraglutide on cardiac function in patients with type 2 diabetes mellitus: Randomized placebo-controlled trial. Cardiovasc. Diabetol. 2019, 18, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Russo, C.; Jin, Z.; Homma, S.; Rundek, T.; Elkind, M.S.; Sacco, R.L.; Di Tullio, M.R. Effect of Obesity and Overweight on Left Ventricular Diastolic Function: A community-based study in an elderly cohort. J. Am. Coll. Cardiol. 2011, 57, 1368–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ussher, J.R.; Drucker, D.J. Cardiovascular Actions of Incretin-Based Therapies. Circ. Res. 2014, 114, 1788–1803. [Google Scholar] [CrossRef] [Green Version]
- Oldgren, J.; Laurila, S.; Åkerblom, A.; Latva-Rasku, A.; Rebelos, E.; Isackson, H.; Saarenhovi, M.; Eriksson, O.; Heurling, K.; Johansson, E.; et al. Effects of 6weeks of treatment with dapagliflozin, a sodiumglucose co-transporter-2 inhibitor, on myocardial function and metabolism in patients with type 2 diabetes: A randomized, placebo-controlled, exploratory study. Diabetes Obes. Metab. 2021, 23, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [Green Version]
- Del Olmo-Garcia, M.I.; Merino-Torres, J.F. GLP-1 Receptor Agonists and Cardiovascular Disease in Patients with Type 2 Diabetes. J. Diabetes Res. 2018, 2018, 4020492. [Google Scholar] [CrossRef] [Green Version]
- Tadic, M.; Sala, C.; Saeed, S.; Grassi, G.; Mancia, G.; Rottbauer, W.; Cuspidi, C. New antidiabetic therapy and HFpEF: Light at the end of tunnel? Heart Fail. Rev. 2021, 27, 1137–1146. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Agent | SGLT2 Inhibitors | GLP-1 Agonists | Combination Therapy | |
|---|---|---|---|---|
| Effects | ||||
| Oxidative stress |  | |  | |
| Inflammation | | | | |
| Endothelial dysfunction | | | | |
| Natriuresis |  |  | | |
| Diuresis | |  | | |
| Plaque stability | | | | |
| Blood pressure | | | | |
| Body weight | |  | | |
| Lipid profile | | | | |
| Epicardial fat | | | | |
| Myocardial fibrosis | | | | |
| Insulin resistance | | | | |
| Beta cell function | | | | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belli, M.; Barone, L.; Bellia, A.; Sergi, D.; Lecis, D.; Prandi, F.R.; Milite, M.; Galluccio, C.; Muscoli, S.; Romeo, F.; et al. Treatment of HFpEF beyond the SGLT2-Is: Does the Addition of GLP-1 RA Improve Cardiometabolic Risk and Outcomes in Diabetic Patients? Int. J. Mol. Sci. 2022, 23, 14598. https://doi.org/10.3390/ijms232314598
Belli M, Barone L, Bellia A, Sergi D, Lecis D, Prandi FR, Milite M, Galluccio C, Muscoli S, Romeo F, et al. Treatment of HFpEF beyond the SGLT2-Is: Does the Addition of GLP-1 RA Improve Cardiometabolic Risk and Outcomes in Diabetic Patients? International Journal of Molecular Sciences. 2022; 23(23):14598. https://doi.org/10.3390/ijms232314598
Chicago/Turabian StyleBelli, Martina, Lucy Barone, Alfonso Bellia, Domenico Sergi, Dalgisio Lecis, Francesca Romana Prandi, Marialucia Milite, Chiara Galluccio, Saverio Muscoli, Francesco Romeo, and et al. 2022. "Treatment of HFpEF beyond the SGLT2-Is: Does the Addition of GLP-1 RA Improve Cardiometabolic Risk and Outcomes in Diabetic Patients?" International Journal of Molecular Sciences 23, no. 23: 14598. https://doi.org/10.3390/ijms232314598
APA StyleBelli, M., Barone, L., Bellia, A., Sergi, D., Lecis, D., Prandi, F. R., Milite, M., Galluccio, C., Muscoli, S., Romeo, F., & Barillà, F. (2022). Treatment of HFpEF beyond the SGLT2-Is: Does the Addition of GLP-1 RA Improve Cardiometabolic Risk and Outcomes in Diabetic Patients? International Journal of Molecular Sciences, 23(23), 14598. https://doi.org/10.3390/ijms232314598