
Autoimmune Neutropenia and Immune-Dysregulation in a Patient Carrying a TINF2 Variant
 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
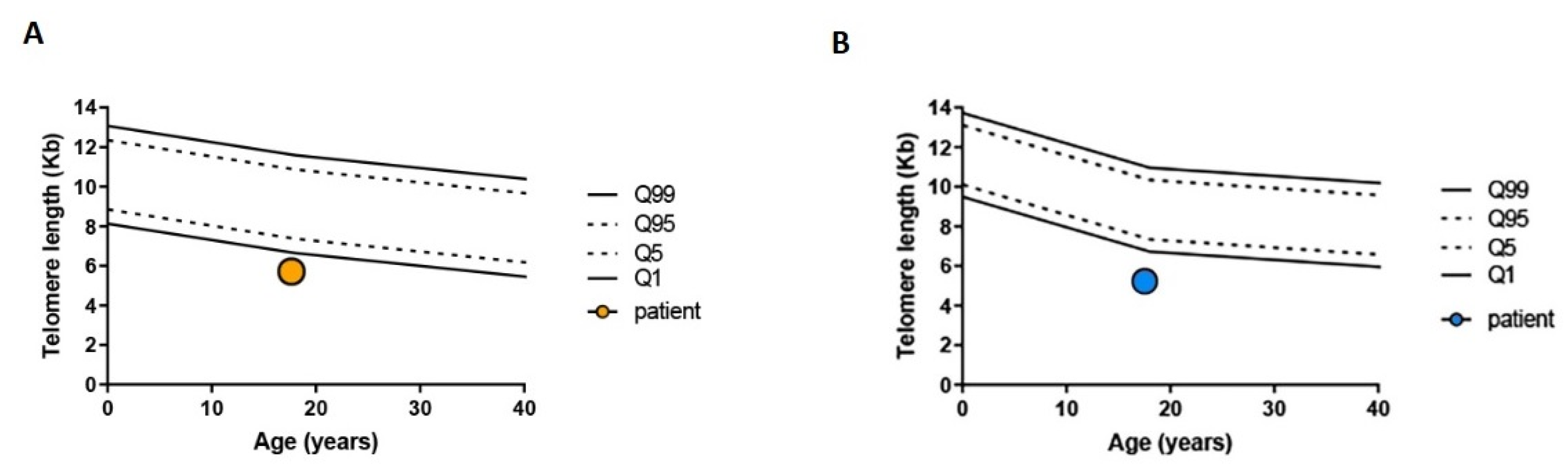
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shimamura, A.; Alter, B.P. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010, 24, 101–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leguit, R.J.; Tweel, J.G.V.D. The pathology of bone marrow failure. Histopathology 2010, 57, 655–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, H.; Nakanishi, K.; Kojima, S. Inherited bone marrow failure syndromes in 2012. Int. J. Hematol. 2013, 97, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Sayour, E.J.; Mousallem, T.; Van Mater, D.; Wang, E.; Martin, P.; Buckley, R.H.; Barfield, R.C. Bone marrow transplantation for CVID-like humoral immune deficiency associated with red cell aplasia. Pediatr. Blood Cancer 2016, 63, 1856–1859. [Google Scholar] [CrossRef]
- Sklarz, T.; Hurwitz, S.N.; Stanley, N.L.; Juusola, J.; Bagg, A.; Babushok, D.V. Aplastic anemia in a patient with CVID due to NFKB1 haploinsufficiency. Mol. Case Stud. 2020, 6, a005769. [Google Scholar] [CrossRef]
- Korthof, E.T.; Svahn, J.; de Latour, R.P.; Terranova, P.; Moins-Teisserenc, H.; Socié, G.; Soulier, J.; Kok, M.; Bredius, R.G.; van Tol, M.; et al. Immunological profile of Fanconi anemia: A multicentric retrospective analysis of 61 patients. Am. J. Hematol. 2013, 88, 472–476. [Google Scholar] [CrossRef]
- Jyonouchi, S.; Forbes, L.; Ruchelli, E.; Sullivan, K. Dyskeratosis congenita: A combined immunodeficiency with broad clinical spectrum-a single-center pediatric experience. Pediatr. Allergy Immunol. 2011, 22, 313–319. [Google Scholar] [CrossRef]
- Myers, K.C.; Sauter, S.; Zhang, X.; Bleesing, J.J.; Davies, S.M.; Wells, S.I.; Mehta, P.A.; Kumar, A.; Marmer, D.; Marsh, R.; et al. Impaired immune function in children and adults with Fanconi anemia. Pediatr. Blood Cancer 2017, 64, e26599. [Google Scholar] [CrossRef] [Green Version]
- Iskander, D.; Roberts, I.; Rees, C.; Szydlo, R.; Alikian, M.; Neale, M.; Harrington, Y.; Kelleher, P.; Karadimitris, A.; De La Fuente, J. Impaired cellular and humoral immunity is a feature of Diamond-Blackfan anaemia; experience of 107 unselected cases in the United Kingdom. Br. J. Haematol. 2019, 186, 321–326. [Google Scholar] [CrossRef]
- Wagner, C.L.; Hanumanthu, V.S.; Talbot, C.C., Jr.; Abraham, R.S.; Hamm, D.; Gable, D.L.; Kanakry, C.G.; Applegate, C.D.; Siliciano, J.; Jackson, J.B.; et al. Short telomere syndromes cause a primary T cell immunodeficiency. J. Clin. Investig. 2018, 128, 5222–5234. [Google Scholar] [CrossRef]
- Sekinaka, Y.; Mitsuiki, N.; Imai, K.; Yabe, M.; Yabe, H.; Mitsui-Sekinaka, K.; Honma, K.; Takagi, M.; Arai, A.; Yoshida, K.; et al. Common Variable Immunodeficiency Caused by FANC Mutations. J. Clin. Immunol. 2017, 37, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Allenspach, E.J.; Bellodi, C.; Jeong, D.; Kopmar, N.; Nakamura, T.; Ochs, H.D.; Ruggero, D.; Skoda-Smith, S.; Shimamura, A.; Torgerson, T.R. Common variable immunodeficiency as the initial presentation of dyskeratosis congenita. J. Allergy Clin. Immunol. 2013, 132, 223–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirwan, M.; Dokal, I. Dyskeratosis congenita: A genetic disorder of many faces. Clin. Genet. 2008, 73, 103–112. [Google Scholar] [CrossRef]
- Farruggia, P.; Fioredda, F.; Puccio, G.; Porretti, L.; Lanza, T.; Ramenghi, U.; Ferro, F.; Macaluso, A.; Barone, A.; Bonanomi, S.; et al. Autoimmune neutropenia of infancy: Data from the Italian neutropenia registry. Am. J. Hematol. 2015, 90, E221–E222. [Google Scholar] [CrossRef] [PubMed]
- Dufour, C.; Miano, M.; Fioredda, F. Old and new faces of neutropenia in children. Haematologica 2016, 101, 789–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fioredda, F.; Rotulo, G.A.; Farruggia, P.; Dagliano, F.; Pillon, M.; Trizzino, A.; Notarangelo, L.; Luti, L.; Lanza, T.; Terranova, P.; et al. Late-onset and long-lasting autoimmune neutropenia: An analysis from the Italian Neutropenia Registry. Blood Adv. 2020, 4, 5644–5649. [Google Scholar] [CrossRef] [PubMed]
- Fioredda, F.; Beccaria, A.; Turrini, E.; Casartelli, P.; Coviello, D.; Maffei, M.; Lanciotti, M.; Lupia, M.; Terranova, P.; Grossi, A.; et al. TACI variants as underlying condition in autoimmune neutropenia: Description of four cases. Am. J. Hematol. 2022, 97, E328–E331. [Google Scholar] [CrossRef]
- Farruggia, P.; Fioredda, F.; Puccio, G.; Onofrillo, D.; Russo, G.; Barone, A.; Bonanomi, S.; Boscarol, G.; Finocchi, A.; Ghilardi, R.; et al. Idiopathic neutropenia of infancy: Data from the Italian Neutropenia Registry. Am. J. Hematol. 2019, 94, 216–222. [Google Scholar] [CrossRef] [Green Version]
- Fioredda, F.; Calvillo, M.; Bonanomi, S.; Coliva, T.; Tucci, F.; Farruggia, P.; Pillon, M.; Martire, B.; Ghilardi, R.; Ramenghi, U.; et al. Congenital and acquired neutropenia consensus guidelines on diagnosis from the Neutropenia Committee of the Marrow Failure Syndrome Group of the AIEOP (Associazione Italiana Emato-Oncologia Pediatrica). Pediatr. Blood Cancer 2011, 57, 10–17. [Google Scholar] [CrossRef]
- Grossi, A.; Miano, M.; Lanciotti, M.; Fioredda, F.; Guardo, D.; Palmisani, E.; Terranova, P.; Santamaria, G.; Caroli, F.; Caorsi, R.; et al. Targeted NGS Yields Plentiful Ultra-Rare Variants in Inborn Errors of Immunity Patients. Genes 2021, 12, 1299. [Google Scholar] [CrossRef]
- Miano, M.; Lanciotti, M.; Giardino, S.; Dufour, C. Ser245Tyr TINF2 mutation in a long-term survivor after a second myeloablative SCT following late graft failure for Aplastic Anaemia. Blood Cells Mol. Dis. 2015, 55, 187–188. [Google Scholar] [CrossRef] [PubMed]
- Lauhasurayotin, S.; Cuvelier, G.D.; Klaassen, R.J.; Fernandez, C.V.; Pastore, Y.D.; Abish, S.; Rayar, M.; Steele, M.; Jardine, L.; Breakey, V.R.; et al. Reanalysing genomic data by normalized coverage values uncovers CNVs in bone marrow failure gene panels. NPJ Genom. Med. 2019, 4, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, D.; Ameli, A.; Prokopenko, D.; Chen, H.; Kho, A.T.; Parker, M.M.; Morrow, J.; Hobbs, B.; Liu, Y.; Beaty, T.H.; et al. Whole exome sequencing analysis in severe chronic obstructive pulmonary disease. Hum. Mol. Genet. 2018, 27, 3801–3812. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, T.W.; van der Vis, J.J.; van Oosterhout, M.F.M.; van Es, H.W.; van Kessel, D.A.; Grutters, J.C.; van Moorsel, C.H.M. TINF2Gene Mutation in a Patient with Pulmonary Fibrosis. Case Rep. Pulmonol. 2016, 2016, 1310862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miano, M.; Grossi, A.; Dell’Orso, G.; Lanciotti, M.; Fioredda, F.; Palmisani, E.; Lanza, T.; Guardo, D.; Beccaria, A.; Ravera, S.; et al. Genetic screening of children with marrow failure. The role of primary Immunodeficiencies. Am. J. Hematol. 2021, 96, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, E.; Witzel, M.; Racek, T.; Puchałka, J.; Hollizeck, S.; Greif-Kohistani, N.; Kotlarz, D.; Horny, H.-P.; Feederle, R.; Schmidt, H.; et al. Myb-like, SWIRM, and MPN domains 1 (MYSM1) deficiency: Genotoxic stress-associated bone marrow failure and developmental aberrations. J. Allergy Clin. Immunol. 2017, 140, 1112–1119. [Google Scholar] [CrossRef] [Green Version]
- Oleaga-Quintas, C.; de Oliveira-Júnior, E.B.; Rosain, J.; Rapaport, F.; Deswarte, C.; Guérin, A.; Sajjath, S.M.; Zhou, Y.J.; Marot, S.; Lozano, C.; et al. Inherited GATA2 Deficiency Is Dominant by Haploinsufficiency and Displays Incomplete Clinical Penetrance. J. Clin. Immunol. 2021, 41, 639–657. [Google Scholar] [CrossRef]
- Lee, P.Y. Vasculopathy, Immunodeficiency, and Bone Marrow Failure: The Intriguing Syndrome Caused by Deficiency of Adenosine Deaminase 2. Front. Pediatr. 2018, 6, 282. [Google Scholar] [CrossRef] [Green Version]
- Carrillo, J.; Calvete, O.; Pintado-Berninches, L.; Manguan-García, C.; Navarro, J.S.; Arias-Salgado, E.G.; Sastre, L.; Guenechea, G.; Granados, E.L.; de Villartay, J.; et al. Mutations in XLF/NHEJ1/Cernunnos gene results in downregulation of telomerase genes expression and telomere shortening. Hum. Mol. Genet. 2017, 26, 1900–1914. [Google Scholar] [CrossRef]
- Al-Marhoobi, R.; Al-Musalhi, M.; Naseem, S.-U.; Wali, Y.; Alsayegh, A.; Al-Tamemi, S. Combined Immunodeficiency, Hemolytic Anemia, and Growth Retardation Secondary to a Homozygous Mutation in the NHEJ1 Gene. J. Pediatr. Hematol. 2020, 42, 333–335. [Google Scholar] [CrossRef]
- Vulliamy, T.; Beswick, R.; Kirwan, M.; Hossain, U.; Walne, A.; Dokal, I. Telomere length measurement can distinguish pathogenic from non-pathogenic variants in the shelterin component, TIN2. Clin. Genet. 2012, 81, 76–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walne, A.J.; Vulliamy, T.; Beswick, R.; Kirwan, M.; Dokal, I. TINF2 mutations result in very short telomeres: Analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood 2008, 112, 3594–3600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez-Rodrigues, F.; Masri, N.; Chouery, E.; Diamond, C.; Jalkh, N.; Vicente, A.; Kajigaya, S.; Abillama, F.; Bejjani, N.; Serhal, W.; et al. A novel homozygous RTEL1 variant in a consanguineous Lebanese family: Phenotypic heterogeneity and disease anticipation. Qual. Life Res. 2019, 138, 1323–1330. [Google Scholar] [CrossRef]
- Dell’Orso, G.; Grossi, A.; Penco, F.; Caorsi, R.; Palmisani, E.; Terranova, P.; Schena, F.; Lupia, M.; Ricci, E.; Montalto, S.; et al. Case Report: Deficiency of Adenosine Deaminase 2 Presenting With Overlapping Features of Autoimmune Lymphoproliferative Syndrome and Bone Marrow Failure. Front. Immunol. 2021, 12, 754029. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Hanumanthu, V.S.; Strong, M.A.; DeZern, A.E.; Stanley, S.E.; Takemoto, C.M.; Danilova, L.; Applegate, C.D.; Bolton, S.G.; Mohr, D.W.; et al. Diagnostic utility of telomere length testing in a hospital-based setting. Proc. Natl. Acad. Sci. USA 2018, 115, E2358–E2365. [Google Scholar] [CrossRef] [Green Version]
- E Stanley, S.; Armanios, M. The short and long telomere syndromes: Paired paradigms for molecular medicine. Curr. Opin. Genet. Dev. 2015, 33, 19. [Google Scholar] [CrossRef] [Green Version]
- Dietz, A.C.; Orchard, P.J.; Baker, K.S.; Giller, R.H.; Savage, S.; Alter, B.P.; Tolar, J. Disease-specific hematopoietic cell transplantation: Nonmyeloablative conditioning regimen for dyskeratosis congenita. Bone Marrow Transplant. 2011, 46, 98–104. [Google Scholar] [CrossRef]
- Miano, M.; Porta, F.; Locatelli, F.; Miniero, R.; La Nasa, G.; Di Bartolomeo, P.; Giardini, C.; Messina, C.; Balduzzi, A.; Testi, A.M.; et al. Unrelated donor marrow transplantation for inborn errors. Bone Marrow Transplant. 1998, 21, S37–S41. [Google Scholar]
- Parry, E.M.; Alder, J.K.; Qi, X.; Chen, J.J.-L.; Armanios, M. Syndrome complex of bone marrow failure and pulmonary fibrosis predicts germline defects in telomerase. Blood 2011, 117, 5607–5611. [Google Scholar] [CrossRef] [Green Version]
- Fioredda, F.; Iacobelli, S.; Korthof, E.T.; Knol, C.; Van Biezen, A.; Bresters, D.; Veys, P.; Yoshimi, A.; Fagioli, F.; Mats, B.; et al. Outcome of haematopoietic stem cell transplantation in dyskeratosis congenita. Br. J. Haematol. 2018, 183, 110–118. [Google Scholar] [CrossRef]


{kind=link}
| Results | Reference Range | |
|---|---|---|
| Haemoglobin | 13.3 g/dL | 11.5–16.5 |
| White blood cells | 2080/mmc | 4000–9800 |
| Neutrophyls | 150/mmc | 2100–6430 |
| Lymphocytes | 1140/mmc | 1000–2800 |
| Platelets | 148.000/mmc | 150.000–450.000 |
| Vitamin B12 | 816 pg/mL | 191–663 |
| IgA | 201 mg/dL | 70–400 |
| IgM | 67 mg/dL | 40–230 |
| IgG | 1166 mg/dL | 700–1600 |
| IgG 1 | 696 mg/dL | 370–1280 |
| IgG 2 | 602 mg/dL | 106–610 |
| IgG 3 | 36.9 mg/dL | 18–163 |
| IgG 4 | 12.2 mg/dL | 4–230 |
| Folic acid | 4.1 ng/mL | 4.6–18.7 |
| Results | Reference Range | |
|---|---|---|
| CD3+ | 80.8% (921/mmc) | 59–70% |
| CD3+CD4+ | 39% | 37–50 |
| CD3+CD8+ | 20.9% | 20–27 |
| CD3+HLA DR+ | 8.0% | 4–6 |
| CD19+ | 11.6% (132/mmc) | 9–13% |
| CD16+CD56+CD3- | 6.8% (78/mmc) | 12–16% |
| CD16+CD56+CD3+ | 2.0% | 3.3–4.6 |
| CD4+/CD8+ ratio | 1.9 | 1.5–2.2 |
| CD25+ | 15.6% | NA |
| CD3+CD4+CD25br+CD45RA- | 0.8% | 0.6–0.8 |
| CD3+CD45RA+ | 32.0% | 24–33 |
| CD3+CD45RO+ | 44.6% | 36–49 |
| CD3+TCR α/β+ | 63.7% | 54–74 |
| CD3+TCR γ/δ+ | 17.0% | 3.3–4.6 |
| CD3+TCR α/β+CD4-CD8- | 3.9% | <1.5% of total lymphocytes |
| CD3+TCR α/β+CD4-CD8- B220+ (%on total α/β+) | 34.0% | <60.0 |
| CD19+CD27+ | 21.5% | >15.0 |
| CD3+CD25+/CD3+ HLA DR+ ratio | 1.8 | >1.0 |
| Target ID | Coverage Design | Mean Coverage Run | Target ID | Coverage Design | Mean Coverage Run |
|---|---|---|---|---|---|
| AP3B1 | 94.5 | 332.8 | NRAS | 100.0 | 369.6 |
| CARD11 | 99.9 | 315.2 | PIK3CD | 98.4 | 274.9 |
| CASP10 | 99.3 | 342.1 | PIK3R1 | 99.7 | 445.1 |
| CASP8 | 99.4 | 396.4 | PRKCD | 99.7 | 366.2 |
| CD19 | 100.0 | 298.2 | RAB27A | 100.0 | 440.5 |
| CD20 | 88.4 | 397.9 | RAC2 | 100.0 | 288.2 |
| CD40 | 100.0 | 413.2 | RPL11 | 100.0 | 406.6 |
| CD40LG | 99.3 | 477.0 | RPL26 | 100.0 | 437.0 |
| CSF3R | 100.0 | 351.8 | RPL35A | 100.0 | 471.8 |
| CTC1 * | 93.3 | 375.5 | RPL5 | 100.0 | 370.8 |
| CTLA4 | 100.0 | 374.0 | RPS10 | 100.0 | 373.3 |
| CXCR4 | 100.0 | 395.1 | RPS17 | 99.1 | 142.3 |
| DKC1 * | 99.5 | 386.0 | RPS19 | 100.0 | 318.8 |
| ELANE | 90.6 | 154.3 | RPS24 | 97.9 | 383.8 |
| FADD | 92.8 | 236.5 | RPS26 | 100.0 | 301.6 |
| FAS | 99.4 | 401.1 | RPS7 | 100.0 | 375.3 |
| FASLG | 100.0 | 355.4 | RTEL1 * | 97.9 | 267.8 |
| G6PC | 99.0 | 410.6 | SBDS | 100.0 | 368.3 |
| GFI1 | 97.5 | 218.6 | SLC37A4 | 99.4 | 334.0 |
| HAX1 | 100,0 | 426.9 | TAZ | 84.0 | 362.5 |
| ITK | 98,4 | 354.0 | TERT * | 88.7 | 214.9 |
| JAGN1 | 100.0 | 354.0 | TINF2 * | 100.0 | 405.3 |
| KRAS | 100.0 | 403.7 | TNFRSF13B | 89.4 | 417.8 |
| LAMTOR2 | 88.8 | 309.0 | TNFRSF13C | 33.9 | 304.5 |
| LRBA | 99.94 | 334.6 | USB1 | 100.0 | 379.2 |
| LYST | 100.0 | 385.0 | VPS13B | 96.5 | 368.3 |
| MAGT1 | 100,0 | 359.2 | VPS45 | 96.7 | 370.7 |
| NHP2 * | 100,0 | 307.3 | WAS | 86.0 | 320.9 |
| NOP10 * | 100,0 | 407.0 | WRAP53* | 98.8 | 380.7 |
| Results | Reference Range | |
|---|---|---|
| CFU-E/BFU-E | 35 | 27–81 |
| CFU-GEMM | 0 | 0–10 |
| CFU-GM | 33 | 33–100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chianucci, B.; Grossi, A.; Dell'Orso, G.; Palmisani, E.; Lanciotti, M.; Terranova, P.; Pierri, F.; Lupia, M.; Arcuri, L.; Laurino, M.; et al. Autoimmune Neutropenia and Immune-Dysregulation in a Patient Carrying a TINF2 Variant. Int. J. Mol. Sci. 2022, 23, 14535. https://doi.org/10.3390/ijms232314535
Chianucci B, Grossi A, Dell'Orso G, Palmisani E, Lanciotti M, Terranova P, Pierri F, Lupia M, Arcuri L, Laurino M, et al. Autoimmune Neutropenia and Immune-Dysregulation in a Patient Carrying a TINF2 Variant. International Journal of Molecular Sciences. 2022; 23(23):14535. https://doi.org/10.3390/ijms232314535
Chicago/Turabian StyleChianucci, Benedetta, Alice Grossi, Gianluca Dell'Orso, Elena Palmisani, Marina Lanciotti, Paola Terranova, Filomena Pierri, Michela Lupia, Luca Arcuri, Marica Laurino, and et al. 2022. "Autoimmune Neutropenia and Immune-Dysregulation in a Patient Carrying a TINF2 Variant" International Journal of Molecular Sciences 23, no. 23: 14535. https://doi.org/10.3390/ijms232314535