
IL-33 and the Cytokine Storm in COVID-19: From a Potential Immunological Relationship towards Precision Medicine

 ,
,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Biology of IL-33
3. The Role of IL-33 Signalling in COVID-19 Inflammatory Status
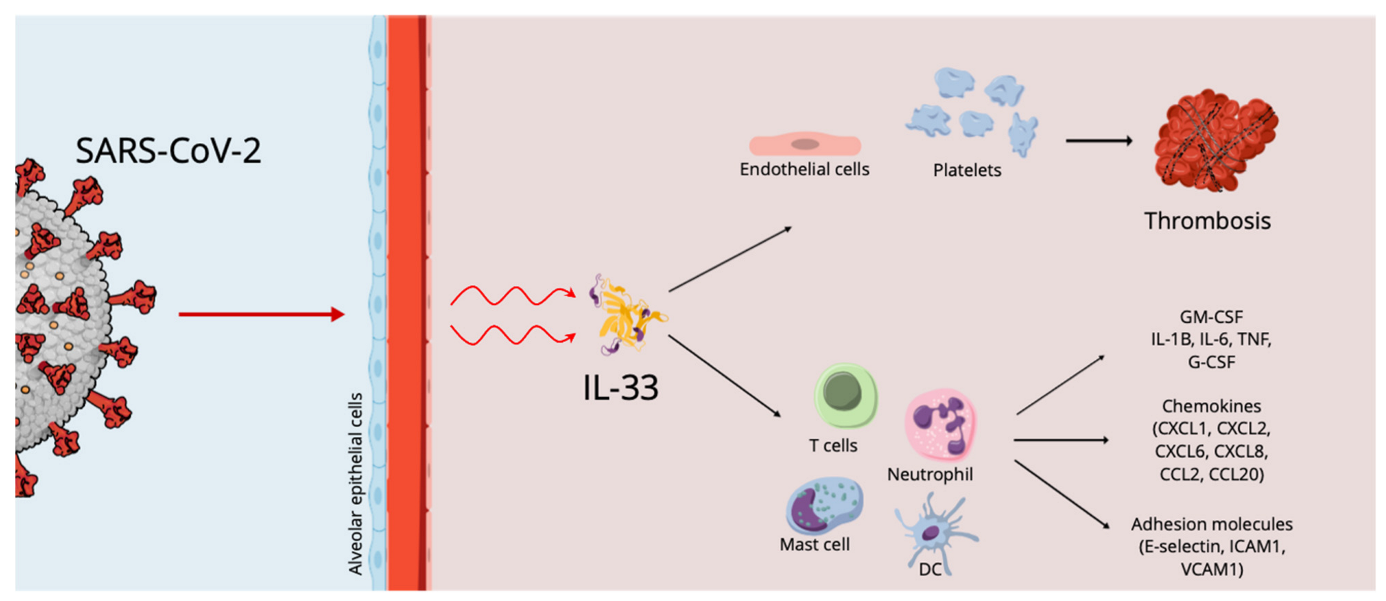
Cytokine Storm, IL-33 Effects and Thrombosis in COVID 19 Infection
4. Discussion and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; McGoogan, J.M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.S.; Kim, J.Y.; Kim, M.-C.; Park, S.Y.; Kim, B.-N.; Bae, S.; Cha, H.H.; Jung, J.; Kim, M.-J.; Li, M.J.; et al. Factors of Severity in Patients With COVID-19: Cytokine/Chemokine Concentrations, Viral Load, and Antibody Responses. Am. J. Trop. Med. Hyg. 2020, 1036, 2412–2418. [Google Scholar] [CrossRef] [PubMed]
- Gavriilaki, E.; Brodsky, R.A. Severe COVID-19 Infection and Thrombotic Microangiopathy: Success Does Not Come Easily. Br. J. Haematol. 2020, 189, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiao, M.; Zhang, S.; Xia, P.; Cao, W.; Jiang, W.; Chen, H.; Ding, X.; Zhao, H.; Zhanh, H.; et al. Coagulopathy and Antiphospholipid Antibodies in Patients With COVID-19. N. Engl. J. Med. 2020, 382, 38. [Google Scholar] [CrossRef]
- Verity, R.; Okell, L.C.; Dorigatti, I.; Winskill, P.; Whittaker, C.; Imai, N.; Cuomo-Dannenburg, G.; Thompson, H.; Walker, P.G.T.; Fu, H.; et al. Estimates of the Severity of Coronavirus Disease 2019: A Model-Based Analysis. Lancet Infect Dis. 2020, 20, 669–677. [Google Scholar] [CrossRef]
- Sandler, C.X.; Wyller, V.B.B.; Moss-Morris, R.; Buchwald, D.; Crawley, E.; Hautvast, J.; Katz, B.J.; Knoop, H.; Little, P.; Taylor, R.; et al. Long COVID and Post-Infective Fatigue Syndrome: A Review. Open Forum. Infect Dis. 2021, 8, ofab440. [Google Scholar] [CrossRef] [PubMed]
- Stavem, K.; Ghanima, W.; Olsen, M.K.; Gilboe, H.M. Prevalence and Determinants of Fatigue After COVID-19 in Non-Hospitalized Subjects: A Population-Based Study. Int. J. Environ. Res. Public. Health 2021, 18, 2030. [Google Scholar] [CrossRef]
- Montazersaheb, S.; Hosseiniyan Khatibi, S.M.; Hejazi, M.S.; Tarhriz, V.; Farjami, A.; Sorbeni, F.G.; Farahzadi, R.; Ghasemnejad, T. COVID-19 infection: An overview on cytokine storm and related interventions. Virol. J. 2022, 19, 92. [Google Scholar] [CrossRef]
- Yang, M. Cell Pyroptosis, a Potential Pathogenic Mechanism of 2019-nCoV Infection. SSRN J. 2020. Available online: https://ssrn.com/abstract=3527420 (accessed on 19 November 2022).
- Tavakolpour, S.; Rakhshandehroo, T.; Wei, E.X.; Rashidian, M. Lymphopenia during the COVID-19 infection: What it shows and what can be learned. Immunol. Lett. 2020, 225, 31. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Barton, L.M.; Duval, E.; Stroberg, E.; Gosh, S.; Mukhopadhyay, S. COVID-19 autopsies, Oklahoma, U.S.A. Am. J. Clin. Pathol. 2020, 153, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Park, M.D. Macrophages: A Trojan horse in COVID-19? Nat. Rev. Immunol. 2020, 20, 351. [Google Scholar] [CrossRef]
- Murdaca, G.; Greco, M.; Tonacci, A.; Negrini, S.; Borro, M.; Puppo, S.; Gangemi, S. IL-33/IL-31 Axis in Immune-Mediated and Allergic Diseases. Int. J. Mol. Sci. 2019, 20, 5856. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.B.; Savage, A.K.; Locksley, R.M. Interleukin-33 in tissue homeostasis, injury, and inflammation. Immunity 2015, 42, 1005–1019. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.P. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol. Rev. 2018, 281, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Smithgall, M.D.; Comeau, M.R.; Yoon, B.R.; Kaufman, D.; Armitage, R.; Smith, D.E. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int. Immunol. 2008, 20, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, E.; Van, L.P.; Samson, M.; Diem, S.; Barra, A.; Roga, S.; Gombert, J.-M.; Schneider, E.; Dy, M.; Gourdy, P.; et al. The pro-Th2 cytokine IL-33 directly interacts with invariant NKT and NK cells to induce IFN- gamma production. Eur. J. Immunol. 2009, 39, 1046–1055. [Google Scholar] [CrossRef]
- Alves-Filho, J.C.; Sonego, F.; Souto, F.O.; Freitas, A.; Verri, W.A., Jr.; Auxiliadora-Martins, M.; Basile-Filho, A.; McKenzie, A.N.; Xu, D.; Cunha, F.Q.; et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat. Med. 2010, 16, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Carriere, V.; Roussel, L.; Ortega, N.; Lacorre, D.-A.; Americh, L.; Aguilar, L.; Bouche, G.; Girard, J.-P. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 282–287. [Google Scholar] [CrossRef]
- Roussel, L.; Erard, M.; Cayrol, C.; Girard, J.P. Molecular mimicry between IL-33 and KSHV for attachment to chromatin through the H2A–H2B acidic pocket. EMBO Rep. 2008, 9, 1006–1012. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef]
- Cayrol, C.; Duval, A.; Schmitt, P.; Roga, S.; Camus, M.; Stella, A.; Burlet- Schiltz, O.; Gonzalez-de-Peredo, A.; Girard, J.-P. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat. Immunol. 2018, 19, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Lefrancais, E.; Roga, S.; Gautier, V.; Gonzalez-de-Peredo, A.; Monsarrat, B.; Girard, J.-P.; Cayrol, C. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc. Natl. Acad. Sci. USA 2012, 109, 1673–1678. [Google Scholar] [CrossRef] [PubMed]
- Lefrancais, E.; Duval, A.; Mirey, E.; Roga, S.; Espinosa, S.; Cayrol, C.; Girard, J.-P. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc. Natl. Acad. Sci. USA 2014, 11, 15502–15507. [Google Scholar] [CrossRef]
- Scott, I.C.; Majithiya, J.B.; Sanden, C.; Thornton, P.; Sanders, P.N.; Moore, T.; Guscott, M.; Corkill, D.J.; Erjefalt, J.S.; Cohen, E.S. Interleukin-33 is activated by allergen- and necrosis-associated proteolytic activities to regulate its alarmin activity during epithelial damage. Sci. Rep. 2018, 8, 3363. [Google Scholar] [CrossRef]
- Pichery, M.; Mirey, E.; Mercier, P.; Lefrancais, E.; Dujardin, A.; Ortega, N.; Girard, J.-P. Endogenous IL-33 Is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues. in situ analysis using a novel Il-33-LacZ gene trap reporter strain. J. Immunol. 2012, 188, 3488–3495. [Google Scholar] [CrossRef] [PubMed]
- Sundnes, O.; Pietka, W.; Loos, T.; Sponheim, J.; Rankin, A.L.; Pflanz, S.; Bertelsen, V.; Sitek, J.C.; Hol, J.; Haraldsen, G.; et al. Epidermal Expression and Regulation of Interleukin-33 during Homeostasis and Inflammation: Strong Species Differences. J. Investig. Dermatol. 2015, 135, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Muto, T.; Kawagoe, T.; Matsumoto, M.; Sasaki, Y.; Matsushita, K.; Taki, Y.; Futatsugi-Yumikura, S.; Tsutsui, H.; Ishii, K.J.; et al. Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc. Natl. Acad. Sci. USA 2012, 109, 3451–3456. [Google Scholar] [CrossRef] [PubMed]
- Byers, D.E.; Alexander-Brett, J.; Patel, A.C.; Agapov, E.; Dang-Vu, G.; Jin, X.; Wu, K.; You, Y.; Alevy, Y.; Girard, J.-P.; et al. Long-term IL-33-producing epithelial progenitor cells in chronic obstructive lung disease. J. Clin. Investig. 2013, 123, 3967–3982. [Google Scholar] [CrossRef] [PubMed]
- Kearley, J.; Silver, J.S.; Sanden, C.; Liu, Z.; Berlin, A.A.; White, N.; Mori, M.; Pham, T.-H.; Ward, C.K.; Criner, G.J.; et al. Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin-33-dependent response to infection. Immunity 2015, 42, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Girard, J.P. Interleukin-33 (IL-33): A critical review of its biology and the mechanisms involved in its release as a potent extracellular cytokine. Cytokine 2022, 156, 155891. [Google Scholar] [CrossRef]
- Murdaca, G.; Di Gioacchino, M.; Greco, M.; Borro, M.; Paladin, F.; Petrarca, C.; Gangemi, S. Basophils and Mast Cells in CVID-19 pathogenesis. Cells 2021, 10, 2754. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Di Gioacchino, M.; Tonacci, A.; Musolino, C. Immunopathology of SARS-CoV-2 Infection: Immune Cells and Mediators, Prognostic Factors, and Immune-Therapeutic Implications. Int. J. Mol. Sci. 2020, 21, 4782. [Google Scholar] [CrossRef] [PubMed]
- Di Salvo, E.; Di Gioacchino, M.; Tonacci, A.; Casciaro, M.; Gangemi, S. Alarmins, COVID-19 and comorbidities. Ann. Med. 2021, 53, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Ge, Y.; Sun, J. IL-33 in COVID-19: Friend or foe? Cell. Mol. Immunol. 2021, 18, 1602–1604. [Google Scholar] [CrossRef]
- Stanczak, M.A.; Sanin, D.E.; Apostolova, P.; Nerz, G.; Lampaki, D.; Hofmann, M.; Steimann, D.; Krohn-Grimberghe, M.; Thimme, R.; Mittler, G.; et al. IL-33 expression in response to SARS-CoV-2 correlates with seropositivity in COVID-19 convalescent individuals. Nat. Commun. 2021, 12, 2133. [Google Scholar] [CrossRef] [PubMed]
- Markovic, S.S.; Jovanovic, M.; Gajovic, N.; Jurisevic, M.; Arsenijevic, N.; Jovanovic, M.; Jovanovic, M.; Mijailovic, Z.; Lukic, S.; Zornic, N.; et al. IL 33 Correlates With COVID-19 Severity, Radiographic and Clinical Finding. Front. Med. 2021, 8, 749569. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Sánchez, R.; Lepe-Balsalobre, E.; Viloria-Peñas, M. Prognostic factors for the severity of SARS-CoV-2 infection. Adv. Lab. Med. 2021, 2, 253–258. [Google Scholar] [CrossRef]
- Meidaninikjeh, S.; Sabouni, N.; Marzouni, H.Z.; Bengar, S.; Khalili, A.; Jafari, R. Monocytes and macrophages in COVID-19: Friends and foes. Life Sci. 2021, 269, 119010. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, M. Immunopathogenesis of SARS-CoV-2-induced pneumonia:lessons from influenza virus infection. Inflamm. Regen. 2020, 40, 39. [Google Scholar] [CrossRef] [PubMed]
- Murdaca, G.; Paladin, F.; Tonacci, A.; Borro, M.; Greco, M.; Gerosa, A.; Isola, S.; Allegra, A.; Gangemi, S. Involvement of Il-33 in the Pathogenesis and Prognosis of Major Respiratory Viral Infections: Future Perspectives for Personalized Therapy. Biomedicines 2022, 10, 715. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Hong, X.Y.; Li, Y.; Chen, W.; Ye, G.; Li, Y.; Luo, Y. Serum-soluble ST2 as a novel biomarker reflecting inflammatory status and illness severity in patients with COVID-19. Biomark. Med. 2020, 14, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Cristinziano, L.; Poto, R.; Criscuolo, G.; Ferrara, A.L.; Galdiero, M.R.; Modestino, L.; Loffredo, S.; de Paulis, A.; Marone, G.; Spadaro, G.; et al. IL-33 and Superantigenic Activation of Human Lung Mast Cells Induce the Release of Angiogenic and Lymphangiogenic Factors. Cells 2021, 10, 145. [Google Scholar] [CrossRef] [PubMed]
- Gaurav, R.; Anderson, D.R.; Radio, S.J.; Bailey, K.L.; England, B.R.; Mikuls, T.R.; Thiele, G.M.; Strah, H.M.; Romberger, D.J.; Wyatt, T.A.; et al. IL-33 Depletion in COVID-19 Lungs. Chest 2021, 160, 1656–1659. [Google Scholar] [CrossRef] [PubMed]
- Opal, S.M. Interactions between coagulation and inflammation. Scand. J. Infect. Dis. 2003, 35, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Wifi, M.N.; Morad, M.A.; El Sheemy, R.; Abdeen, N.; Afify, S.; Abdalgaber, M.; Abdellatef, A.; Zaghloul, M.; Alboraie, M.; El-Kassas, M. Hemostatic system and COVID-19 crosstalk: A review of the available evidence. World J. Methodol. 2022, 12, 331–349. [Google Scholar] [CrossRef]
- Nappi, F.; Nappi, P.; Gambardella, I.; Avtaar Singh, S.S. Thromboembolic Disease and Cardiac Thrombotic Complication in COVID-19: A Systematic Review. Metabolites 2022, 12, 889. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Innao, V.; Allegra, A.G.; Musolino, C. Coagulopathy and thromboembolic events in patients with SARS-CoV-2 infection: Pathogenesis and management strategies. Ann. Hematol. 2020, 99, 1953–1965. [Google Scholar] [CrossRef] [PubMed]
- Conway, E.M.; Mackman, N.; Warren, R.Q.; Wolberg, A.S.; Mosnier, L.O.; Campbell, R.A.; Gralinski, L.E.; Rondina, M.T.; van de Veerdonk, F.L.; Hoffmeister, K.M.; et al. Understanding COVID-19-associated coagulopathy. Nat. Rev. Immunol. 2022, 22, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Vecchié, A.; Dagna, L.; Martinod, K.; Dixon, D.L.; Van Tassell, B.W.; Dentali, F.; Montecucco, F.; Massberg, S.; Levi, M.; et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 2021, 21, 319–329. [Google Scholar] [CrossRef] [PubMed]
- McFadyen, J.D.; Stevens, H.; Peter, K. The Emerging Threat of (Micro)Thrombosis in COVID-19 and Its Therapeutic Implications. Circ. Res. 2020, 127, 571–587. [Google Scholar] [CrossRef] [PubMed]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Braunstein, E.M.; Yuan, X.; Yu, J.; Alexander, A.; Chen, H.; Gavriilaki, E.; Alluri, R.; Streiff, M.B.; Petri, M.; et al. Complement activity and complement regulatory gene mutations are associated with thrombosis in APS and CAPS. Blood 2020, 135, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Gurbel, P.A.; Tantry, U.S. In Vitro Evidence for the Role of Cytokine Storm in the Generation of Stent Thrombosis in COVID-19 Patients. Cardiovasc. Revasc. Med. 2022, 35, 139–140. [Google Scholar] [CrossRef]
- Demyanets, S.; Konya, V.; Kastl, S.P.; Kaun, C.; Rauscher, S.; Niessner, A.; Pentz, R.; Pfaffenberger, S.; Rychli, K.; Lemberger, C.E.; et al. Interleukin-33 induces expression of adhesion molecules and inflammatory activation in human endothelial cells and in human atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Hayakawa, M.; Ozaki, H.; Takezako, N.; Obata, H.; Ibaraki, N.; Tsuru, T.; Tominaga, S.; Yanagisawa, K. ST2 gene expression is proliferation-dependent and its ligand, IL-33, induces inflammatory reaction in endothelial cells. Mol. Cell. Biochem. 2010, 335, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Choi, H.J.; Min, J.K.; Pyun, B.J.; Maeng, Y.S.; Park, H.; Kim, J.; Kim, Y.M.; Kwon, Y.G. Interleukin-33 induces angiogenesis and vascular permeability through ST2/TRAF6-mediated endothelial nitric oxide production. Blood 2009, 114, 3117–3126. [Google Scholar] [CrossRef] [PubMed]
- Stojkovic, S.; Kaun, C.; Heinz, M.; Krychtiuk, K.A.; Rauscher, S.; Lemberger, C.E.; de Martin, R.; Gröger, M.; Petzelbauer, P.; Huk, I.; et al. Interleukin-33 induces urokinase in human endothelial cells--possible impact on angiogenesis. J. Thromb. Haemost. 2014, 12, 948–957. [Google Scholar] [CrossRef] [PubMed]
- Demyanets, S.; Speidl, W.S.; Tentzeris, I.; Jarai, R.; Katsaros, K.M.; Farhan, S.; Krychtiuk, K.A.; Wonnerth, A.; Weiss, T.W.; Huber, K.; et al. Soluble ST2 and interleukin-33 levels in coronary artery disease: Relation to disease activity and adverse outcome. PLoS ONE. 2014, 9, e95055. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, O.S.; Narayan, H.K.; Khan, S.Q.; Kelly, D.; Quinn, P.A.; Squire, I.B.; Davies, J.E.; Ng, L.L. Pre-discharge risk stratification in unselected STEMI: Is there a role for ST2 or its natural ligand IL-33 when compared with contemporary risk markers? Int. J. Cardiol. 2013, 167, 2182–2188. [Google Scholar] [CrossRef] [PubMed]
- Demyanets, S.; Tentzeris, I.; Jarai, R.; Katsaros, K.M.; Farhan, S.; Wonnerth, A.; Weiss, T.W.; Wojta, J.; Speidl, W.S.; Huber, K. An increase of interleukin-33 serum levels after coronary stent implantation is associated with coronary in-stent restenosis. Cytokine 2014, 67, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Stojkovic, S.; Kaun, C.; Basilio, J.; Rauscher, S.; Hell, L.; Krychtiuk, K.A.; Bonstingl, C.; de Martin, R.; Gröger, M.; Ay, C.; et al. Tissue factor is induced by interleukin-33 in human endothelial cells: A new link between coagulation and inflammation. Sci. Rep. 2016, 6, 25171. [Google Scholar] [CrossRef] [PubMed]
- Stojkovic, S.; Thulin, Å.; Hell, L.; Thaler, B.; Rauscher, S.; Baumgartner, J.; Gröger, M.; Ay, C.; Demyanets, S.; Neumayer, C.; et al. IL-33 stimulates the release of procoagulant microvesicles from human monocytes and differentially increases tissue factor in human monocyte subsets. Thromb. Haemost. 2017, 117, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Zeyda, M.; Wernly, B.; Demyanets, S.; Kaun, C.; Hammerle, M.; Hantusch, B.; Schranz, M.; Neuhofer, A.; Itariu, B.K.; Keck, M.; et al. Severe obesity increases adipose tissue expression of interleukin-33 and its receptor ST2, both predominantly detectable in endothelial cells of human adipose tissue. Int. J. Obes. 2013, 37, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Van Hinsbergh, V.W. Endothelium-role in regulation of coagulation and inflammation. Semin. Immunopathol. 2012, 34, 93–106. [Google Scholar] [CrossRef]
- Demyanets, S.; Stojkovic, S.; Huber, K.; Wojta, J. The Paradigm Change of IL-33 in Vascular Biology. Int. J. Mol. Sci. 2021, 22, 13288. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Luo, J.; Li, J.; Kim, G.; Stewart, A.; Huang, Y.; Wu, C. Intestinal IL-33 promotes platelet activity for neutrophil recruitment during acute inflammation. Blood 2022, 139, 1878–1891. [Google Scholar] [CrossRef]
- Pariser, D.N.; Hilt, Z.T.; Ture, S.K.; Blick-Nitko, S.K.; Looney, M.R.; Cleary, S.J.; Roman-Pagan, E.; Saunders, J., II; Georas, S.N.; Veazey, J.; et al. Lung megakaryocytes are immune modulatory cells. J. Clin. Investig. 2021, 131, e137377. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The Trinity of COVID-19: Immunity, Inflammation and Intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tang, J.; Yang, X.; Zanvit, P.; Cui, K.; Ku, W.L.; Jin, W.; Zhang, D.; Goldberg, N.; Cain, A.; et al. TGF-β induces ST2 and programs ILC2 development. Nat. Commun. 2020, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Zizzo, G.; Cohen, P.L. Imperfect storm: Is interleukin-33 the Achilles heel of COVID-19? Lancet Rheumatol. 2020, 2, e779–e790. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Burn, T.N.; Weaver, L.; Rood, J.E.; Chu, N.; Bodansky, A. Genetic deficiency of interferon-γ reveals interferon-γ-independent manifestations of murine hemophagocytic lymphohistiocytosis. Arthritis Rheumatol. 2020, 72, 335–347. [Google Scholar] [CrossRef]
- Chan, B.C.; Lam, C.W.K.; Tam, L.S.; Wong, C.K. IL33: Roles in Allergic Inflammation and Therapeutic Perspectives. Front. Immunol. 2019, 10, 364. [Google Scholar] [CrossRef]
- Hirahara, K.; Mato, N.; Hagiwara, K.; Nakayama, T. The pathogenicity of IL-33 on steroid-resistant eosinophilic inflammation via the activation of memory-type ST2(+) CD4(+) T cells. J. Leukoc. Biol. 2018, 104, 895–901. [Google Scholar] [CrossRef]
- Saglani, S.; Lui, S.; Ullmann, N.; Campbell, G.A.; Sherburn, R.T.; Mathie, S.A.; Denney, L.; Bossley, C.J.; Oates, T.; Walker, S.A.; et al. IL-33 promotes airway remodeling in pediatric patients with severe steroid-resistant asthma. J. Allergy Clin. Immunol. 2013, 132, 676–685. [Google Scholar] [CrossRef]
- Balato, A.; Di Caprio, R.; Canta, L.; Mattii, M.; Lembo, S.; Raimondo, A.; Schiattarella, M.; Balato, N.; Ayala, F. IL-33 is regulated by TNF-alpha in normal and psoriatic skin. Arch. Dermatol. Res. 2014, 306, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Yanagawa, Y.; Matsumoto, M.; Togashi, H. Adrenoceptor-mediated enhancement of interleukin-33 production by dendritic cells. Brain Behav. Immun. 2011, 25, 1427–1433. [Google Scholar] [CrossRef] [PubMed]
- Nabe, T. Interleukin (IL)-33: New therapeutic target for atopic diseases. J. Pharmacol. Sci. 2014, 126, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Thio, C.L.; Chi, P.Y.; Lai, A.C.; Chang, Y.J. Regulation of type 2 innate lymphoid cell-dependent airway hyperreactivity by butyrate. J. Allergy Clin. Immunol. 2018, 142, 1867–1883. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furci, F.; Murdaca, G.; Allegra, A.; Gammeri, L.; Senna, G.; Gangemi, S. IL-33 and the Cytokine Storm in COVID-19: From a Potential Immunological Relationship towards Precision Medicine. Int. J. Mol. Sci. 2022, 23, 14532. https://doi.org/10.3390/ijms232314532
Furci F, Murdaca G, Allegra A, Gammeri L, Senna G, Gangemi S. IL-33 and the Cytokine Storm in COVID-19: From a Potential Immunological Relationship towards Precision Medicine. International Journal of Molecular Sciences. 2022; 23(23):14532. https://doi.org/10.3390/ijms232314532
Chicago/Turabian StyleFurci, Fabiana, Giuseppe Murdaca, Alessandro Allegra, Luca Gammeri, Gianenrico Senna, and Sebastiano Gangemi. 2022. "IL-33 and the Cytokine Storm in COVID-19: From a Potential Immunological Relationship towards Precision Medicine" International Journal of Molecular Sciences 23, no. 23: 14532. https://doi.org/10.3390/ijms232314532
APA StyleFurci, F., Murdaca, G., Allegra, A., Gammeri, L., Senna, G., & Gangemi, S. (2022). IL-33 and the Cytokine Storm in COVID-19: From a Potential Immunological Relationship towards Precision Medicine. International Journal of Molecular Sciences, 23(23), 14532. https://doi.org/10.3390/ijms232314532