

Ononitol Monohydrate—A Glycoside Potentially Inhibit HT-115 Human Colorectal Cancer Cell Proliferation through COX-2/PGE-2 Inflammatory Axis Regulations

 ,
,  , and
, and 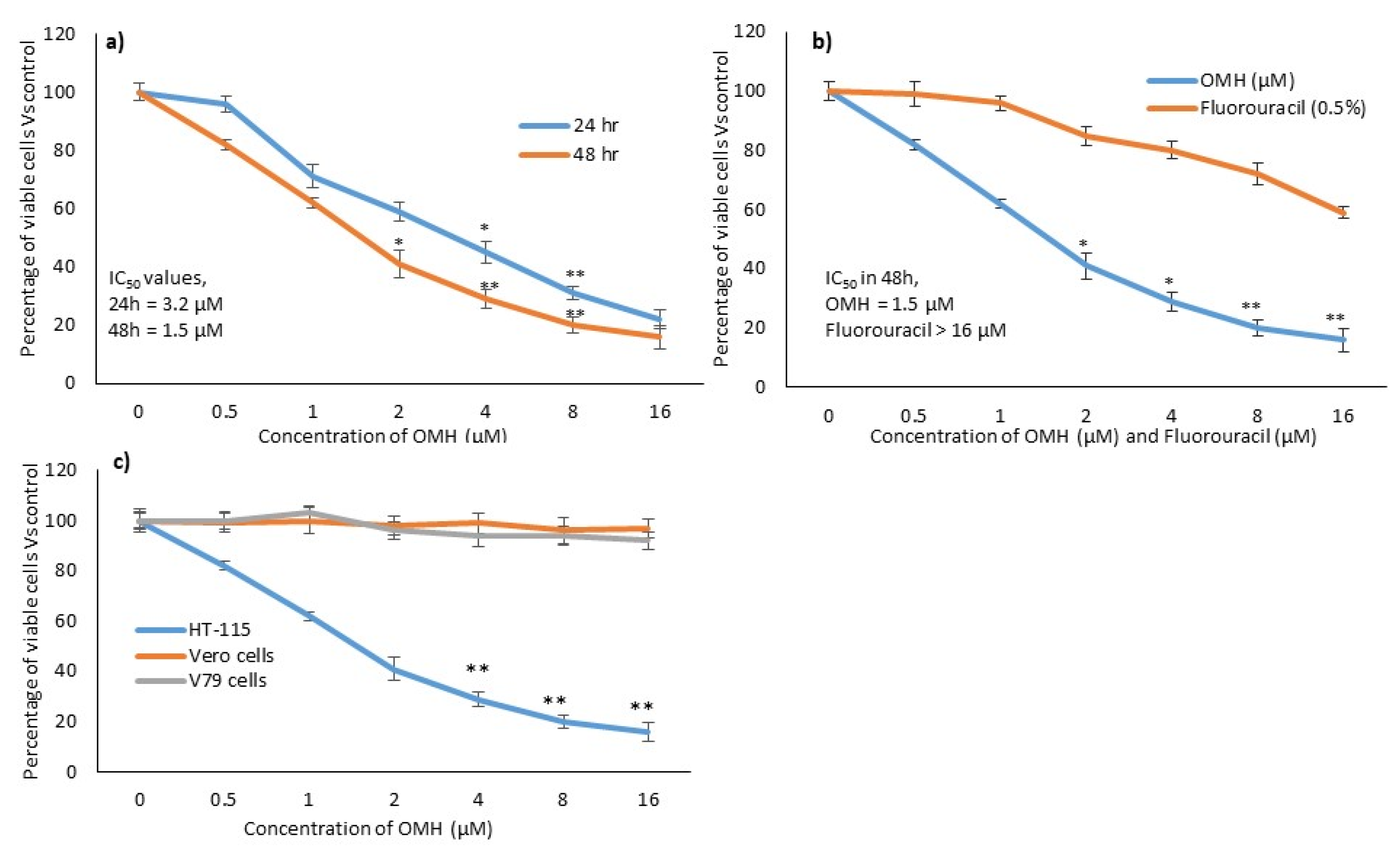{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. In Vitro Cytotoxic Effect of OMH
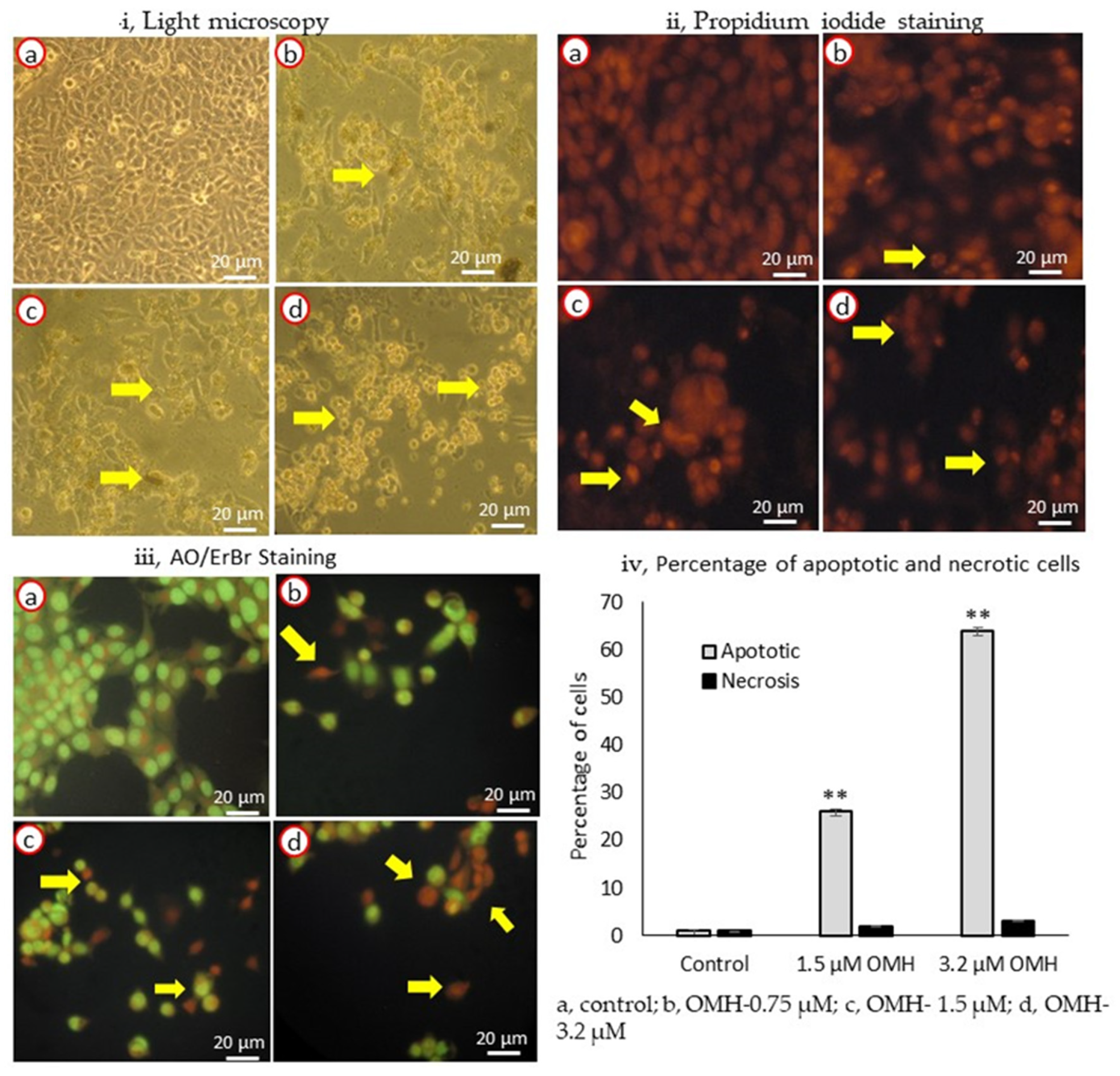
2.2. Effect of OMH on Cell and Nuclear Morphology
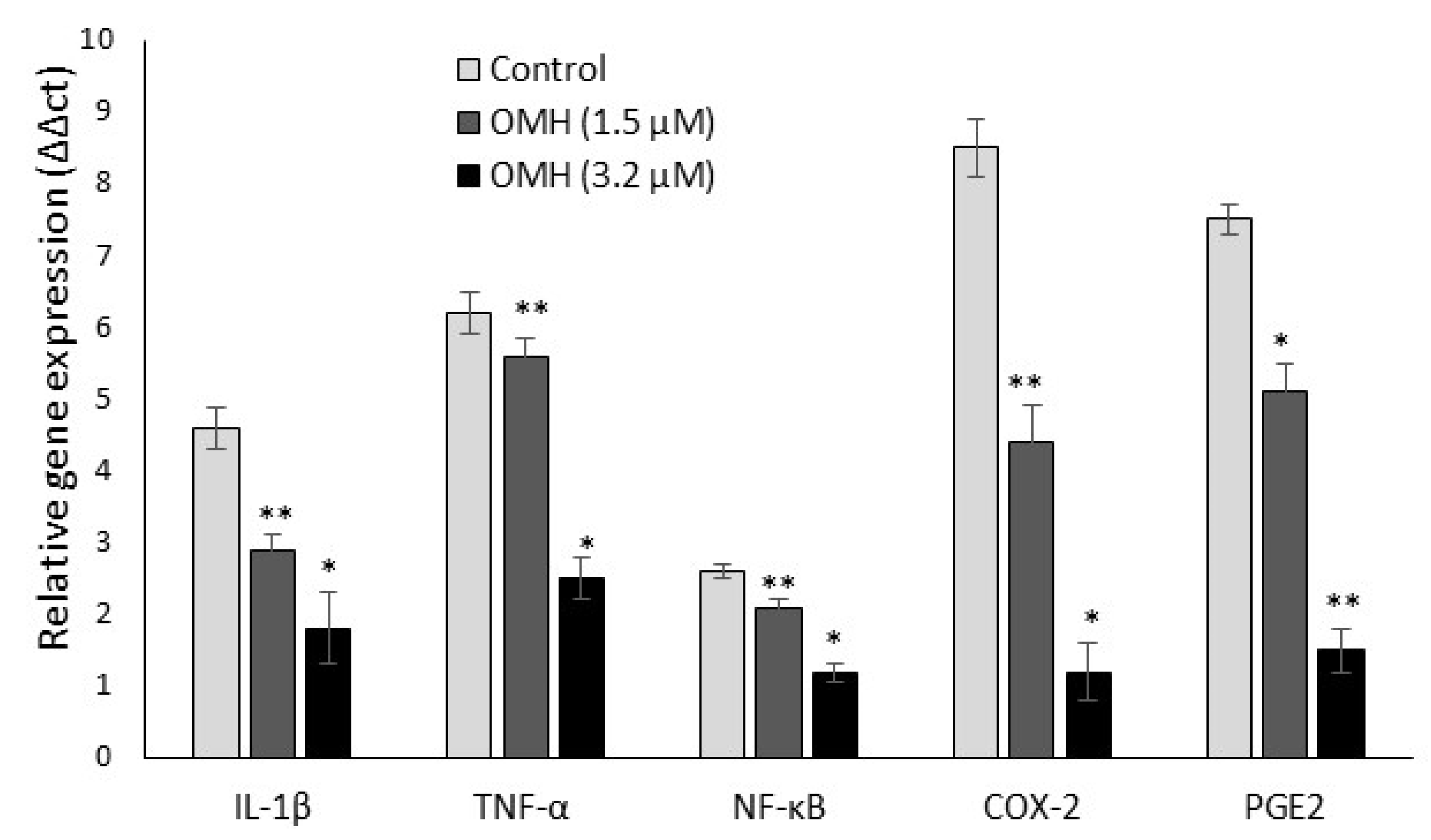
2.3. Effect of OMH on Inflamamtory and Protumorigenic Gene Expression Levels
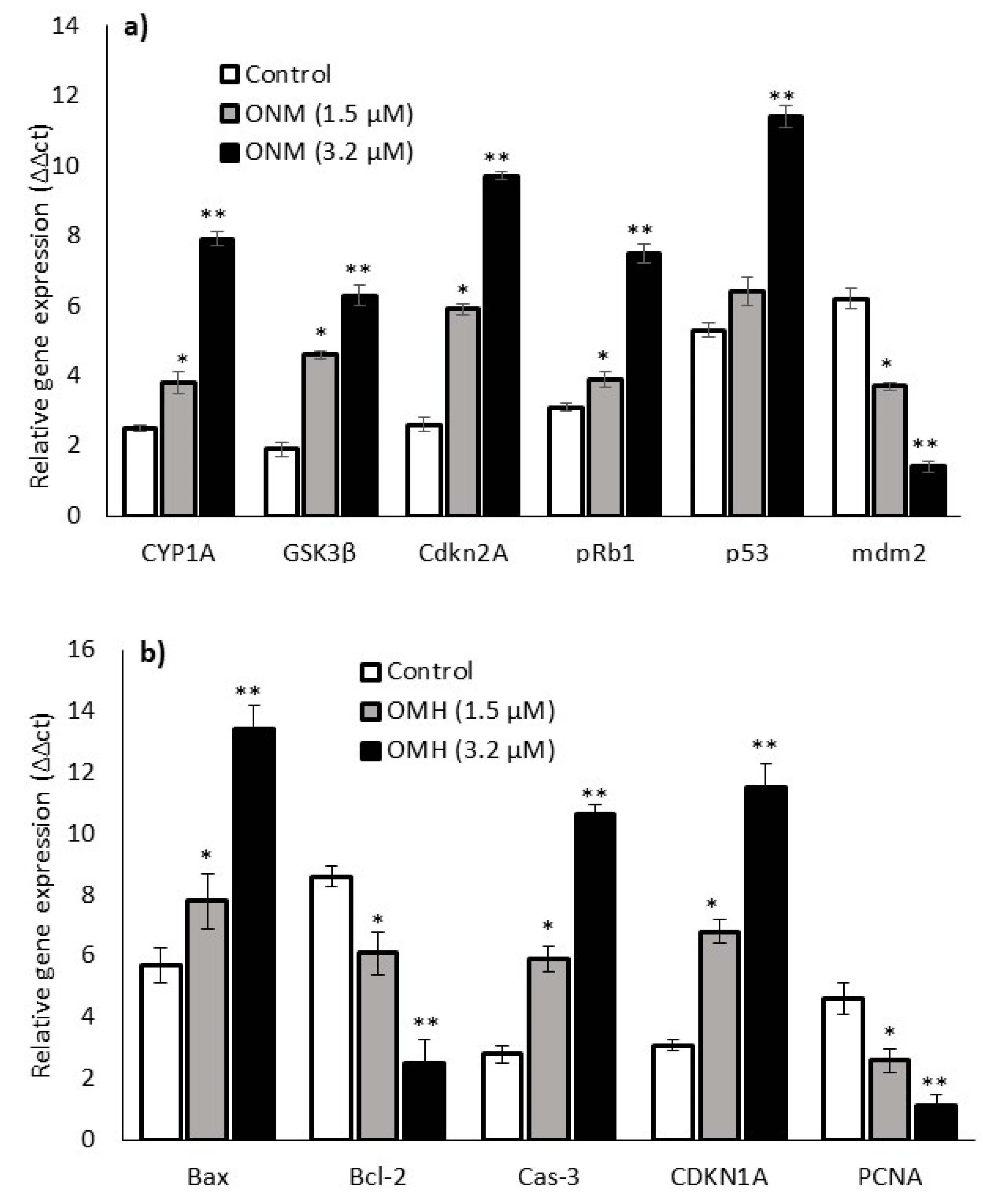
2.4. Effect of OMH on Oxidative Stress and Apoptotic Gene Expression
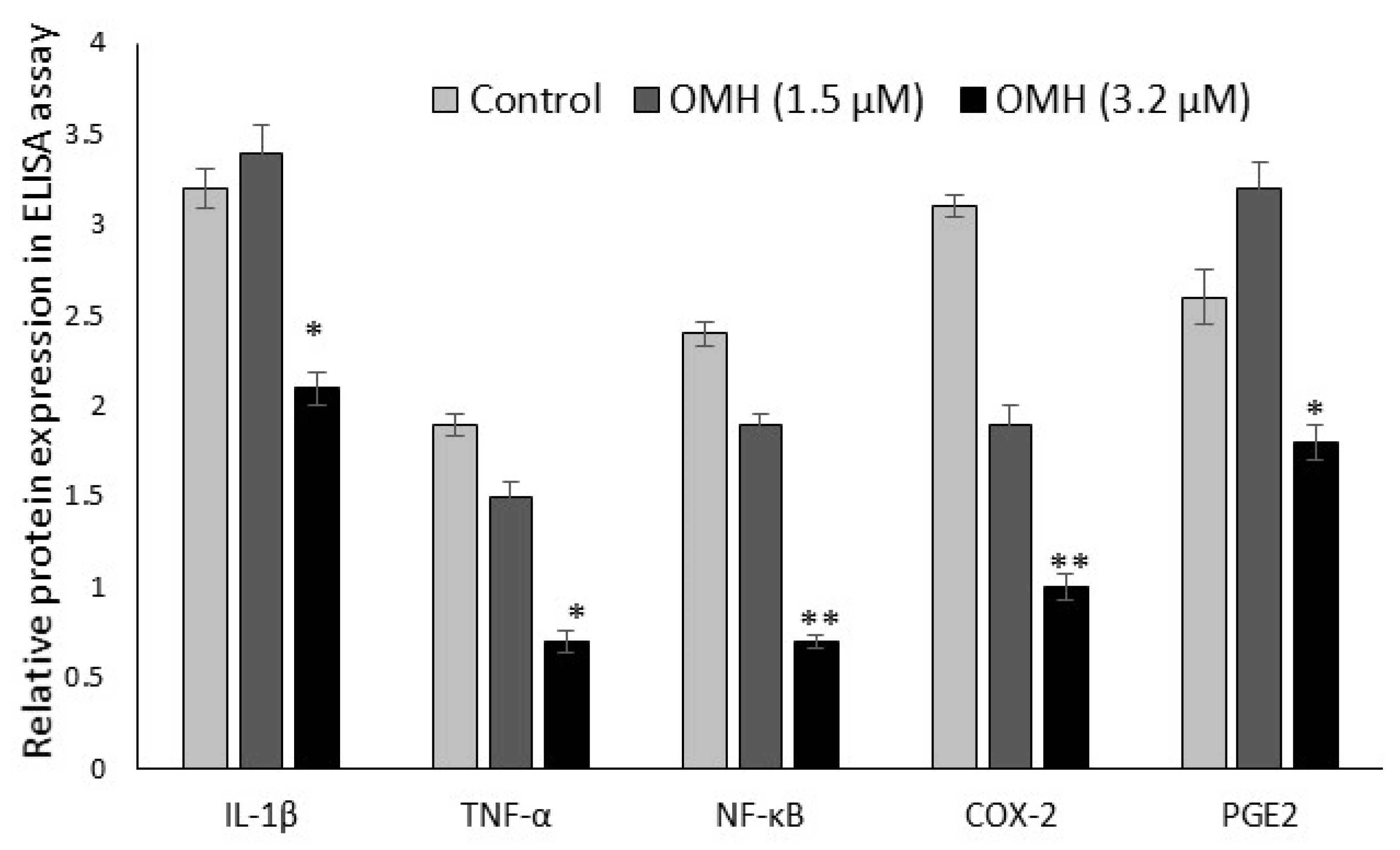
2.5. Effect of OMH on Protein Expression Levels
3. Discussion
4. Materials and Method
4.1. Cell Lines and Molecular Biology Chemicals
4.2. Cell Culture
4.3. Extraction of Cassia Tora (L.) and Isolation of Ononitol Monohydrate
4.4. Cytotoxicity Assay
4.5. Cell and Nuclear Morphology
4.6. Quantitative Polymerase Chain Reaction (qPCR) Analysis
4.7. Inflammatory Cytokine Assay Using ELISA Method
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, D.; Dubois, R.N. The role of COX- in intestinal inflammation and colorectal cancer. Oncogene 2010, 29, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation and cancer: The good, the bad and the ugly. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Rowland, I.R. The role of the gastrointestinal microbiota in colorectal cancer. Curr. Pharm. Des. 2009, 15, 1524–1527. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Roelofs, H.M.; Te Morsche, R.H.; van Heumen, B.W.; Nagengast, F.M.; Peters, W.H.M. Over-expression of COX-2 mRNA in colorectal cancer. BMC Gastroenterol. 2014, 14, 9005. [Google Scholar] [CrossRef]
- Chandrasekharan, N.V.; Simmons, D.L. The cyclooxygenases. Genome Biol. 2004, 5, 241. [Google Scholar] [CrossRef]
- Chandrasekharan, N.V.; Dai, H.; Roos, K.L.; Evanson, N.K.; Tomsik, J.; Elton, T.S.; Simmons, D.L. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: Cloning, structure and expression. Proc. Natl. Acad. Sci. USA 2002, 99, 13926–13931. [Google Scholar] [CrossRef]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef]
- Tessner, T.G.; Muhale, F.; Riehl, T.E.; Anant, S.; Stenson, W.F. Prostaglandin E2 reduces radiation-induced epithelial apoptosis through a mechanism involving AKT activation and bax translocation. J. Clin. Investig. 2004, 114, 1676–1685. [Google Scholar] [CrossRef]
- Castellone, M.D.; Teramoto, H.; Williams, B.O.; Druey, K.M.; Gutkind, J.S. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science 2005, 310, 1504–1510. [Google Scholar] [CrossRef]
- Brown, J.R.; Dubois, R.N. COX-2: A molecular target for colorectal cancer prevention. J. Clin. Oncol. 2005, 23, 2840–2855. [Google Scholar] [CrossRef] [PubMed]
- Arber, N.; Eagle, C.J.; Spicak, J.; Rácz, I.; Dite, P.; Hajer, J.; Zavoral, M.; Lechuga, M.J.; Gerletti, P.; Tang, J.; et al. Celecoxib for the prevention of colorectal adenomatous polyps. N. Engl. J. Med. 2006, 355, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, K.M.; Sheahan, K.; O’Donoghue, D.P.; MacSweeney, F.; Conroy, R.M.; Fitzgerald, D.J.; Murray, F.E. The relationship between cyclooxygenase-2 expression and colorectal cancer. J. Am. Med. Assoc. 1999, 282, 1254–1257. [Google Scholar] [CrossRef]
- Kawamori, T.; Uchiya, N.; Sugimura, T.; Wakabayashi, K. Enhancement of colon carcinogenesis by prostaglandin E2 administration. Carcinogenesis 2003, 24, 985–990. [Google Scholar] [CrossRef]
- Pugh, S.; Thomas, G.A. Patients with adenomatous polyps and carcinomas have increased colonic mucosal prostaglandin E2. Gut 1994, 35, 675–678. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, M.; Ignacimuthu, S.; Agastian, P. Potential hepatoprotective activity of ononitol monohydrate isolated from Cassia tora L. on carbon tetrachloride induced hepatotoxicity in wistar rats. Phytomedicine 2009, 16, 891–895. [Google Scholar] [CrossRef]
- Cesquini, M.; Torsoni, M.A.; Stoppa, S.R.; Ogo, S.H. t-BOOH induced oxidative damage in sickle red blood cells and the role of flavonoids. Biomed. Pharmacother. 2003, 57, 124–129. [Google Scholar] [CrossRef]
- Chen, J.; Xia, Y.; Sui, X.; Peng, Q.; Zhang, T.; Li, J.; Zhang, J. Steviol, a natural product inhibits proliferation of the gastrointestinal cancer cells intensively. Oncotarget 2018, 9, 26299–26308. [Google Scholar] [CrossRef]
- Calderón-Montaño, J.M.; Burgos-Morón, E.; Orta, M.L.; Maldonado-Navas, D.; García-Domínguez, I.; López-Lázaro, M. Evaluating the Cancer Therapeutic Potential of Cardiac Glycosides. BioMed Res. Int. 2014, 9, 794930. [Google Scholar] [CrossRef]
- Subash-Babu, P.; Alshatwi, A.A. Ononitol monohydrate enhances PRDM16 & UCP-1 expression, mitochondrial biogenesis and insulin sensitivity via STAT6 and LTB4R in maturing adipocytes. Biomed. Pharmacother. 2018, 99, 375–383. [Google Scholar] [PubMed]
- Karikas, G.A. Anticancer and chemopreventing natural products: Some biochemical and therapeutic aspects. J. Balk. Union Oncol. 2010, 15, 627–638. [Google Scholar]
- Owczarek, K.; Hrabec, E.; Fichna, J.; Sosnowska, D.; Koziołkiewicz, M.; Szymański, J.; Lewandowska, U. Flavanols from Japanese quince (Chaenomeles japonica) fruit suppress expression of cyclooxygenase-2, metalloproteinase-9, and nuclear factor-kappaB in human colon cancer cells. Acta Biochim. Pol. 2017, 64, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Shen, S.C.; Chow, J.M.; Ko, C.H.; Tseng, S.W. Flavone inhibition of tumor growth via apoptosis in vitro and in vivo. Int. J. Oncol. 2004, 25, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Kaeid, S.L.; Shukla, G. Probiotics (Lactobacillus acidophilus and Lactobacillus rhamnosus GG) in conjunction with celecoxib (selective cox-2 inhibitor) modulated DMH-induced early experimental colon carcinogenesis. Nutr. Cancer 2018, 70, 1–10. [Google Scholar]
- Eberhart, C.E.; Coffey, R.J.; Radhika, A.; Giardiello, F.M.; Ferrenbach, S.; DuBois, R.N. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 1994, 107, 1183–1188. [Google Scholar] [CrossRef]
- Qi-Bing, W.; Guo-Ping, S. Expression of COX-2 and HER-2 in colorectal cancer and their correlation. World J. Gastroenterol. 2015, 21, 6206–6214. [Google Scholar]
- Elder, D.J.; Baker, J.A.; Banu, N.A.; Moorghen, M.; Paraskeva, C. Human colorectal adenomas demonstrate a size dependent increase in epithelial cyclooxygenase-2 expression. J. Pathol. 2002, 198, 428–434. [Google Scholar] [CrossRef]
- Jacoby, R.F.; Seibert, K.; Cole, C.E.; Kelloff, G.; Lubet, R.A. The cyclooxygenase-2 inhibitor celecoxib is a potent preventive and therapeutic agent in the min mouse model of adenomatous polyposis. Cancer Res. 2000, 60, 5040–5044. [Google Scholar]
- Colicos, M.; Dash, P. Apoptotic morphology of dentate gyrus granule cells following experimental cortical impact injury in rats: Possible role in spatial memory deficits. Brain Res. 1996, 739, 120–131. [Google Scholar] [CrossRef]
- Li, X.; Kong, L.; Liao, S.; Lu, J.; Ma, L.; Long, X. The expression and significance of feces cyclooxygensae-2 mRNA in colorectal cancer and colorectal adenomas. Saudi J. Gastroenterol. 2017, 23, 28–33. [Google Scholar] [PubMed]
- Gungor, H.; Ilhan, N.; Eroksuz, H. The effectiveness of cyclooxygenase-2 inhibitors and evaluation of angiogenesis in the model of experimental colorectal cancer. Biomed. Pharmacother. 2018, 102, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, J. Initiator caspases in apoptosis signaling pathways. Apoptosis 2002, 7, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Michael, O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar]
- Chen, Y.; Wang, Z.Y.; Wu, H. P14ARF deficiency and its correlation with over expression of p53/MDM2 in sporadic vestibular schwannomas. Eur. Arch. Oto-Rhino-Laryngol. 2015, 272, 2227–2234. [Google Scholar] [CrossRef]
- Pomerantz, J.; Schreiber–Agus, N.; Liégeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.W.; et al. The Ink4a tumor suppressor gene product, p19, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef]
- Jayaraman, L.; Prives, C. Covalent and noncovalent modifiers of the p53 protein. Cell Mol. Life Sci. 1999, 55, 76–87. [Google Scholar] [CrossRef]
- Yun, N.; Kim, C.; Cha, H.; Park, W.J.; Shibayama, H.; Park, I.S.; Oh, Y.J. Caspase-3-mediated cleavage of picot in apoptosis. Biochem. Biophys. Res. Commun. 2013, 432, 533–538. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Leite, M.; Quinta-Costa, M.; Leite, P.S.; Guimaraes, J.E. Critical evaluation of techniques to detect and measure cell death–study in a model of UV radiation of the leukaemic cell line HL60. Anal. Cell. Pathol. 1999, 19, 139–151. [Google Scholar] [CrossRef]
- Yuan, J.S.; Reed, A.; Chen, F.; Stewart, C.N. Statistical analysis of real–time PCR data. BMC Bioinform. 2006, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y. Analysis of variance (ANOVA) comparing means of more than two groups. Restor. Dent. Endod. 2014, 39, 74–77. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subash-Babu, P.; Aladel, A.; Almanaa, T.N.; AlSedairy, S.A.; Alshatwi, A.A. Ononitol Monohydrate—A Glycoside Potentially Inhibit HT-115 Human Colorectal Cancer Cell Proliferation through COX-2/PGE-2 Inflammatory Axis Regulations. Int. J. Mol. Sci. 2022, 23, 14440. https://doi.org/10.3390/ijms232214440
Subash-Babu P, Aladel A, Almanaa TN, AlSedairy SA, Alshatwi AA. Ononitol Monohydrate—A Glycoside Potentially Inhibit HT-115 Human Colorectal Cancer Cell Proliferation through COX-2/PGE-2 Inflammatory Axis Regulations. International Journal of Molecular Sciences. 2022; 23(22):14440. https://doi.org/10.3390/ijms232214440
Chicago/Turabian StyleSubash-Babu, Pandurangan, Alanoud Aladel, Taghreed N. Almanaa, Sahar Abdulaziz AlSedairy, and Ali A. Alshatwi. 2022. "Ononitol Monohydrate—A Glycoside Potentially Inhibit HT-115 Human Colorectal Cancer Cell Proliferation through COX-2/PGE-2 Inflammatory Axis Regulations" International Journal of Molecular Sciences 23, no. 22: 14440. https://doi.org/10.3390/ijms232214440
APA StyleSubash-Babu, P., Aladel, A., Almanaa, T. N., AlSedairy, S. A., & Alshatwi, A. A. (2022). Ononitol Monohydrate—A Glycoside Potentially Inhibit HT-115 Human Colorectal Cancer Cell Proliferation through COX-2/PGE-2 Inflammatory Axis Regulations. International Journal of Molecular Sciences, 23(22), 14440. https://doi.org/10.3390/ijms232214440