

Testing Serum Albumins and Cyclodextrins as Potential Binders of the Mycotoxin Metabolites Alternariol-3-Sulfate, Alternariol-9-Monomethylether and Alternariol-9-Monomethylether-3-Sulfate

 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
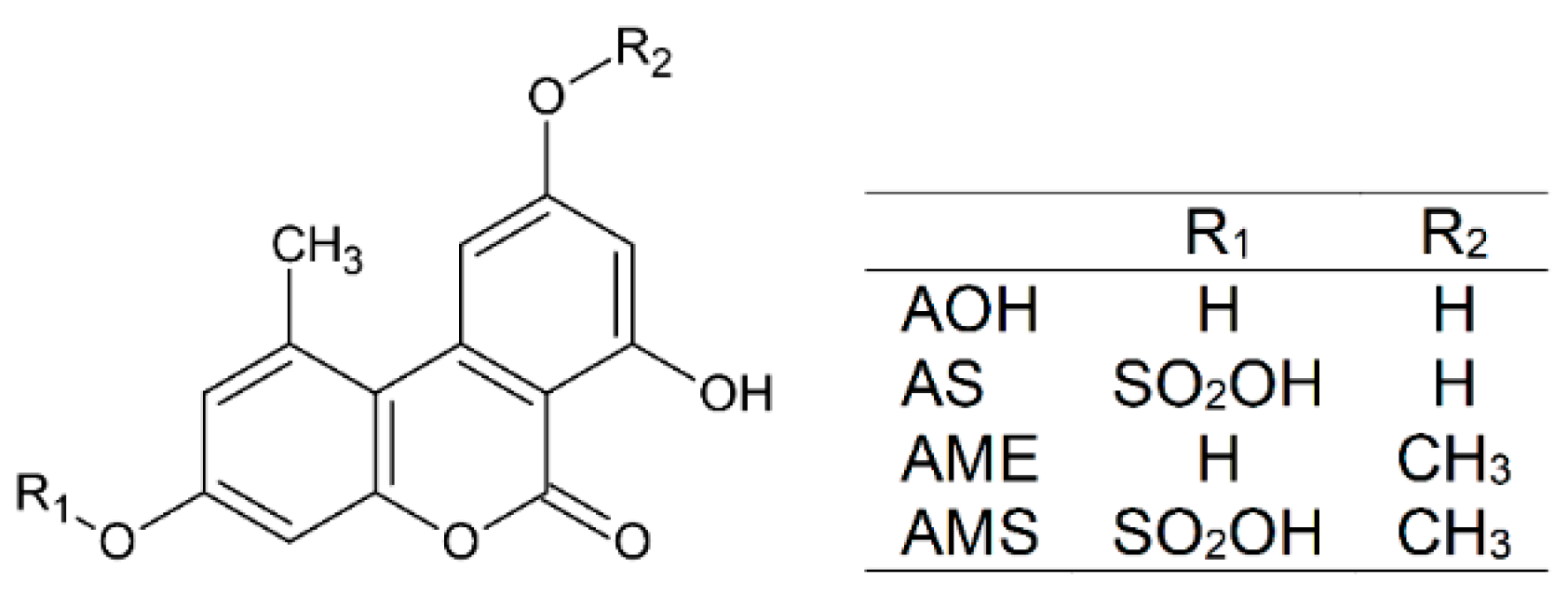
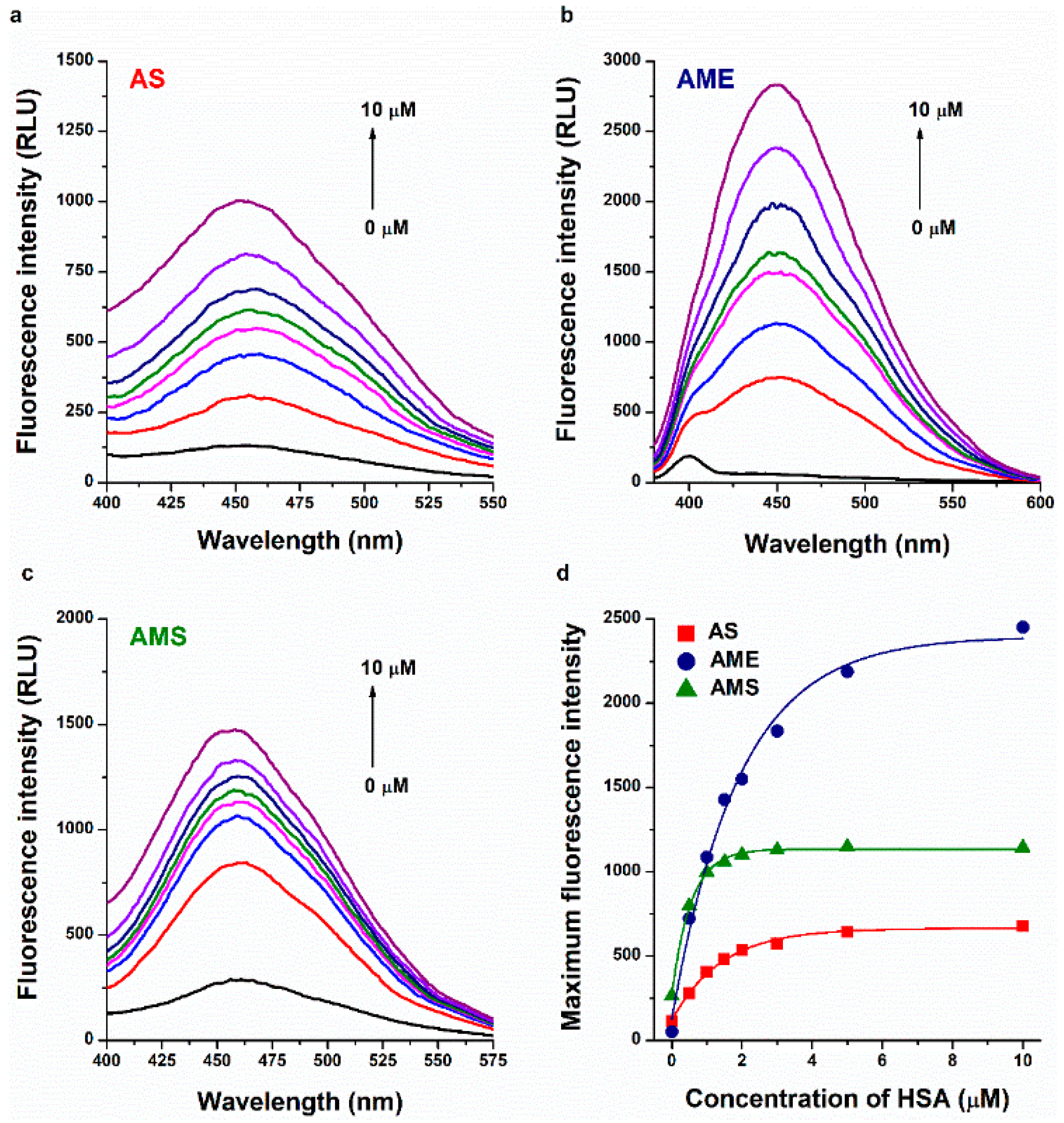
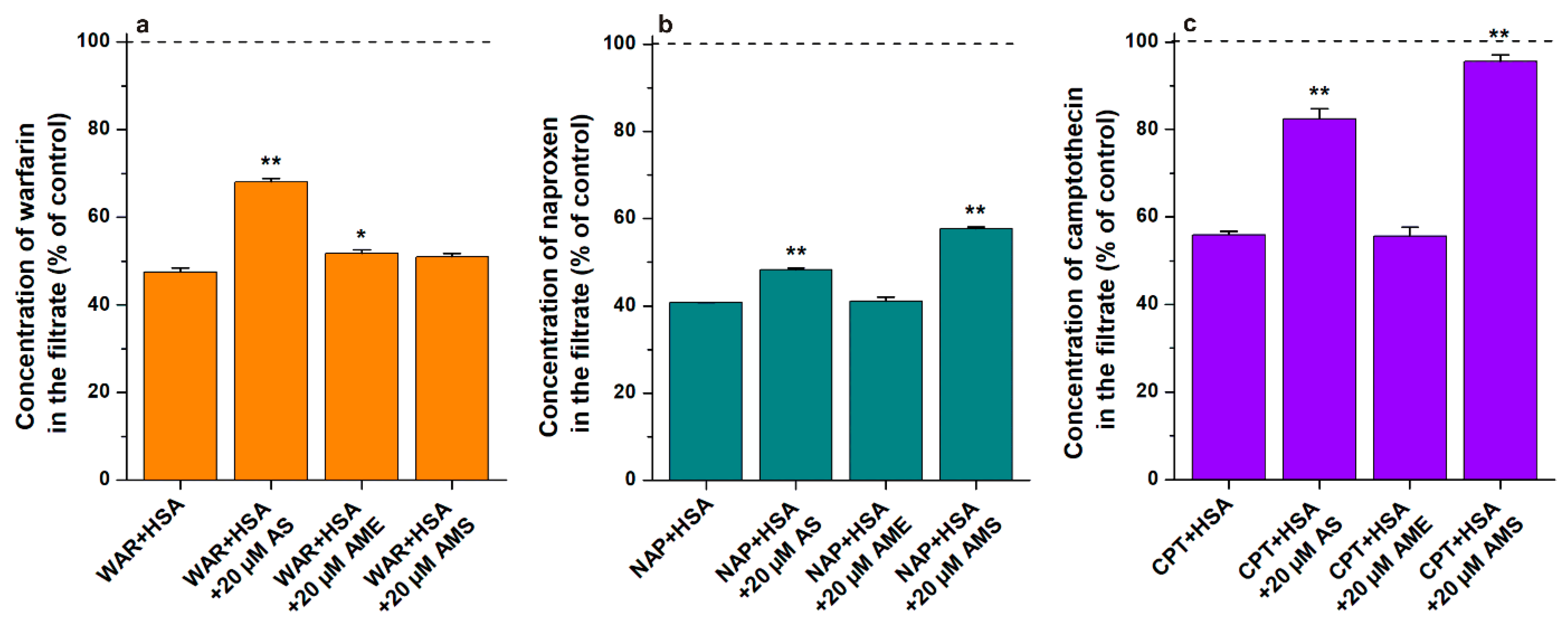
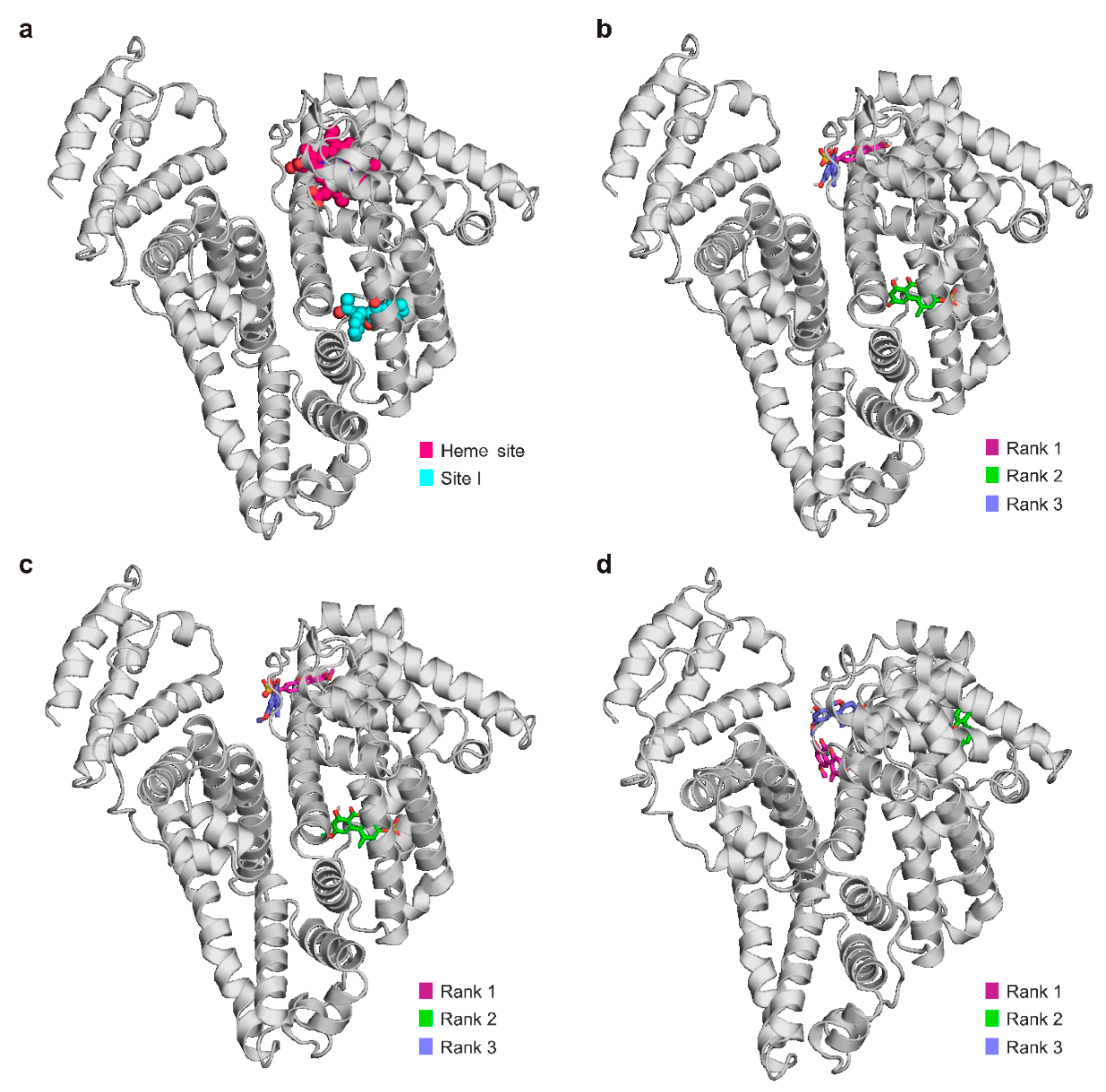
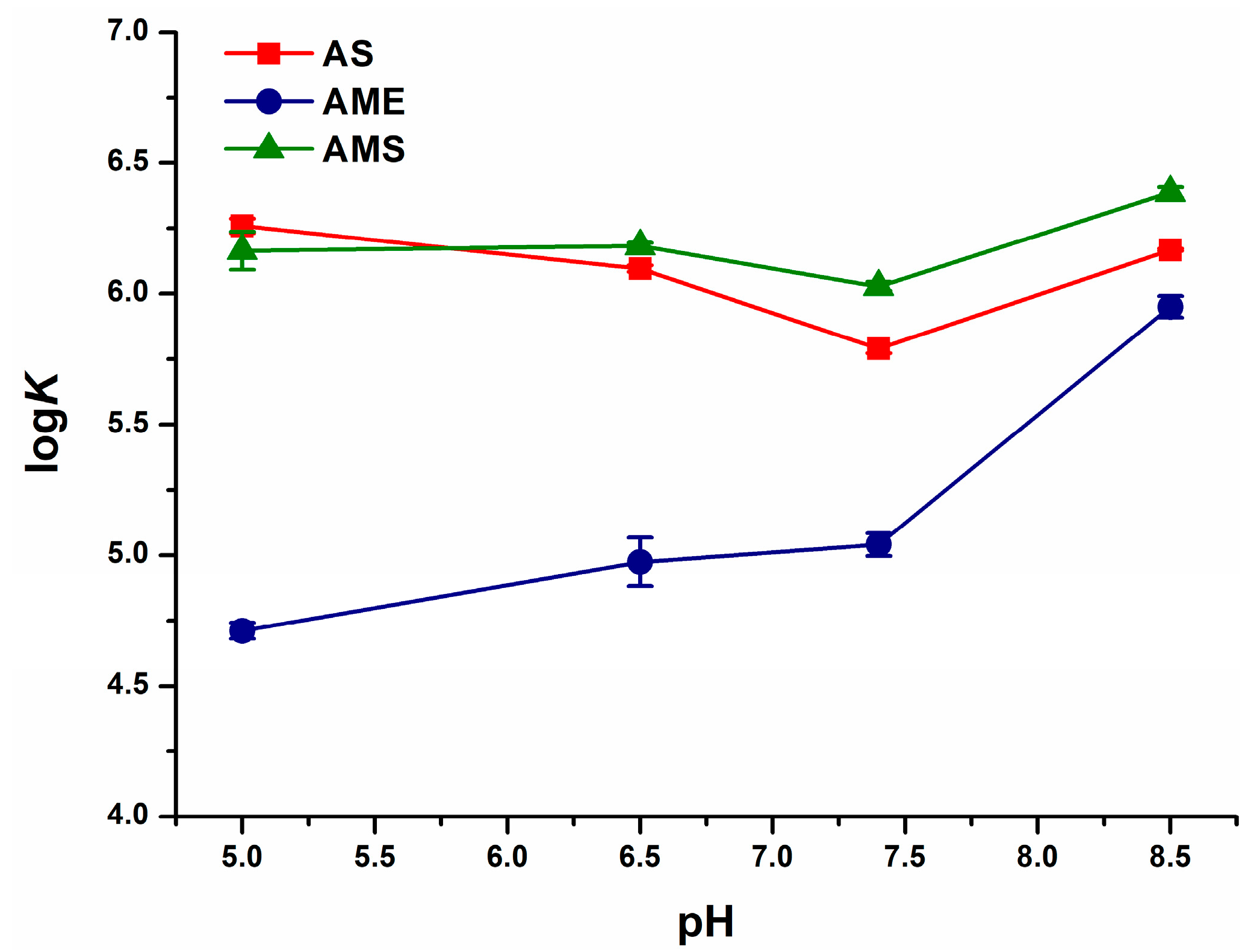
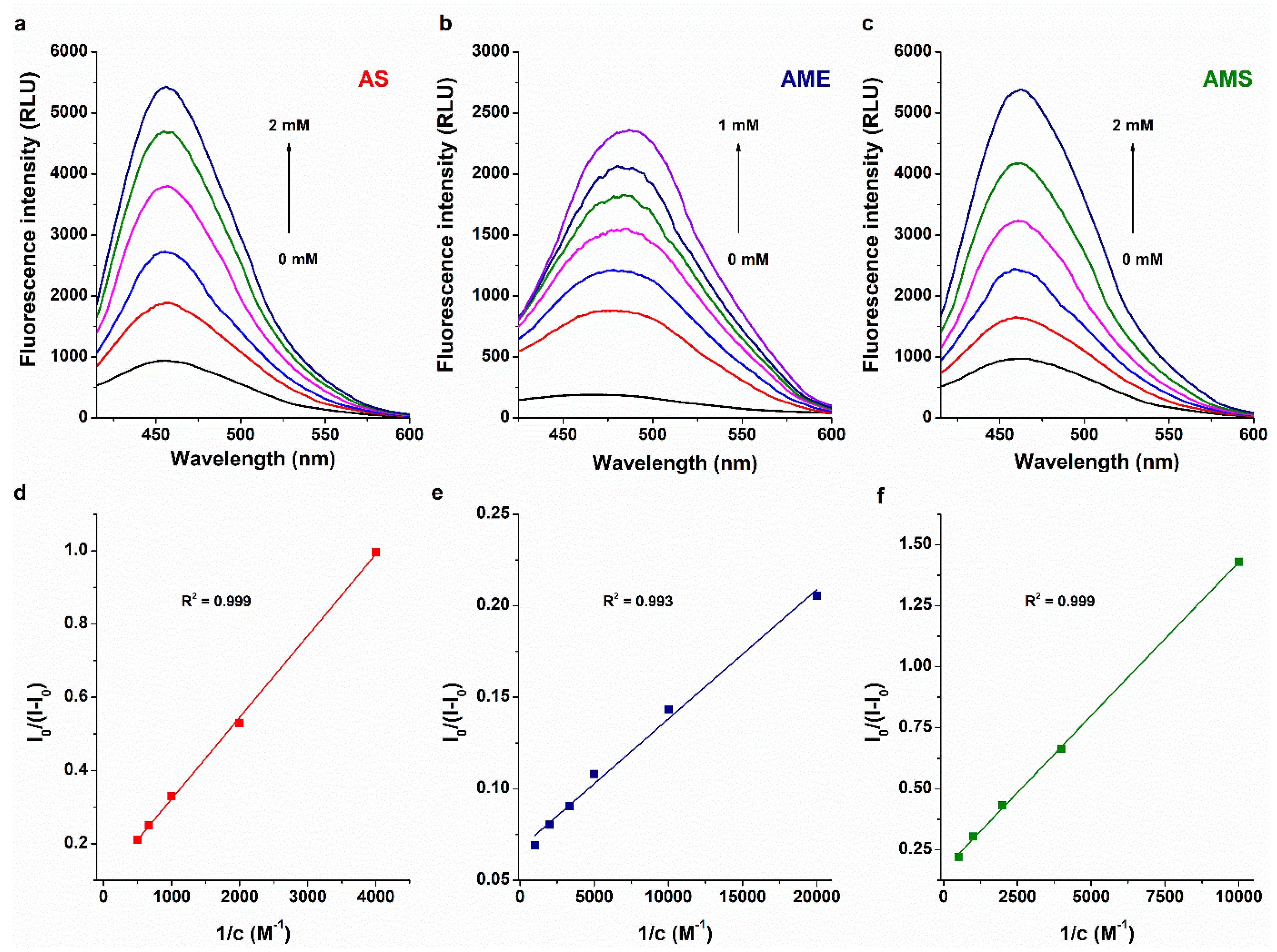
2.1. Interaction of AS, AME, and AMS with Serum Albumins
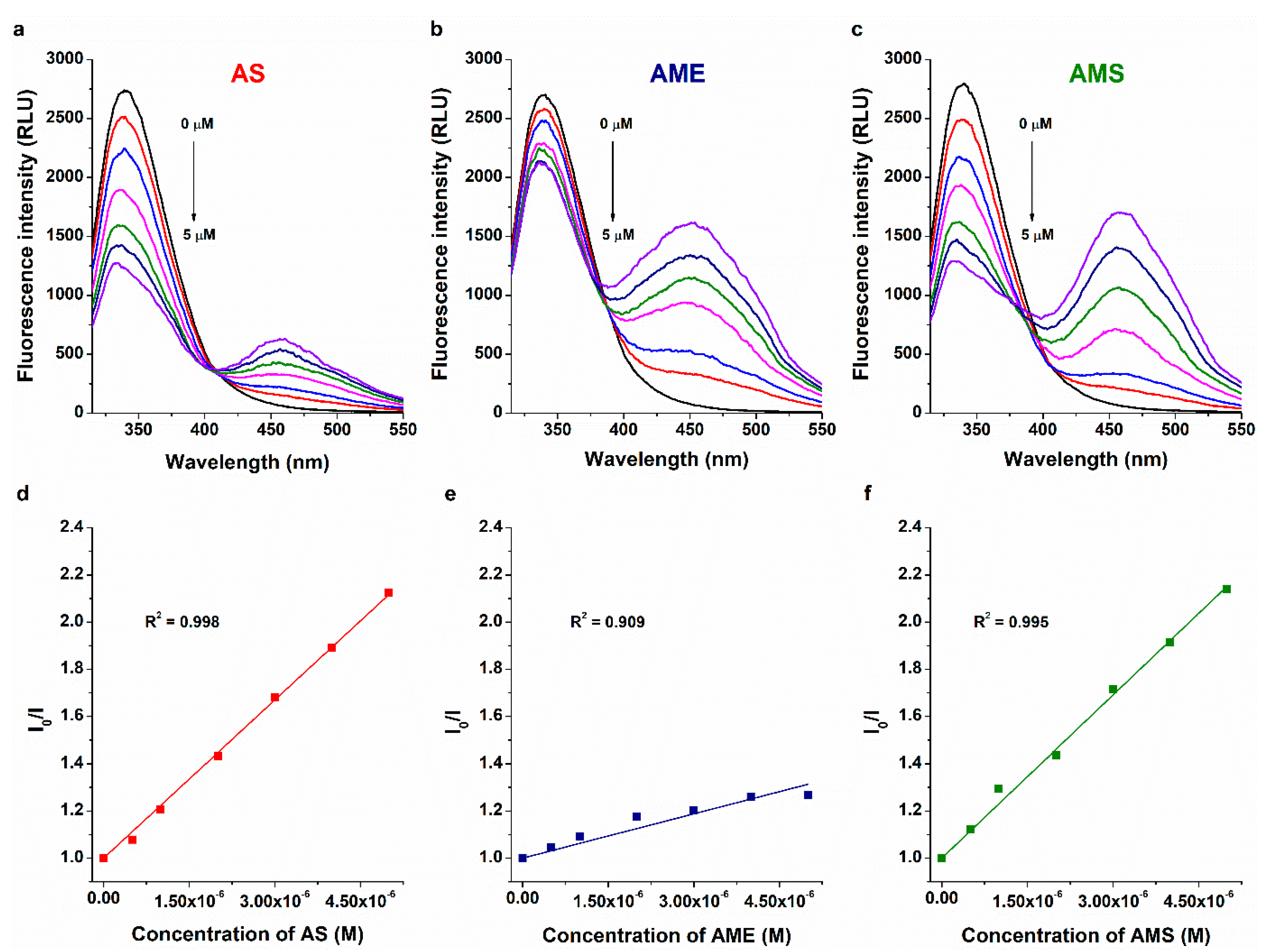
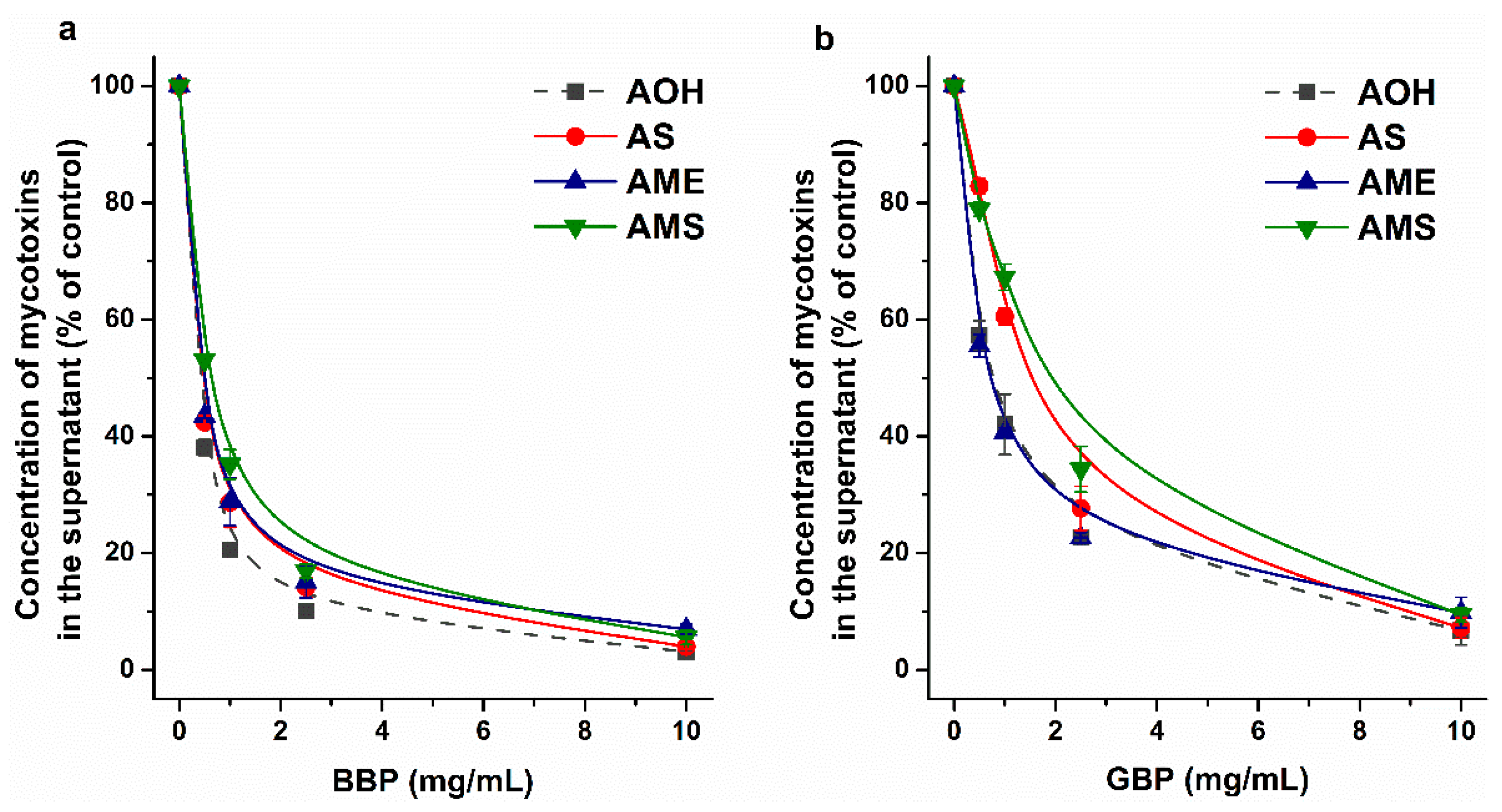
2.2. Interaction of AS, AME, and AMS with Cyclodextrins
3. Materials and Methods
3.1. Reagents
3.2. Spectroscopic Studies
3.3. Ultracentrifugation Studies
3.4. Ultrafiltration Studies
3.5. Modeling Studies
3.6. Extraction of AS, AME, and AMS by CD Bead Polymers
3.7. HPLC Analyses
3.8. Statistical Analyses
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- European Food Safety Authority; Arcella, D.; Eskola, M.; Gómez Ruiz, J.A. Dietary Exposure Assessment to Alternaria Toxins in the European Population. EFSA J. 2016, 14, 4654. [Google Scholar] [CrossRef]
- EFSA Panel on Contaminants in the Food Chain (CONTAM). Scientific Opinion on the Risks for Animal and Public Health Related to the Presence of Alternaria Toxins in Feed and Food. EFSA J. 2011, 9, 2407. [Google Scholar] [CrossRef]
- Solhaug, A.; Eriksen, G.S.; Holme, J.A. Mechanisms of Action and Toxicity of the Mycotoxin Alternariol: A Review. Basic Clin. Pharmacol. Toxicol. 2016, 119, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, F.; Crews, C.; Dall’Asta, C.; Saeger, S.D.; Haesaert, G.; Karlovsky, P.; Oswald, I.P.; Seefelder, W.; Speijers, G.; Stroka, J. Masked Mycotoxins: A Review. Mol. Nutr. Food Res. 2013, 57, 165–186. [Google Scholar] [CrossRef]
- Walravens, J.; Mikula, H.; Rychlik, M.; Asam, S.; Devos, T.; Njumbe Ediage, E.; Diana Di Mavungu, J.; Jacxsens, L.; Van Landschoot, A.; Vanhaecke, L.; et al. Validated UPLC-MS/MS Methods To Quantitate Free and Conjugated Alternaria Toxins in Commercially Available Tomato Products and Fruit and Vegetable Juices in Belgium. J. Agric. Food Chem. 2016, 64, 5101–5109. [Google Scholar] [CrossRef]
- Puntscher, H.; Kütt, M.-L.; Skrinjar, P.; Mikula, H.; Podlech, J.; Fröhlich, J.; Marko, D.; Warth, B. Tracking Emerging Mycotoxins in Food: Development of an LC-MS/MS Method for Free and Modified Alternaria Toxins. Anal. Bioanal. Chem. 2018, 410, 4481–4494. [Google Scholar] [CrossRef]
- Puntscher, H.; Cobankovic, I.; Marko, D.; Warth, B. Quantitation of Free and Modified Alternaria Mycotoxins in European Food Products by LC-MS/MS. Food Control 2019, 102, 157–165. [Google Scholar] [CrossRef]
- Burkhardt, B.; Pfeiffer, E.; Metzler, M. Absorption and Metabolism of the Mycotoxins Alternariol and Alternariol-9-Methyl Ether in Caco-2 Cells in Vitro. Mycotoxin Res. 2009, 25, 149–157. [Google Scholar] [CrossRef]
- Puntscher, H.; Hankele, S.; Tillmann, K.; Attakpah, E.; Braun, D.; Kütt, M.-L.; Del Favero, G.; Aichinger, G.; Pahlke, G.; Höger, H.; et al. First Insights into Alternaria Multi-Toxin in Vivo Metabolism. Toxicol. Lett. 2019, 301, 168–178. [Google Scholar] [CrossRef]
- Peters, T. All about Albumin: Biochemistry, Genetics, and Medical Applications; Academic Press: San Diego, CA, USA, 1996; ISBN 978-0-12-552110-9. [Google Scholar]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human Serum Albumin: From Bench to Bedside. Mol. Aspects Med. 2012, 33, 209–290. [Google Scholar] [CrossRef]
- Sugio, S.; Kashima, A.; Mochizuki, S.; Noda, M.; Kobayashi, K. Crystal Structure of Human Serum Albumin at 2.5 Å Resolution. Protein Eng. Des. Sel. 1999, 12, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. The Characterization of Two Specific Drug Binding Sites on Human Serum Albumin. Mol. Pharmacol. 1975, 11, 824–832. [Google Scholar] [PubMed]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. Further Characterization of Specific Drug Binding Sites on Human Serum Albumin. Mol. Pharmacol. 1976, 12, 1052–1061. [Google Scholar] [PubMed]
- Zsila, F. Subdomain IB Is the Third Major Drug Binding Region of Human Serum Albumin: Toward the Three-Sites Model. Mol. Pharm. 2013, 10, 1668–1682. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.-Y.; Chen, Y.-C. Selective Enrichment of Ochratoxin A Using Human Serum Albumin Bound Magnetic Beads as the Concentrating Probes for Capillary Electrophoresis/Electrospray Ionization-Mass Spectrometric Analysis. J. Chromatogr. A 2007, 1159, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Galaverna, G.; Dall’Asta, C.; Corradini, R.; Dossena, A.; Marchelli, R. Cyclodextrins as Selectors for Mycotoxin Recognition. World Mycotoxin J. 2008, 1, 397–406. [Google Scholar] [CrossRef]
- Ueda, H.; Ou, D.; Endo, T.; Nagase, H.; Tomono, K.; Nagai, T. Evaluation of a Sulfobutyl Ether β-Cyclodextrin as a Solubilizing/Stabilizing Agent for Several Drugs. Drug Dev. Ind. Pharm. 1998, 24, 863–867. [Google Scholar] [CrossRef]
- Keating, G.M. Sugammadex: A Review of Neuromuscular Blockade Reversal. Drugs 2016, 76, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Stella, V.J.; He, Q. Cyclodextrins. Toxicol. Pathol. 2008, 36, 30–42. [Google Scholar] [CrossRef]
- Jansook, P.; Ogawa, N.; Loftsson, T. Cyclodextrins: Structure, Physicochemical Properties and Pharmaceutical Applications. Int. J. Pharm. 2018, 535, 272–284. [Google Scholar] [CrossRef]
- Morin-Crini, N.; Crini, G. Environmental Applications of Water-Insoluble β-Cyclodextrin–Epichlorohydrin Polymers. Prog. Polym. Sci. 2013, 38, 344–368. [Google Scholar] [CrossRef]
- Morin-Crini, N.; Winterton, P.; Fourmentin, S.; Wilson, L.D.; Fenyvesi, É.; Crini, G. Water-Insoluble β-Cyclodextrin–Epichlorohydrin Polymers for Removal of Pollutants from Aqueous Solutions by Sorption Processes Using Batch Studies: A Review of Inclusion Mechanisms. Prog. Polym. Sci. 2018, 78, 1–23. [Google Scholar] [CrossRef]
- Moulahcene, L.; Skiba, M.; Senhadji, O.; Milon, N.; Benamor, M.; Lahiani-Skiba, M. Inclusion and Removal of Pharmaceutical Residues from Aqueous Solution Using Water-Insoluble Cyclodextrin Polymers. Chem. Eng. Res. Des. 2015, 97, 145–158. [Google Scholar] [CrossRef]
- Fliszár-Nyúl, E.; Lemli, B.; Kunsági-Máté, S.; Szente, L.; Poór, M. Interactions of Mycotoxin Alternariol with Cyclodextrins and Its Removal from Aqueous Solution by Beta-Cyclodextrin Bead Polymer. Biomolecules 2019, 9, 428. [Google Scholar] [CrossRef] [PubMed]
- Fliszár-Nyúl, E.; Szabó, Á.; Szente, L.; Poór, M. Extraction of Mycotoxin Alternariol from Red Wine and from Tomato Juice with Beta-Cyclodextrin Bead Polymer. J. Mol. Liq. 2020, 319, 114180. [Google Scholar] [CrossRef]
- Appell, M.; Jackson, M.A. Sorption of Ochratoxin A from Aqueous Solutions Using β-Cyclodextrin-Polyurethane Polymer. Toxins 2012, 4, 98–109. [Google Scholar] [CrossRef]
- Poór, M.; Faisal, Z.; Zand, A.; Bencsik, T.; Lemli, B.; Kunsági-Máté, S.; Szente, L. Removal of Zearalenone and Zearalenols from Aqueous Solutions Using Insoluble Beta-Cyclodextrin Bead Polymer. Toxins 2018, 10, 216. [Google Scholar] [CrossRef]
- Murai, S.; Imajo, S.; Maki, Y.; Takahashi, K.; Hattori, K. Adsorption and Recovery of Nonionic Surfactants by β-Cyclodextrin Polymer. J. Colloid Interface Sci. 1996, 183, 118–123. [Google Scholar] [CrossRef]
- Fliszár-Nyúl, E.; Lemli, B.; Kunsági-Máté, S.; Dellafiora, L.; Dall’Asta, C.; Cruciani, G.; Pethő, G.; Poór, M. Interaction of Mycotoxin Alternariol with Serum Albumin. Int. J. Mol. Sci. 2019, 20, 2352. [Google Scholar] [CrossRef]
- Fliszár-Nyúl, E.; Lemli, B.; Kunsági-Máté, S.; Poór, M. Effects of Microenvironmental Changes on the Fluorescence Signal of Alternariol: Magnesium Induces Strong Enhancement in the Fluorescence of the Mycotoxin. Int. J. Mol. Sci. 2021, 22, 8692. [Google Scholar] [CrossRef]
- Dobretsov, G.E.; Syrejschikova, T.I.; Smolina, N.V. On Mechanisms of Fluorescence Quenching by Water. Biophysics 2014, 59, 183–188. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of Equilibria in Solution. Determination of Equilibrium Constants with the HYPERQUAD Suite of Programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- van de Weert, M. Fluorescence Quenching to Study Protein-Ligand Binding: Common Errors. J. Fluoresc. 2010, 20, 625–629. [Google Scholar] [CrossRef] [PubMed]
- van de Weert, M.; Stella, L. Fluorescence Quenching and Ligand Binding: A Critical Discussion of a Popular Methodology. J. Mol. Struct. 2011, 998, 144–150. [Google Scholar] [CrossRef]
- Fliszár-Nyúl, E.; Faisal, Z.; Skaper, R.; Lemli, B.; Bayartsetseg, B.; Hetényi, C.; Gömbös, P.; Szabó, A.; Poór, M. Interaction of the Emerging Mycotoxins Beauvericin, Cyclopiazonic Acid, and Sterigmatocystin with Human Serum Albumin. Biomolecules 2022, 12, 1106. [Google Scholar] [CrossRef]
- Petitpas, I.; Bhattacharya, A.A.; Twine, S.; East, M.; Curry, S. Crystal Structure Analysis of Warfarin Binding to Human Serum Albumin: ANATOMY OF DRUG SITE I. J. Biol. Chem. 2001, 276, 22804–22809. [Google Scholar] [CrossRef]
- Wardell, M.; Wang, Z.; Ho, J.X.; Robert, J.; Ruker, F.; Ruble, J.; Carter, D.C. The Atomic Structure of Human Methemalbumin at 1.9 Å. Biochem. Biophys. Res. Commun. 2002, 291, 813–819. [Google Scholar] [CrossRef]
- Fliszár-Nyúl, E.; Bock, I.; Csepregi, R.; Szente, L.; Szabó, I.; Csenki, Z.; Poór, M. Testing the Protective Effects of Cyclodextrins vs. Alternariol-Induced Acute Toxicity in HeLa Cells and in Zebrafish Embryos. Environ. Toxicol. Pharmacol. 2022, 95, 103965. [Google Scholar] [CrossRef]
- Vahl, K.; Kahlert, H.; von Mühlen, L.; Albrecht, A.; Meyer, G.; Behnert, J. Determination of the Titratable Acidity and the PH of Wine Based on Potentiometric Flow Injection Analysis. Talanta 2013, 111, 134–139. [Google Scholar] [CrossRef]
- Fachin, D.; Van Loey, A.M.; Ly Nguyen, B.; Verlent, I.; Indrawati; Hendrickx, M.E. Inactivation Kinetics of Polygalacturonase in Tomato Juice. Innov. Food Sci. Emerg. Technol. 2003, 4, 135–142. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, NY, USA, 2006; ISBN 978-0-387-31278-1. [Google Scholar]
- Benesi, H.A.; Hildebrand, J.H. A Spectrophotometric Investigation of the Interaction of Iodine with Aromatic Hydrocarbons. J. Am. Chem. Soc. 1949, 71, 2703–2707. [Google Scholar] [CrossRef]
- Boulton, D.W.; Walle, U.K.; Walle, T. Extensive Binding of the Bioflavonoid Quercetin to Human Plasma Proteins. J. Pharm. Pharmacol. 1998, 50, 243–249. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef]
- Stewart, J.J.P. MOPAC2016, Stewart Computational Chemistry, Colorado Springs, CO, USA. 2016. Available online: http://OpenMOPAC.net (accessed on 31 October 2022).
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods VI: More Modifications to the NDDO Approximations and Re-Optimization of Parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, J.; Marsili, M. Iterative Partial Equalization of Orbital Electronegativity—A Rapid Access to Atomic Charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Mohos, V.; Fliszár-Nyúl, E.; Lemli, B.; Zsidó, B.Z.; Hetényi, C.; Mladěnka, P.; Horký, P.; Pour, M.; Poór, M. Testing the Pharmacokinetic Interactions of 24 Colonic Flavonoid Metabolites with Human Serum Albumin and Cytochrome P450 Enzymes. Biomolecules 2020, 10, 409. [Google Scholar] [CrossRef]
- Hetényi, C.; van der Spoel, D. Blind Docking of Drug-Sized Compounds to Proteins with up to a Thousand Residues. FEBS Lett. 2006, 580, 1447–1450. [Google Scholar] [CrossRef]
- Hetényi, C.; van der Spoel, D. Toward Prediction of Functional Protein Pockets Using Blind Docking and Pocket Search Algorithms. Protein Sci. 2011, 20, 880–893. [Google Scholar] [CrossRef]
- Zsidó, B.Z.; Börzsei, R.; Szél, V.; Hetényi, C. Determination of Ligand Binding Modes in Hydrated Viral Ion Channels to Foster Drug Design and Repositioning. J. Chem. Inf. Model. 2021, 61, 4011–4022. [Google Scholar] [CrossRef]
- Zsidó, B.Z.; Balog, M.; Erős, N.; Poór, M.; Mohos, V.; Fliszár-Nyúl, E.; Hetényi, C.; Nagane, M.; Hideg, K.; Kálai, T.; et al. Synthesis of Spin-Labelled Bergamottin: A Potent CYP3A4 Inhibitor with Antiproliferative Activity. Int. J. Mol. Sci. 2020, 21, 508. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HSA logKSV (±SEM) | BSA logKSV (±SEM) | PSA logKSV (±SEM) | RSA logKSV (±SEM) | |
|---|---|---|---|---|
| AS | 5.44 ± 0.07 | 5.60 ± 0.01 | 5.57 ± 0.02 | 5.85 ± 0.03 |
| AME | 4.86 ± 0.05 | 4.93 ± 0.05 | 4.73 ± 0.06 | 5.15 ± 0.06 |
| AMS | 5.42 ± 0.06 | 5.81 ± 0.01 | 5.60 ± 0.02 | 5.92 ± 0.03 |
| HSA logK (±SEM) | BSA logK (±SEM) | PSA logK (±SEM) | RSA logK (±SEM) | |
|---|---|---|---|---|
| AS | 5.61 ± 0.11 | 5.79 ± 0.02 | 5.78 ± 0.03 | 6.08 ± 0.03 |
| AME | 4.94 ± 0.07 | 5.04 ± 0.04 | 4.83 ± 0.05 | 5.29 ± 0.07 |
| AMS | 5.58 ± 0.08 | 6.03 ± 0.02 | 5.78 ± 0.03 | 6.13 ± 0.05 |
| SBECD | SGD | |||
|---|---|---|---|---|
| logK (±SEM) BH-Plot | logK (±SEM) Hyperquad | logK (±SEM) BH-Plot | logK (±SEM) Hyperquad | |
| AS | 3.02 ± 0.01 | 3.02 ± 0.03 | 2.83 ± 0.01 | 2.99 ± 0.03 |
| AME | 3.46 ± 0.01 | 3.56 ± 0.01 | 3.95 ± 0.01 | 4.06 ± 0.01 |
| AMS | 2.96 ± 0.01 | 3.08 ± 0.01 | 2.95 ± 0.05 | 3.21 ± 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lemli, B.; Vilmányi, P.; Fliszár-Nyúl, E.; Zsidó, B.Z.; Hetényi, C.; Szente, L.; Poór, M. Testing Serum Albumins and Cyclodextrins as Potential Binders of the Mycotoxin Metabolites Alternariol-3-Sulfate, Alternariol-9-Monomethylether and Alternariol-9-Monomethylether-3-Sulfate. Int. J. Mol. Sci. 2022, 23, 14353. https://doi.org/10.3390/ijms232214353
Lemli B, Vilmányi P, Fliszár-Nyúl E, Zsidó BZ, Hetényi C, Szente L, Poór M. Testing Serum Albumins and Cyclodextrins as Potential Binders of the Mycotoxin Metabolites Alternariol-3-Sulfate, Alternariol-9-Monomethylether and Alternariol-9-Monomethylether-3-Sulfate. International Journal of Molecular Sciences. 2022; 23(22):14353. https://doi.org/10.3390/ijms232214353
Chicago/Turabian StyleLemli, Beáta, Péter Vilmányi, Eszter Fliszár-Nyúl, Balázs Zoltán Zsidó, Csaba Hetényi, Lajos Szente, and Miklós Poór. 2022. "Testing Serum Albumins and Cyclodextrins as Potential Binders of the Mycotoxin Metabolites Alternariol-3-Sulfate, Alternariol-9-Monomethylether and Alternariol-9-Monomethylether-3-Sulfate" International Journal of Molecular Sciences 23, no. 22: 14353. https://doi.org/10.3390/ijms232214353
APA StyleLemli, B., Vilmányi, P., Fliszár-Nyúl, E., Zsidó, B. Z., Hetényi, C., Szente, L., & Poór, M. (2022). Testing Serum Albumins and Cyclodextrins as Potential Binders of the Mycotoxin Metabolites Alternariol-3-Sulfate, Alternariol-9-Monomethylether and Alternariol-9-Monomethylether-3-Sulfate. International Journal of Molecular Sciences, 23(22), 14353. https://doi.org/10.3390/ijms232214353