
Renal Fibrosis in Lupus Nephritis

 ,
,  ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Renal Fibrogenesis
2.1. Myofibroblasts and EMT
2.2. Macrophages
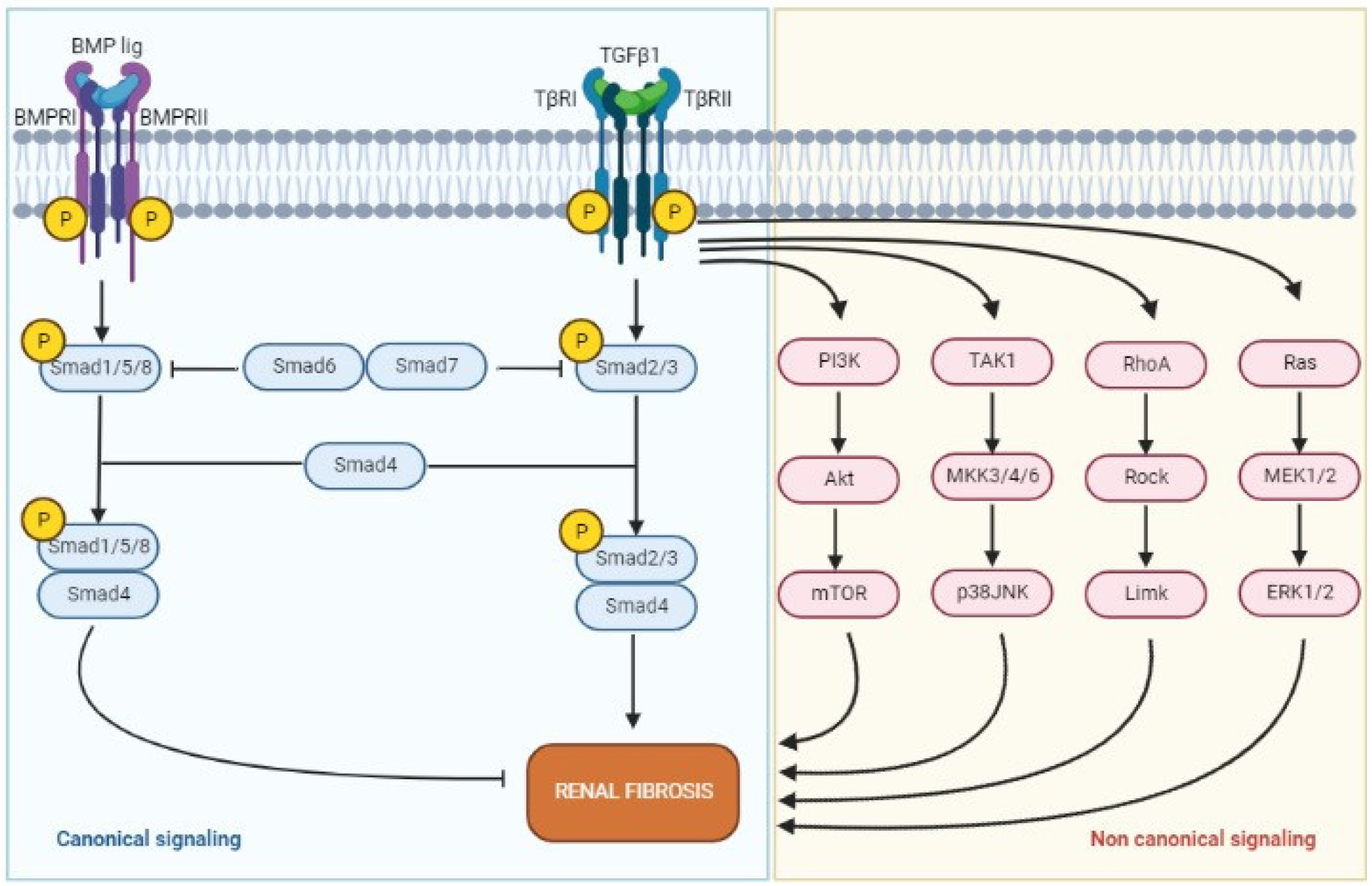
2.3. Transforming Growth Factor-β
2.4. Hypoxia
2.5. Towards CKD
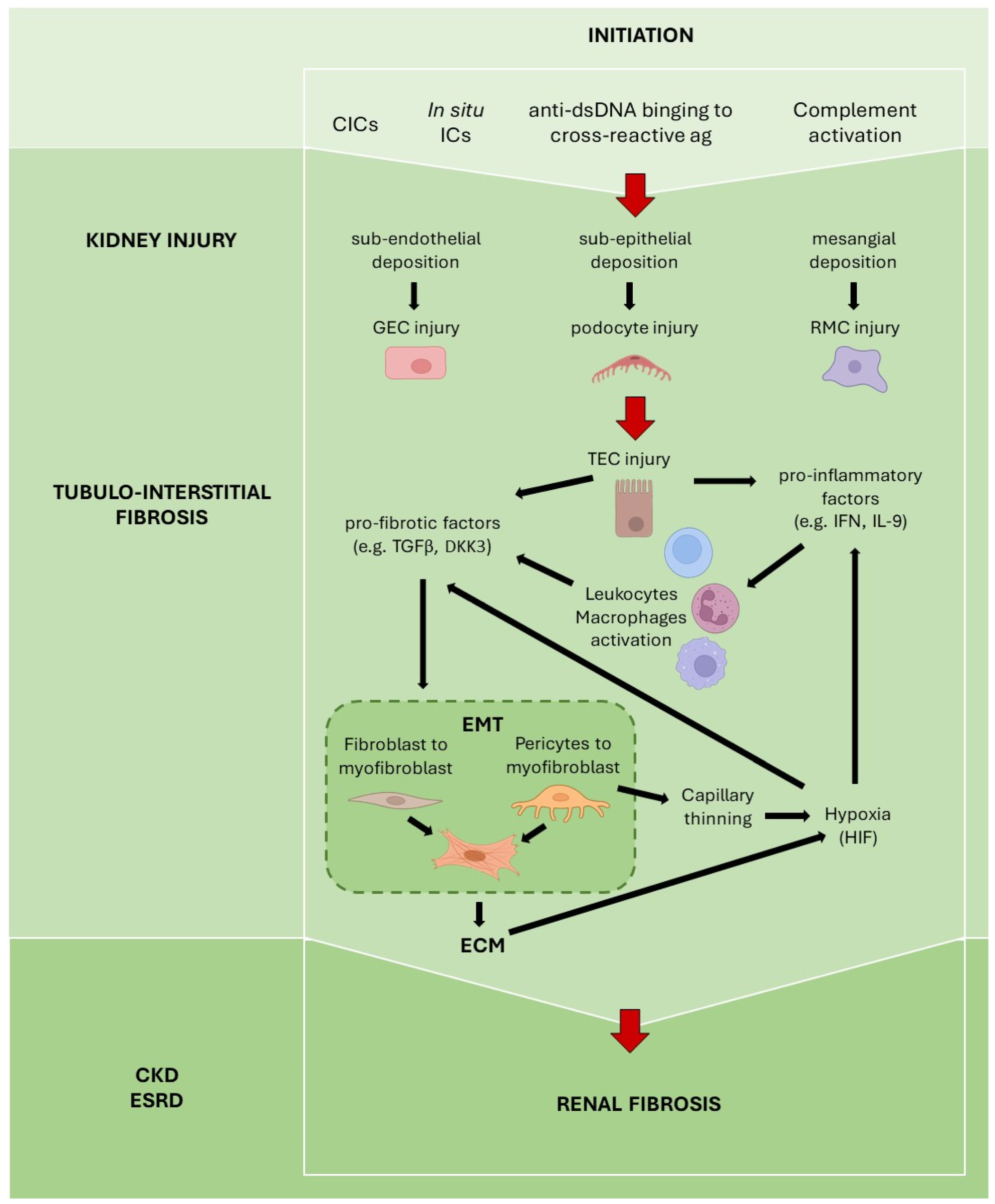
3. Fibrosis in Lupus Nephritis
3.1. Detrimental Role of Autoantibodies
3.2. Tubular Epithelial Cells
3.3. Type 1 Interferon
3.4. TGF-β
3.5. Complement System
3.6. Dickkopf-Related Protein 3
4. Histological Classification—IF/TA as a Prognostic Factor in LN
5. Future Targeted Therapies in Renal Fibrosis
5.1. Anti-Fibrotic Drugs
5.2. Chimeric Antigen Receptor T Cells
5.3. Other Agents Targeting Fibrosis in Kidney Diseases and LN
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wynn, T.A. Common and Unique Mechanisms Regulate Fibrosis in Various Fibroproliferative Diseases. J. Clin. Investig. 2007, 117, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.; Selman, M. Matrix Metalloproteases in Aberrant Fibrotic Tissue Remodeling. Proc. Am. Thorac. Soc. 2006, 3, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Distler, J.H.W.; Györfi, A.H.; Ramanujam, M.; Whitfield, M.L.; Königshoff, M.; Lafyatis, R. Shared and Distinct Mechanisms of Fibrosis. Nat. Rev. Rheumatol. 2019. [CrossRef] [PubMed]
- Wynn, T.A. Cellular and Molecular Mechanisms of Fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Renal Fibrosis: New Insights into the Pathogenesis and Therapeutics. Kidney Int. 2006, 69, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef]
- Romagnani, P.; Remuzzi, G.; Glassock, R.; Levin, A.; Jager, K.J.; Tonelli, M.; Massy, Z.; Wanner, C.; Anders, H.J. Chronic Kidney Disease. Nat. Rev. Dis. Prim. 2017, 3, 17088. [Google Scholar] [CrossRef]
- Boor, P.; Floege, J. Renal Allograft Fibrosis: Biology and Therapeutic Targets. Am. J. Transplant. 2015, 15, 863–886. [Google Scholar] [CrossRef]
- Maria, N.I.; Davidson, A. Protecting the Kidney in Systemic Lupus Erythematosus: From Diagnosis to Therapy. Nat. Rev. Rheumatol. 2020, 16, 255–267. [Google Scholar] [CrossRef]
- Anders, H.J.; Saxena, R.; Zhao, M.; Parodis, I.; Salmon, J.E.; Mohan, C. Lupus Nephritis. Nat. Rev. Dis. Prim. 2020, 6, 7. [Google Scholar] [CrossRef]
- Bajema, I.M.; Wilhelmus, S.; Alpers, C.E.; Bruijn, J.A.; Colvin, R.B.; Cook, H.T.; D’Agati, V.D.; Ferrario, F.; Haas, M.; Jennette, J.C.; et al. Revision of the International Society of Nephrology/Renal Pathology Society Classification for Lupus Nephritis: Clarification of Definitions, and Modified National Institutes of Health Activity and Chronicity Indices. Kidney Int. 2018, 93, 789–796. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Toews, G.B.; White, E.S.; Lynch, J.P., 3rd; Martinez, F.J. Mechanisms of Pulmonary Fibrosis. Annu. Rev. Med. 2004, 55, 395–417. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and Function of Myofibroblasts in Kidney Fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef]
- Falke, L.L.; Gholizadeh, S.; Goldschmeding, R.; Kok, R.J.; Nguyen, T.Q. Diverse Origins of the Myofibroblast—Implications for Kidney Fibrosis. Nat. Rev. Nephrol. 2015, 11, 233–244. [Google Scholar] [CrossRef]
- Henderson, N.C.; Arnold, T.D.; Katamura, Y.; Giacomini, M.M.; Rodriguez, J.D.; McCarty, J.H.; Pellicoro, A.; Raschperger, E.; Betsholtz, C.; Ruminski, P.G.; et al. Targeting of Av Integrin Identifies a Core Molecular Pathway That Regulates Fibrosis in Several Organs. Nat. Med. 2013, 19, 1617–1624. [Google Scholar] [CrossRef]
- Liu, Y. Epithelial to Mesenchymal Transition in Renal Fibrogenesis: Pathologic Significance, Molecular Mechanism, and Therapeutic Intervention. J. Am. Soc. Nephrol. 2004, 15, 1–12. [Google Scholar] [CrossRef]
- Schunk, S.J.; Floege, J.; Fliser, D.; Speer, T. WNT-β-Catenin Signalling—A Versatile Player in Kidney Injury and Repair. Nat. Rev. Nephrol. 2021, 17, 172–184. [Google Scholar] [CrossRef]
- Hu, L.; Ding, M.; He, W. Emerging Therapeutic Strategies for Attenuating Tubular EMT and Kidney Fibrosis by Targeting Wnt/β-Catenin Signaling. Front. Pharmacol. 2022, 12, 830340. [Google Scholar] [CrossRef]
- Tang, P.M.-K.; Nikolic-Paterson, D.J.; Lan, H.Y. Macrophages: Versatile Players in Renal Inflammation and Fibrosis. Nat. Rev. Nephrol. 2019, 15, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Nikolic-Paterson, D.J.; Wang, S.; Lan, H.Y. Macrophages Promote Renal Fibrosis through Direct and Indirect Mechanisms. Kidney Int. Suppl. 2014, 4, 34–38. [Google Scholar] [CrossRef]
- Kim, M.-G.; Kim, S.C.; Ko, Y.S.; Lee, H.Y.; Jo, S.-K.; Cho, W. The Role of M2 Macrophages in the Progression of Chronic Kidney Disease Following Acute Kidney Injury. PLoS ONE 2015, 10, e0143961. [Google Scholar] [CrossRef] [PubMed]
- Kushiyama, T.; Oda, T.; Yamada, M.; Higashi, K.; Yamamoto, K.; Sakurai, Y.; Miura, S.; Kumagai, H. Alteration in the Phenotype of Macrophages in the Repair of Renal Interstitial Fibrosis in Mice. Nephrology 2011, 16, 522–535. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-G.; Lee, C.-R.; Kim, M.-G.; Kim, G.; Shin, H.M.; Jeon, Y.-H.; Yang, S.H.; Kim, D.K.; Joo, K.W.; Choi, E.Y.; et al. Kidney Residency of VISTA-Positive Macrophages Accelerates Repair from Ischemic Injury. Kidney Int. 2020, 97, 980–994. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.G.; Yun, D.; Kang, C.L.; Hong, M.; Hwang, J.; Moon, K.C.; Jeong, C.W.; Kwak, C.; Kim, D.K.; Oh, K.H.; et al. Kidney VISTA Prevents IFN-γ/IL-9 Axis–Mediated Tubulointerstitial Fibrosis after Acute Glomerular Injury. J. Clin. Investig. 2022, 132, 1–15. [Google Scholar] [CrossRef] [PubMed]
- ElTanbouly, M.A.; Schaafsma, E.; Noelle, R.J.; Lines, J.L. VISTA: Coming of Age as a Multi-Lineage Immune Checkpoint. Clin. Exp. Immunol. 2020, 200, 120–130. [Google Scholar] [CrossRef]
- Ceeraz, S.; Sergent, P.A.; Plummer, S.F.; Schned, A.R.; Pechenick, D.; Burns, C.M.; Noelle, R.J. VISTA Deficiency Accelerates the Development of Fatal Murine Lupus Nephritis. Arthritis Rheumatol. 2017, 69, 814–825. [Google Scholar] [CrossRef]
- Wang, L.; Le Mercier, I.; Putra, J.; Chen, W.; Liu, J.; Schenk, A.D.; Nowak, E.C.; Suriawinata, A.A.; Li, J.; Noelle, R.J. Disruption of the Immune-Checkpoint VISTA Gene Imparts a Proinflammatory Phenotype with Predisposition to the Development of Autoimmunity. Proc. Natl. Acad. Sci. USA 2014, 111, 14846–14851. [Google Scholar] [CrossRef]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The Master Regulator of Fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Wu, C.-F.; Chiang, W.-C.; Lai, C.-F.; Chang, F.-C.; Chen, Y.-T.; Chou, Y.-H.; Wu, T.-H.; Linn, G.R.; Ling, H.; Wu, K.-D.; et al. Transforming Growth Factor β-1 Stimulates Profibrotic Epithelial Signaling to Activate Pericyte-Myofibroblast Transition in Obstructive Kidney Fibrosis. Am. J. Pathol. 2013, 182, 118–131. [Google Scholar] [CrossRef]
- Gu, Y.-Y.; Dou, J.-Y.; Huang, X.-R.; Liu, X.-S.; Lan, H.-Y. Transforming Growth Factor-β and Long Non-Coding RNA in Renal Inflammation and Fibrosis. Front. Physiol. 2021, 12, 684236. [Google Scholar] [CrossRef]
- Yamamoto, T.; Noble, N.A.; Cohen, A.H.; Nast, C.C.; Hishida, A.; Gold, L.I.; Border, W.A. Expression of Transforming Growth Factor-Beta Isoforms in Human Glomerular Diseases. Kidney Int. 1996, 49, 461–469. [Google Scholar] [CrossRef]
- Epstein, F.H. Oxygen and Renal Metabolism. Kidney Int. 1997, 51, 381–385. [Google Scholar] [CrossRef]
- Fu, Q.; Colgan, S.P.; Shelley, C.S. Hypoxia: The Force That Drives Chronic Kidney Disease. Clin. Med. Res. 2016, 14, 15–39. [Google Scholar] [CrossRef]
- Lee, J.W.; Ko, J.; Ju, C.; Eltzschig, H.K. Hypoxia Signaling in Human Diseases and Therapeutic Targets. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Higgins, D.F.; Kimura, K.; Bernhardt, W.M.; Shrimanker, N.; Akai, Y.; Hohenstein, B.; Saito, Y.; Johnson, R.S.; Kretzler, M.; Cohen, C.D.; et al. Hypoxia Promotes Fibrogenesis in Vivo via HIF-1 Stimulation of Epithelial-to-Mesenchymal Transition. J. Clin. Investig. 2007, 117, 3810–3820. [Google Scholar] [CrossRef]
- Vesey, D.A.; Cheung, C.W.Y.; Cuttle, L.; Endre, Z.A.; Gobé, G.; Johnson, D.W. Interleukin-1beta Induces Human Proximal Tubule Cell Injury, Alpha-Smooth Muscle Actin Expression and Fibronectin Production. Kidney Int. 2002, 62, 31–40. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, L.; Bai, M.; Liu, M.; Wei, L.; Yang, Z.; Qian, Q.; Ning, X.; Sun, S. Hypoxia-Induced HE4 in Tubular Epithelial Cells Promotes Extracellular Matrix Accumulation and Renal Fibrosis via NF-ΚB. FASEB J. 2020, 34, 2554–2567. [Google Scholar] [CrossRef]
- Wei, X.; Zhu, X.; Jiang, L.; Huang, X.; Zhang, Y.; Zhao, D.; Du, Y. Recent Advances in Understanding the Role of Hypoxia-Inducible Factor 1α in Renal Fibrosis. Int. Urol. Nephrol. 2020, 52, 1287–1295. [Google Scholar] [CrossRef]
- Helal, I.; Fick-Brosnahan, G.M.; Reed-Gitomer, B.; Schrier, R.W. Glomerular Hyperfiltration: Definitions, Mechanisms and Clinical Implications. Nat. Rev. Nephrol. 2012, 8, 293–300. [Google Scholar] [CrossRef]
- Kriz, W.; Lemley, K.v. A Potential Role for Mechanical Forces in the Detachment of Podocytes and the Progression of CKD. J. Am. Soc. Nephrol. 2015, 26, 258–269. [Google Scholar] [CrossRef]
- Rupérez, M.; Lorenzo, O.; Blanco-Colio, L.M.; Esteban, V.; Egido, J.; Ruiz-Ortega, M. Connective Tissue Growth Factor Is a Mediator of Angiotensin II-Induced Fibrosis. Circulation 2003, 108, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Tampe, D.; Schridde, L.; Korsten, P.; Ströbel, P.; Zeisberg, M.; Hakroush, S.; Tampe, B. Different Patterns of Kidney Fibrosis Are Indicative of Injury to Distinct Renal Compartments. Cells 2021, 10, 2014. [Google Scholar] [CrossRef] [PubMed]
- Piek, A.; de Boer, R.A.; Silljé, H.H.W. The Fibrosis-Cell Death Axis in Heart Failure. Heart Fail. Rev. 2016, 21, 199–211. [Google Scholar] [CrossRef]
- Kaul, A.; Gordon, C.; Crow, M.K.; Touma, Z.; Urowitz, M.B.; Van Vollenhoven, R.; Ruiz-Irastorza, G.; Hughes, G. Systemic Lupus Erythematosus. Nat. Rev. Dis. Prim. 2016, 2, 16039. [Google Scholar] [CrossRef] [PubMed]
- Almaani, S.; Meara, A.; Rovin, B.H. Update on Lupus Nephritis. Clin. J. Am. Soc. Nephrol. 2017, 12, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Rijnink, E.C.; Teng, Y.K.O.; Wilhelmus, S.; Almekinders, M.; Wolterbeek, R.; Cransberg, K.; Bruijn, J.A.; Bajema, I.M. Clinical and Histopathologic Characteristics Associated with Renal Outcomes in Lupus Nephritis. Clin. J. Am. Soc. Nephrol. 2017, 12, 734–737. [Google Scholar] [CrossRef]
- Park, D.J.; Choi, S.E.; Xu, H.; Kang, J.H.; Lee, K.E.; Lee, J.S.; Choi, Y.D.; Lee, S.S. Chronicity Index, Especially Glomerular Sclerosis, Is the Most Powerful Predictor of Renal Response Following Immunosuppressive Treatment in Patients with Lupus Nephritis. Int. J. Rheum. Dis. 2018, 21, 458–467. [Google Scholar] [CrossRef]
- Obrișcă, B.; Jurubiță, R.; Andronesi, A.; Sorohan, B.; Achim, C.; Bobeica, R.; Gherghiceanu, M.; Mandache, E.; Ismail, G. Histological Predictors of Renal Outcome in Lupus Nephritis: The Importance of Tubulointerstitial Lesions and Scoring of Glomerular Lesions. Lupus 2018, 27, 1455–1463. [Google Scholar] [CrossRef]
- Yung, S.; Ng, C.Y.C.; Ho, S.K.; Cheung, K.F.; Chan, K.W.; Zhang, Q.; Chau, M.K.M.; Chan, T.M. Anti-DsDNA Antibody Induces Soluble Fibronectin Secretion by Proximal Renal Tubular Epithelial Cells and Downstream Increase of TGF-Β1 and Collagen Synthesis. J. Autoimmun. 2015, 58, 111–122. [Google Scholar] [CrossRef]
- Koren, E.; Koscec, M.; Wolfson-Reichlin, M.; Ebling, F.M.; Tsao, B.; Hahn, B.H.; Reichlin, M. Murine and Human Antibodies to Native DNA That Cross-React with the A and D SnRNP Polypeptides Cause Direct Injury of Cultured Kidney Cells. J. Immunol. 1995, 154, 4857–4864. [Google Scholar]
- Chan, T.M.; Cheng, I.K. Identification of Endothelial Cell Membrane Proteins That Bind Anti-DNA Antibodies from Patients with Systemic Lupus Erythematosus by Direct or Indirect Mechanisms. J. Autoimmun. 1997, 10, 433–439. [Google Scholar] [CrossRef]
- Satoskar, A.A.; Brodsky, S.V.; Nadasdy, G.; Bott, C.; Rovin, B.; Hebert, L.; Nadasdy, T. Discrepancies in Glomerular and Tubulointerstitial/Vascular Immune Complex IgG Subclasses in Lupus Nephritis. Lupus 2011, 20, 1396–1403. [Google Scholar] [CrossRef]
- Hong, S.; Healy, H.; Kassianos, A.J. The Emerging Role of Renal Tubular Epithelial Cells in the Immunological Pathophysiology of Lupus Nephritis. Front. Immunol. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Der, E.; Ranabothu, S.; Suryawanshi, H.; Akat, K.M.; Clancy, R.; Morozov, P.; Kustagi, M.; Czuppa, M.; Izmirly, P.; Belmont, H.M.; et al. Single Cell RNA Sequencing to Dissect the Molecular Heterogeneity in Lupus Nephritis. JCI Insight 2017, 2, e93009. [Google Scholar] [CrossRef]
- Der, E.; Suryawanshi, H.; Morozov, P.; Kustagi, M.; Goilav, B.; Ranabothu, S.; Izmirly, P.; Clancy, R.; Belmont, H.M.; Koenigsberg, M.; et al. Tubular Cell and Keratinocyte Single-Cell Transcriptomics Applied to Lupus Nephritis Reveal Type I IFN and Fibrosis Relevant Pathways. Nat. Immunol. 2019, 20, 915–927. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, X.; Hong, Q.; Lin, H.; Zhu, H.; Liu, Q.; Wang, J.; Xie, Y.; Shang, X.; Shi, S.; et al. TIMP-1 Promotes Age-Related Renal Fibrosis through Upregulating ICAM-1 in Human TIMP-1 Transgenic Mice. J. Gerontol. A. Biol. Sci. Med. Sci. 2006, 61, 1130–1143. [Google Scholar] [CrossRef]
- Ling, X.B.; Sigdel, T.K.; Lau, K.; Ying, L.; Lau, I.; Schilling, J.; Sarwal, M.M. Integrative Urinary Peptidomics in Renal Transplantation Identifies Biomarkers for Acute Rejection. J. Am. Soc. Nephrol. 2010, 21, 646–653. [Google Scholar] [CrossRef]
- Ding, X.; Ren, Y.; He, X. IFN-I Mediates Lupus Nephritis From the Beginning to Renal Fibrosis. Front. Immunol. 2021, 12, 8–10. [Google Scholar] [CrossRef]
- Kishimoto, D.; Kirino, Y.; Tamura, M.; Takeno, M.; Kunishita, Y.; Takase-Minegishi, K.; Nakano, H.; Kato, I.; Nagahama, K.; Yoshimi, R.; et al. Dysregulated Heme Oxygenase-1Low M2-like Macrophages Augment Lupus Nephritis via Bach1 Induced by Type I Interferons. Arthritis Res. Ther. 2018, 20, 64. [Google Scholar] [CrossRef]
- López, P.; Scheel-Toellner, D.; Rodríguez-Carrio, J.; Caminal-Montero, L.; Gordon, C.; Suárez, A. Interferon-α-Induced B-Lymphocyte Stimulator Expression and Mobilization in Healthy and Systemic Lupus Erthymatosus Monocytes. Rheumatology 2014, 53, 2249–2258. [Google Scholar] [CrossRef]
- Wright, R.D.; Dimou, P.; Northey, S.J.; Beresford, M.W. Mesangial Cells Are Key Contributors to the Fibrotic Damage Seen in the Lupus Nephritis Glomerulus. J. Inflamm. 2019, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Solé, C.; Moliné, T.; Vidal, M.; Ordi-ros, J.; Cortés-hernández, J. An Exosomal Urinary MiRNA Signature for Early Diagnosis of Renal Fibrosis in Lupus Nephritis. Cells 2019, 8, 773. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, A.; Alexander, R.V.; Zack, D.J. A Review of Complement Activation in SLE. Curr. Rheumatol. Rep. 2021, 23, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, M.; Chiriboga, L.; Zeck, B.; Goilav, B.; Wang, S.; Jimenez, A.L.; Putterman, C.; Schwartz, D.; Pullman, J.; et al. Membrane Attack Complex (MAC) Deposition in Renal Tubules Is Associated with Interstitial Fibrosis and Tubular Atrophy: A Pilot Study. Lupus Sci. Med. 2022, 9, e000576. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C. Function and Biological Roles of the Dickkopf Family of Wnt Modulators. Oncogene 2006, 25, 7469–7481. [Google Scholar] [CrossRef] [PubMed]
- Baetta, R.; Banfi, C. Dkk (Dickkopf) Proteins. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1330–1342. [Google Scholar] [CrossRef]
- Sciascia, S.; Barinotti, A.; Radin, M.; Cecchi, I.; Menegatti, E.; Terzolo, E.; Rossi, D.; Baldovino, S.; Fenoglio, R.; Roccatello, D. Dickkopf Homolog 3 (DKK3) as a Prognostic Marker in Lupus Nephritis: A Prospective Monocentric Experience. J. Clin. Med. 2022, 11, 2977. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, W.; Zhu, M.; Zheng, L.; Xie, L.; Yao, Z.; Zhang, H.; Cao, D.; Lu, B. Lupus Nephritis Pathology Prediction with Clinical Indices. Sci. Rep. 2018, 8, 10231. [Google Scholar] [CrossRef]
- Yu, F.; Wu, L.H.; Tan, Y.; Li, L.H.; Wang, C.L.; Wang, W.K.; Qu, Z.; Chen, M.H.; Gao, J.J.; Li, Z.Y.; et al. Tubulointerstitial Lesions of Patients with Lupus Nephritis Classified by the 2003 International Society of Nephrology and Renal Pathology Society System. Kidney Int. 2010, 77, 820–829. [Google Scholar] [CrossRef]
- Hsieh, C.; Chang, A.; Brandt, D.; Guttikonda, R.; Utset, T.O.; Clark, M.R. Predicting Outcomes of Lupus Nephritis with Tubulointerstitial Inflammation and Scarring. Arthritis Care Res. 2011, 63, 865–874. [Google Scholar] [CrossRef]
- Austin, H.A.; Muenz, L.R.; Joyce, K.M. Diffuse Proliferative Lupus Nephritis: Identification of Specific Pathologic Features Affecting Renal Outcome. Kidney Int. 1984, 25, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.C.; Kashgarian, M.; Moeckel, G. Interstitial Inflammation and Interstitial Fibrosis and Tubular Atrophy Predict Renal Survival in Lupus Nephritis. Clin. Kidney J. 2018, 11, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Broder, A.; Mowrey, W.B.; Khan, H.N.; Jovanovic, B.; Londono-Jimenez, A.; Izmirly, P.; Putterman, C. Tubulointerstitial Damage Predicts End Stage Renal Disease in Lupus Nephritis with Preserved to Moderately Impaired Renal Function: A Retrospective Cohort Study. Semin. Arthritis Rheum. 2018, 47, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Leatherwood, C.; Speyer, C.B.; Feldman, C.H.; D’Silva, K.; Gómez-Puerta, J.A.; Hoover, P.J.; Waikar, S.S.; McMahon, G.M.; Rennke, H.G.; Costenbader, K.H. Clinical Characteristics and Renal Prognosis Associated with Interstitial Fibrosis and Tubular Atrophy (IFTA) and Vascular Injury in Lupus Nephritis Biopsies. Semin. Arthritis Rheum. 2019, 49, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.F.; Mardones, C.; Xipell, M.; Blasco, M.; Solé, M.; Espinosa, G.; García-Herrera, A.; Cervera, R.; Quintana, L.F. The Extent of Tubulointerstitial Inflammation Is an Independent Predictor of Renal Survival in Lupus Nephritis. J. Nephrol. 2021, 34, 1897–1905. [Google Scholar] [CrossRef]
- Moroni, G.; Porata, G.; Raffiotta, F.; Quaglini, S.; Frontini, G.; Sacchi, L.; Binda, V.; Calatroni, M.; Reggiani, F.; Banfi, G.; et al. Beyond ISN/RPS Lupus Nephritis Classification: Adding Chronicity Index to Clinical Variables Predicts Kidney Survival. Kidney360 2022, 3, 122–132. [Google Scholar] [CrossRef]
- Mannon, R.B.; Matas, A.J.; Grande, J.; Leduc, R.; Connett, J.; Kasiske, B.; Cecka, J.M.; Gaston, R.S.; Cosio, F.; Gourishankar, S.; et al. Inflammation in Areas of Tubular Atrophy in Kidney Allograft Biopsies: A Potent Predictor of Allograft Failure. Am. J. Transplant. 2010, 10, 2066–2073. [Google Scholar] [CrossRef]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E.J.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis (CAPACITY): Two Randomised Trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- King, T.E.J.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef]
- Richeldi, L.; Cottin, V.; du Bois, R.M.; Selman, M.; Kimura, T.; Bailes, Z.; Schlenker-Herceg, R.; Stowasser, S.; Brown, K.K. Nintedanib in Patients with Idiopathic Pulmonary Fibrosis: Combined Evidence from the TOMORROW and INPULSIS(®) Trials. Respir. Med. 2016, 113, 74–79. [Google Scholar] [CrossRef]
- Cho, M.E.; Smith, D.C.; Branton, M.H.; Penzak, S.R.; Kopp, J.B. Pirfenidone Slows Renal Function Decline in Patients with Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2007, 2, 906–913. [Google Scholar] [CrossRef]
- Wollin, L.; Maillet, I.; Quesniaux, V.; Holweg, A.; Ryffel, B. Antifibrotic and Anti-Inflammatory Activity of the Tyrosine Kinase Inhibitor Nintedanib in Experimental Models of Lung Fibrosis. J. Pharmacol. Exp. Ther. 2014, 349, 209–220. [Google Scholar] [CrossRef]
- Hostettler, K.E.; Zhong, J.; Papakonstantinou, E.; Karakiulakis, G.; Tamm, M.; Seidel, P.; Sun, Q.; Mandal, J.; Lardinois, D.; Lambers, C.; et al. Anti-Fibrotic Effects of Nintedanib in Lung Fibroblasts Derived from Patients with Idiopathic Pulmonary Fibrosis. Respir. Res. 2014, 15, 157. [Google Scholar] [CrossRef]
- Kuwana, M.; Azuma, A. Nintedanib: New Indication for Systemic Sclerosis-Associated Interstitial Lung Disease. Mod. Rheumatol. 2020, 30, 225–231. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.F.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef]
- Liu, F.; Wang, L.; Qi, H.; Wang, J.; Wang, Y.; Jiang, W.; Xu, L.; Liu, N.; Zhuang, S. Nintedanib, a Triple Tyrosine Kinase Inhibitor, Attenuates Renal Fibrosis in Chronic Kidney Disease. Clin. Sci. 2017, 131, 2125–2143. [Google Scholar] [CrossRef]
- Fujita, T.; Nakagawa, H.; Yokota, T.; Umetani, J.; Nagawa, D.; Nakata, M.; Narita-Kinjo, I.; Murakami, R.; Shimada, M.; Nakamura, N.; et al. Nintedanib-Induced Renal Thrombotic Microangiopathy. Case Reports Nephrol. Dial. 2021, 11, 227–232. [Google Scholar] [CrossRef]
- Feng, L.; Li, W.; Chao, Y.; Huan, Q.; Lu, F.; Yi, W.; Jun, W.; Binbin, C.; Na, L.; Shougang, Z. Synergistic Inhibition of Renal Fibrosis by Nintedanib and Gefitinib in a Murine Model of Obstructive Nephropathy. Kidney Dis. 2021, 7, 34–49. [Google Scholar] [CrossRef]
- Rurik, J.G.; Tombácz, I.; Yadegari, A.; Méndez Fernández, P.O.; Shewale, S.V.; Li, L.; Kimura, T.; Soliman, O.Y.; Papp, T.E.; Tam, Y.K.; et al. CAR T Cells Produced in Vivo to Treat Cardiac Injury. Science 2022, 375, 91–96. [Google Scholar] [CrossRef]
- Aghajanian, H.; Kimura, T.; Rurik, J.G.; Hancock, A.S.; Leibowitz, M.S.; Li, L.; Scholler, J.; Monslow, J.; Lo, A.; Han, W.; et al. Targeting Cardiac Fibrosis with Engineered T Cells. Nature 2019, 573, 430–433. [Google Scholar] [CrossRef]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.-J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T Cells Reverse Senescence-Associated Pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Vincenti, F.; Fervenza, F.C.; Campbell, K.N.; Diaz, M.; Gesualdo, L.; Nelson, P.; Praga, M.; Radhakrishnan, J.; Sellin, L.; Singh, A.; et al. A Phase 2, Double-Blind, Placebo-Controlled, Randomized Study of Fresolimumab in Patients With Steroid-Resistant Primary Focal Segmental Glomerulosclerosis. Kidney Int. Rep. 2017, 2, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Chertow, G.M.; Appel, G.B.; Andreoli, S.; Bangalore, S.; Block, G.A.; Chapman, A.B.; Chin, M.P.; Gibson, K.L.; Goldsberry, A.; Iijima, K.; et al. Study Design and Baseline Characteristics of the CARDINAL Trial: A Phase 3 Study of Bardoxolone Methyl in Patients with Alport Syndrome. Am. J. Nephrol. 2021, 52, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Isaka, Y. Targeting TGF-β Signaling in Kidney Fibrosis. Int. J. Mol. Sci. 2018, 19, 2532. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.M.; Li, F.K.; Tang, C.S.; Wong, R.W.; Fang, G.X.; Ji, Y.L.; Lau, C.S.; Wong, A.K.; Tong, M.K.; Chan, K.W.; et al. Efficacy of Mycophenolate Mofetil in Patients with Diffuse Proliferative Lupus Nephritis. Hong Kong-Guangzhou Nephrology Study Group. N. Engl. J. Med. 2000, 343, 1156–1162. [Google Scholar] [CrossRef]
- Appel, G.B.; Contreras, G.; Dooley, M.A.; Ginzler, E.M.; Isenberg, D.; Jayne, D.; Li, L.-S.; Mysler, E.; Sánchez-Guerrero, J.; Solomons, N.; et al. Mycophenolate Mofetil versus Cyclophosphamide for Induction Treatment of Lupus Nephritis. J. Am. Soc. Nephrol. 2009, 20, 1103–1112. [Google Scholar] [CrossRef]
- Chan, T.-M.; Tse, K.-C.; Tang, C.S.-O.; Mok, M.-Y.; Li, F.-K. Long-Term Study of Mycophenolate Mofetil as Continuous Induction and Maintenance Treatment for Diffuse Proliferative Lupus Nephritis. J. Am. Soc. Nephrol. 2005, 16, 1076–1084. [Google Scholar] [CrossRef]
- Hauser, I.A.; Renders, L.; Radeke, H.H.; Sterzel, R.B.; Goppelt-Struebe, M. Mycophenolate Mofetil Inhibits Rat and Human Mesangial Cell Proliferation by Guanosine Depletion. Nephrol. Dial. Transplant. 1999, 14, 58–63. [Google Scholar] [CrossRef]
- Baer, P.C.; Gauer, S.; Hauser, I.A.; Scherberich, J.E.; Geiger, H. Effects of Mycophenolic Acid on Human Renal Proximal and Distal Tubular Cells in Vitro. Nephrol. Dial. Transplant. 2000, 15, 184–190. [Google Scholar] [CrossRef]
- Heinz, C.; Hudde, T.; Heise, K.; Steuhl, K.-P. Antiproliferative Effect of Mycophenolate Mofetil on Cultured Human Tenon Fibroblasts. Graefe’s Arch. Clin. Exp. Ophthalmol. 2002, 240, 408–414. [Google Scholar] [CrossRef]
- Yung, S.; Zhang, Q.; Zhang, C.Z.; Chan, K.W.; Lui, S.L.; Chan, T.M. Anti-DNA Antibody Induction of Protein Kinase C Phosphorylation and Fibronectin Synthesis in Human and Murine Lupus and the Effect of Mycophenolic Acid. Arthritis Rheum. 2009, 60, 2071–2082. [Google Scholar] [CrossRef]
- Yung, S.; Zhang, Q.; Chau, M.K.M.; Chan, T.M. Distinct Effects of Mycophenolate Mofetil and Cyclophosphamide on Renal Fibrosis in NZBWF1/J Mice. Autoimmunity 2015, 48, 471–487. [Google Scholar] [CrossRef]
- Zhang, C.; Chan, C.C.Y.; Cheung, K.F.; Chau, M.K.M.; Yap, D.Y.H.; Ma, M.K.M.; Chan, K.W.; Yung, S.; Chan, T.M. Effect of Mycophenolate and Rapamycin on Renal Fibrosis in Lupus Nephritis. Clin. Sci. 2019, 133, 1721–1744. [Google Scholar] [CrossRef]
- Zhang, C.; Tam, T.W.; Chau, M.K.M.; García Córdoba, C.A.; Yung, S.; Chan, T.M. Effect of Combined Mycophenolate and Rapamycin Treatment on Kidney Fibrosis in Murine Lupus Nephritis. Front. Pharmacol. 2022, 13, 866077. [Google Scholar] [CrossRef]
- Xue, L.; Xu, J.; Lu, W.; Fu, J.; Liu, Z. Iguratimod Alleviates Tubulo-Interstitial Injury in Mice with Lupus. Ren. Fail. 2022, 44, 636–647. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Agents | Clinical Development |
|---|---|
| Pirfenidone |
|
| Fresolimumab, LY2382770 |
|
| FG-3019 (pamrevlumab) |
|
| GCS-100 |
|
| Fingolimod |
|
| Bardoxolone methyl |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sciascia, S.; Cozzi, M.; Barinotti, A.; Radin, M.; Cecchi, I.; Fenoglio, R.; Mancardi, D.; Wilson Jones, G.; Rossi, D.; Roccatello, D. Renal Fibrosis in Lupus Nephritis. Int. J. Mol. Sci. 2022, 23, 14317. https://doi.org/10.3390/ijms232214317
Sciascia S, Cozzi M, Barinotti A, Radin M, Cecchi I, Fenoglio R, Mancardi D, Wilson Jones G, Rossi D, Roccatello D. Renal Fibrosis in Lupus Nephritis. International Journal of Molecular Sciences. 2022; 23(22):14317. https://doi.org/10.3390/ijms232214317
Chicago/Turabian StyleSciascia, Savino, Martina Cozzi, Alice Barinotti, Massimo Radin, Irene Cecchi, Roberta Fenoglio, Daniele Mancardi, Georgia Wilson Jones, Daniela Rossi, and Dario Roccatello. 2022. "Renal Fibrosis in Lupus Nephritis" International Journal of Molecular Sciences 23, no. 22: 14317. https://doi.org/10.3390/ijms232214317
APA StyleSciascia, S., Cozzi, M., Barinotti, A., Radin, M., Cecchi, I., Fenoglio, R., Mancardi, D., Wilson Jones, G., Rossi, D., & Roccatello, D. (2022). Renal Fibrosis in Lupus Nephritis. International Journal of Molecular Sciences, 23(22), 14317. https://doi.org/10.3390/ijms232214317