

Molecular Characterization of the Dual Effect of the GPER Agonist G-1 in Glioblastoma

 and
and
Abstract
1. Introduction
2. Results
2.1. G-1 Elicits an Anti-Tumor Effect in GBM Cell Xenografted Nude Mice
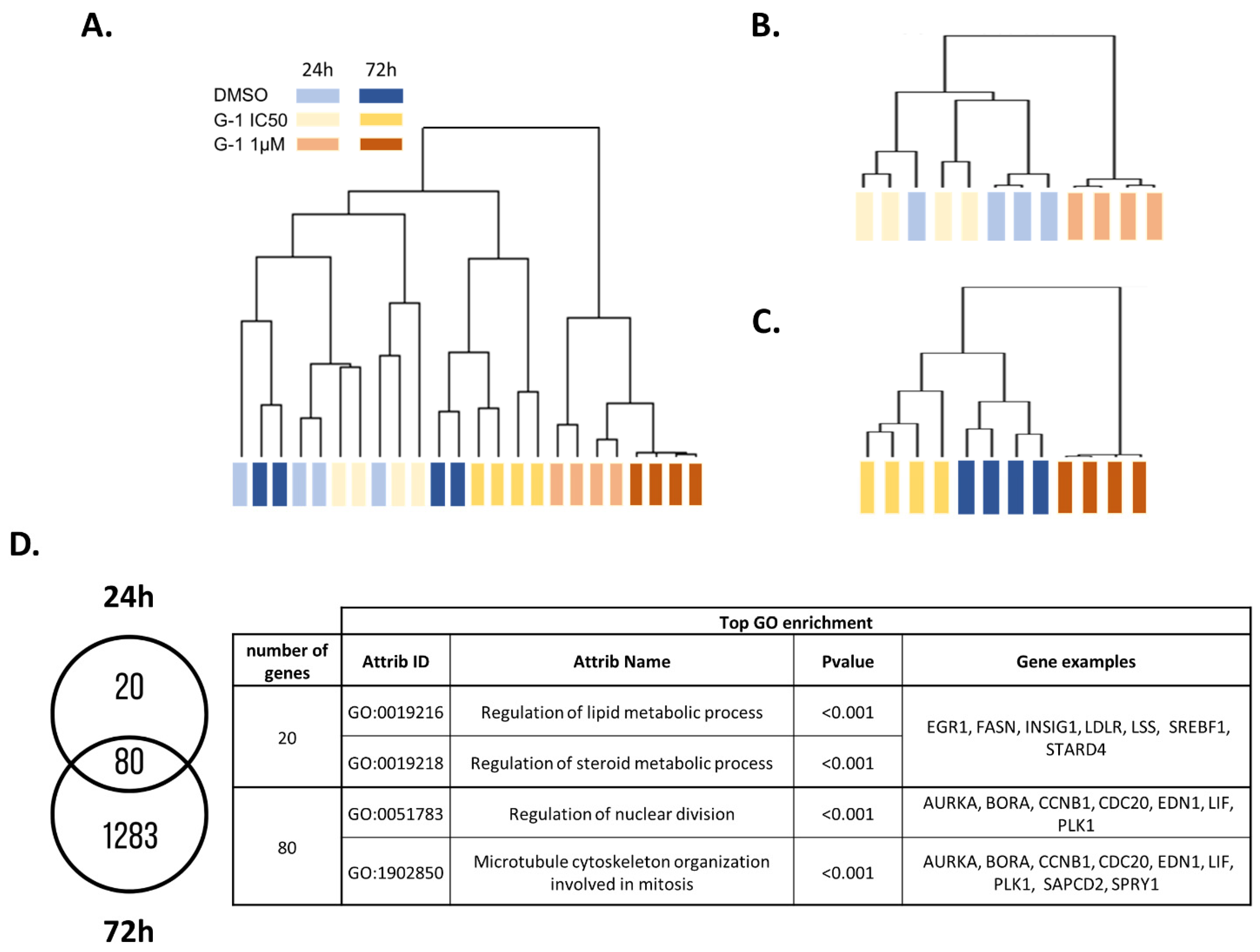
2.2. Transcriptomic Analysis of U251 GBM Cells Exposed to G-1
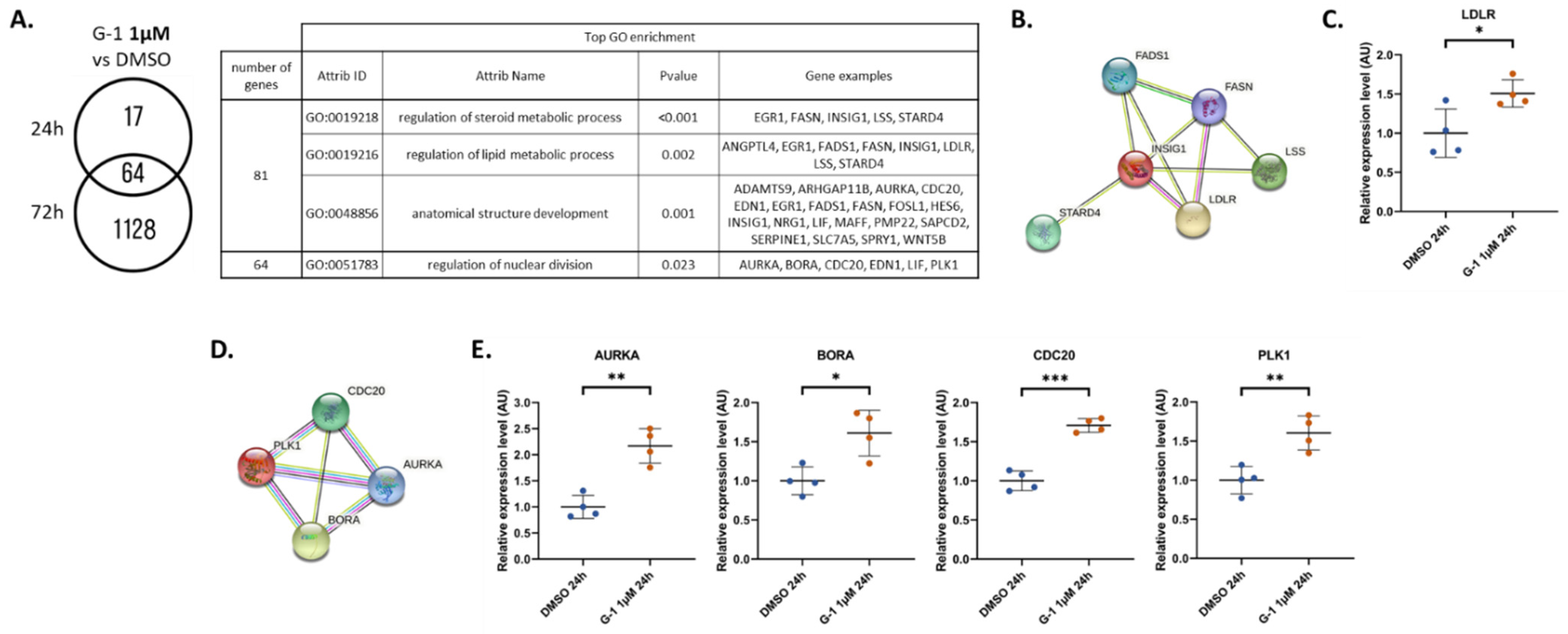
2.3. Identification and Validation of ANGPTL4 Expression as a Marker of G-1 Exposure
2.4. G-1 Impairs Lipid Metabolism and Nuclear Division Pathways
2.5. The Molecular G-1 Signature Is Similar to Microtubule Targeting Agent One
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Transcriptomic Analysis
4.3. Bioinformatic Analysis
4.4. GBM Tumor from Patients
4.5. RT-qPCR
4.6. Animal Xenograft Model and Treatments
4.7. Immunohistochemistry and Hematoxylin and Eosin (H&E) Staining
4.8. Connectivity Map
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Grochans, S.; Cybulska, A.M.; Simińska, D.; Korbecki, J.; Kojder, K.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of Glioblastoma Multiforme-Literature Review. Cancers 2022, 14, 2412. [Google Scholar] [CrossRef] [PubMed]
- Hirtz, A.; Lebourdais, N.; Thomassin, M.; Rech, F.; Dumond, H.; Dubois-Pot-Schneider, H. Identification of Gender- and Subtype-Specific Gene Expression Associated with Patient Survival in Low-Grade and Anaplastic Glioma in Connection with Steroid Signaling. Cancers 2022, 14, 4114. [Google Scholar] [CrossRef] [PubMed]
- Hirtz, A.; Lebourdais, N.; Rech, F.; Bailly, Y.; Vaginay, A.; Smaïl-Tabbone, M.; Dubois-Pot-Schneider, H.; Dumond, H. GPER Agonist G-1 Disrupts Tubulin Dynamics and Potentiates Temozolomide to Impair Glioblastoma Cell Proliferation. Cells 2021, 10, 3438. [Google Scholar] [CrossRef] [PubMed]
- Hazell, G.G.J.; Yao, S.T.; Roper, J.A.; Prossnitz, E.R.; O’Carroll, A.-M.; Lolait, S.J. Localisation of GPR30, a Novel G Protein-Coupled Oestrogen Receptor, Suggests Multiple Functions in Rodent Brain and Peripheral Tissues. J. Endocrinol. 2009, 202, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Mårtensson, U.E.A.; Salehi, S.A.; Windahl, S.; Gomez, M.F.; Swärd, K.; Daszkiewicz-Nilsson, J.; Wendt, A.; Andersson, N.; Hellstrand, P.; Grände, P.-O.; et al. Deletion of the G Protein-Coupled Receptor 30 Impairs Glucose Tolerance, Reduces Bone Growth, Increases Blood Pressure, and Eliminates Estradiol-Stimulated Insulin Release in Female Mice. Endocrinology 2009, 150, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.; Fuchs, I.; Kauselmann, G.; Kern, H.; Zevnik, B.; Andreasen, P.; Schwarz, G.; Altmann, H.; Klewer, M.; Schoor, M.; et al. GPR30 Does Not Mediate Estrogenic Responses in Reproductive Organs in Mice. Biol. Reprod. 2009, 80, 34–41. [Google Scholar] [CrossRef]
- Carmeci, C.; Thompson, D.A.; Ring, H.Z.; Francke, U.; Weigel, R.J. Identification of a Gene (GPR30) with Homology to the G-Protein-Coupled Receptor Superfamily Associated with Estrogen Receptor Expression in Breast Cancer. Genomics 1997, 45, 607–617. [Google Scholar] [CrossRef]
- Feng, Y.; Gregor, P. Cloning of a Novel Member of the G Protein-Coupled Receptor Family Related to Peptide Receptors. Biochem. Biophys. Res. Commun. 1997, 231, 651–654. [Google Scholar] [CrossRef]
- Owman, C.; Blay, P.; Nilsson, C.; Lolait, S.J. Cloning of Human CDNA Encoding a Novel Heptahelix Receptor Expressed in Burkitt’s Lymphoma and Widely Distributed in Brain and Peripheral Tissues. Biochem. Biophys. Res. Commun. 1996, 228, 285–292. [Google Scholar] [CrossRef]
- Shi, Y.; Liu, X.; Zhu, P.; Li, J.; Sham, K.W.Y.; Cheng, S.H.; Li, S.; Zhang, Y.; Cheng, C.H.K.; Lin, H. G-Protein-Coupled Estrogen Receptor 1 Is Involved in Brain Development during Zebrafish (Danio Rerio) Embryogenesis. Biochem. Biophys. Res. Commun. 2013, 435, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, E.; Dun, S.L.; Brailoiu, G.C.; Mizuo, K.; Sklar, L.A.; Oprea, T.I.; Prossnitz, E.R.; Dun, N.J. Distribution and Characterization of Estrogen Receptor G Protein-Coupled Receptor 30 in the Rat Central Nervous System. J. Endocrinol. 2007, 193, 311–321. [Google Scholar] [CrossRef]
- O’Dowd, B.F.; Nguyen, T.; Marchese, A.; Cheng, R.; Lynch, K.R.; Heng, H.H.Q.; Kolakowski, L.F.; George, S.R. Discovery of Three Novel G-Protein-Coupled Receptor Genes. Genomics 1998, 47, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Roque, C.; Mendes-Oliveira, J.; Baltazar, G. G Protein-coupled Estrogen Receptor Activates Cell Type-specific Signaling Pathways in Cortical Cultures: Relevance to the Selective Loss of Astrocytes. J. Neurochem. 2019, 149, 27–40. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Barton, M. The G-Protein-Coupled Estrogen Receptor GPER in Health and Disease. Nat. Rev. Endocrinol. 2011, 7, 715–726. [Google Scholar] [CrossRef]
- Tutzauer, J.; Gonzalez de Valdivia, E.; Swärd, K.; Alexandrakis Eilard, I.; Broselid, S.; Kahn, R.; Olde, B.; Leeb-Lundberg, L.M.F. Ligand-Independent G Protein–Coupled Estrogen Receptor/G Protein–Coupled Receptor 30 Activity: Lack of Receptor-Dependent Effects of G-1 and 17 β -Estradiol. Mol. Pharm. 2021, 100, 271–282. [Google Scholar] [CrossRef]
- Maggiolini, M.; Vivacqua, A.; Fasanella, G.; Recchia, A.G.; Sisci, D.; Pezzi, V.; Montanaro, D.; Musti, A.M.; Picard, D.; Andò, S. The G Protein-Coupled Receptor GPR30 Mediates c-Fos Up-Regulation by 17β-Estradiol and Phytoestrogens in Breast Cancer Cells. J. Biol. Chem. 2004, 279, 27008–27016. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R. Estrogen-Induced Activation of Erk-1 and Erk-2 Requires the G Protein-Coupled Receptor Homolog, GPR30, and Occurs via Trans-Activation of the Epidermal Growth Factor Receptor through Release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef]
- Bologa, C.G.; Revankar, C.M.; Young, S.M.; Edwards, B.S.; Arterburn, J.B.; Kiselyov, A.S.; Parker, M.A.; Tkachenko, S.E.; Savchuck, N.P.; Sklar, L.A.; et al. Virtual and Biomolecular Screening Converge on a Selective Agonist for GPR30. Nat. Chem. Biol. 2006, 2, 207–212. [Google Scholar] [CrossRef]
- Torres-López, L.; Olivas-Aguirre, M.; Villatoro-Gómez, K.; Dobrovinskaya, O. The G-Protein–Coupled Estrogen Receptor Agonist G-1 Inhibits Proliferation and Causes Apoptosis in Leukemia Cell Lines of T Lineage. Front. Cell Dev. Biol. 2022, 10, 811479. [Google Scholar] [CrossRef]
- Wang, C.; Lv, X.; He, C.; Hua, G.; Tsai, M.-Y.; Davis, J.S. The G-Protein-Coupled Estrogen Receptor Agonist G-1 Suppresses Proliferation of Ovarian Cancer Cells by Blocking Tubulin Polymerization. Cell Death Dis. 2013, 4, e869. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Hu, C.; Staquicini, D.I.; Brigman, J.L.; Liu, M.; Mauvais-Jarvis, F.; Pasqualini, R.; Arap, W.; Arterburn, J.B.; Hathaway, H.J.; et al. Preclinical Efficacy of the First-in-Class GPER-Selective Agonist G-1 in Mouse Models of Obesity and Diabetes. Sci. Transl. Med. 2020, 12, eaau5956. [Google Scholar] [CrossRef] [PubMed]
- Camphausen, K.; Purow, B.; Sproull, M.; Scott, T.; Ozawa, T.; Deen, D.F.; Tofilon, P.J. Influence of in Vivo Growth on Human Glioma Cell Line Gene Expression: Convergent Profiles under Orthotopic Conditions. Proc. Natl. Acad. Sci. USA 2005, 102, 8287–8292. [Google Scholar] [CrossRef]
- Pei, Z.; Oey, N.A.; Zuidervaart, M.M.; Jia, Z.; Li, Y.; Steinberg, S.J.; Smith, K.D.; Watkins, P.A. The Acyl-CoA Synthetase “Bubblegum” (Lipidosin): Further characterization and role in neuronal fatty acid β-oxidation *. J. Biol. Chem. 2003, 278, 47070–47078. [Google Scholar] [CrossRef] [PubMed]
- Grube, S.; Dünisch, P.; Freitag, D.; Klausnitzer, M.; Sakr, Y.; Walter, J.; Kalff, R.; Ewald, C. Overexpression of Fatty Acid Synthase in Human Gliomas Correlates with the WHO Tumor Grade and Inhibition with Orlistat Reduces Cell Viability and Triggers Apoptosis. J. Neurooncol. 2014, 118, 277–287. [Google Scholar] [CrossRef]
- Tao, B.-B.; He, H.; Shi, X.; Wang, C.; Li, W.; Li, B.; Dong, Y.; Hu, G.-H.; Hou, L.-J.; Luo, C.; et al. Up-Regulation of USP2a and FASN in Gliomas Correlates Strongly with Glioma Grade. J. Clin. Neurosci. 2013, 20, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Bell, E.H.; Chakravarti, A. Lipid Metabolism Emerges as a Promising Target for Malignant Glioma Therapy. CNS Oncol. 2013, 2, 289–299. [Google Scholar] [CrossRef]
- Yuan, Y.; Shah, N.; Almohaisin, M.I.; Saha, S.; Lu, F. Assessing Fatty Acid-Induced Lipotoxicity and Its Therapeutic Potential in Glioblastoma Using Stimulated Raman Microscopy. Sci. Rep. 2021, 11, 7422. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, D.; Zhang, Q.; Wang, J.; Zhan, J.; Xian, X.; Du, Z.; Wang, X.; Hao, A. Palmitic Acid Affects Proliferation and Differentiation of Neural Stem Cells in Vitro. J. Neurosci. Res. 2014, 92, 574–586. [Google Scholar] [CrossRef]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V.; Ory, D.S.; Schaffer, J.E. Triglyceride Accumulation Protects against Fatty Acid-Induced Lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef]
- Hardy, S.; Langelier, Y.; Prentki, M. Oleate Activates Phosphatidylinositol 3-Kinase and Promotes Proliferation and Reduces Apoptosis of MDA-MB-231 Breast Cancer Cells, Whereas Palmitate Has Opposite Effects. Cancer Res. 2000, 60, 6353–6358. [Google Scholar] [PubMed]
- Tsai, Y.-T.; Wu, A.-C.; Yang, W.-B.; Kao, T.-J.; Chuang, J.-Y.; Chang, W.-C.; Hsu, T.-I. ANGPTL4 Induces TMZ Resistance of Glioblastoma by Promoting Cancer Stemness Enrichment via the EGFR/AKT/4E-BP1 Cascade. Int. J. Mol. Sci. 2019, 20, 5625. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Barton, M. Estrogen Biology: New Insights into GPER Function and Clinical Opportunities. Mol. Cell Endocrinol. 2014, 389, 71–83. [Google Scholar] [CrossRef]
- Lin, S.; Miao, Y.; Zheng, X.; Dong, Y.; Yang, Q.; Yang, Q.; Du, S.; Xu, J.; Zhou, S.; Yuan, T. ANGPTL4 Negatively Regulates the Progression of Osteosarcoma by Remodeling Branched-Chain Amino Acid Metabolism. Cell Death Discov. 2022, 8, 225. [Google Scholar] [CrossRef]
- Galaup, A.; Cazes, A.; Le Jan, S.; Philippe, J.; Connault, E.; Le Coz, E.; Mekid, H.; Mir, L.M.; Opolon, P.; Corvol, P.; et al. Angiopoietin-like 4 Prevents Metastasis through Inhibition of Vascular Permeability and Tumor Cell Motility and Invasiveness. Proc. Natl. Acad. Sci. USA 2006, 103, 18721–18726. [Google Scholar] [CrossRef]
- Ito, Y.; Oike, Y.; Yasunaga, K.; Hamada, K.; Miyata, K.; Matsumoto, S.-I.; Sugano, S.; Tanihara, H.; Masuho, Y.; Suda, T. Inhibition of Angiogenesis and Vascular Leakiness by Angiopoietin-Related Protein 4. Cancer Res. 2003, 63, 6651–6657. [Google Scholar] [PubMed]
- La Paglia, L.; Listì, A.; Caruso, S.; Amodeo, V.; Passiglia, F.; Bazan, V.; Fanale, D. Potential Role of ANGPTL4 in the Cross Talk between Metabolism and Cancer through PPAR Signaling Pathway. PPAR Res. 2017, 2017, 8187235. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Fotovati, A.; Triscott, J.; Chen, J.; Venugopal, C.; Singhal, A.; Dunham, C.; Kerr, J.M.; Verreault, M.; Yip, S.; et al. Polo-like Kinase 1 Inhibition Kills Glioblastoma Multiforme Brain Tumor Cells in Part through Loss of SOX2 and Delays Tumor Progression in Mice. Stem Cells 2012, 30, 1064–1075. [Google Scholar] [CrossRef] [PubMed]
- de Cárcer, G.; Venkateswaran, S.V.; Salgueiro, L.; El Bakkali, A.; Somogyi, K.; Rowald, K.; Montañés, P.; Sanclemente, M.; Escobar, B.; de Martino, A.; et al. Plk1 Overexpression Induces Chromosomal Instability and Suppresses Tumor Development. Nat. Commun. 2018, 9, 3012. [Google Scholar] [CrossRef]
- Rohrberg, J.; Van de Mark, D.; Amouzgar, M.; Lee, J.V.; Taileb, M.; Corella, A.; Kilinc, S.; Williams, J.; Jokisch, M.-L.; Camarda, R.; et al. MYC Dysregulates Mitosis, Revealing Cancer Vulnerabilities. Cell Rep. 2020, 30, 3368–3382.e7. [Google Scholar] [CrossRef]
- Dhyani, P.; Quispe, C.; Sharma, E.; Bahukhandi, A.; Sati, P.; Attri, D.C.; Szopa, A.; Sharifi-Rad, J.; Docea, A.O.; Mardare, I.; et al. Anticancer Potential of Alkaloids: A Key Emphasis to Colchicine, Vinblastine, Vincristine, Vindesine, Vinorelbine and Vincamine. Cancer Cell Int. 2022, 22, 206. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, B.; Lloyd, G.K.; Miller, B.R.; Palladino, M.A.; Kiso, Y.; Hayashi, Y.; Neuteboom, S.T.C. NPI-2358 Is a Tubulin-Depolymerizing Agent: In-Vitro Evidence for Activity as a Tumor Vascular-Disrupting Agent. Anticancer Drugs 2006, 17, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Berges, R.; Baeza-Kallee, N.; Tabouret, E.; Chinot, O.; Petit, M.; Kruczynski, A.; Figarella-Branger, D.; Honore, S.; Braguer, D. End-Binding 1 Protein Overexpression Correlates with Glioblastoma Progression and Sensitizes to Vinca-Alkaloids in Vitro and in Vivo. Oncotarget 2014, 5, 12769–12787. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; He, C.; Huang, C.; Hua, G.; Wang, Z.; Remmenga, S.W.; Rodabough, K.J.; Karpf, A.R.; Dong, J.; Davis, J.S.; et al. G-1 Inhibits Breast Cancer Cell Growth via Targeting Colchicine-Binding Site of Tubulin to Interfere with Microtubule Assembly. Mol. Cancer Ther. 2017, 16, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, P.A.; Hu, J.; Hurren, R.; Wang, X.; Gronda, M.; Sukhai, M.A.; Di Meo, A.; Boss, J.; Ashali, I.; Beheshti Zavareh, R.; et al. The Antihelmintic Flubendazole Inhibits Microtubule Function through a Mechanism Distinct from Vinca Alkaloids and Displays Preclinical Activity in Leukemia and Myeloma. Blood 2010, 115, 4824–4833. [Google Scholar] [CrossRef]
- Bai, R.-Y.; Staedtke, V.; Aprhys, C.M.; Gallia, G.L.; Riggins, G.J. Antiparasitic Mebendazole Shows Survival Benefit in 2 Preclinical Models of Glioblastoma Multiforme. Neuro-Oncology 2011, 13, 974–982. [Google Scholar] [CrossRef]
- Geribaldi-Doldán, N.; Hervás-Corpión, I.; Gómez-Oliva, R.; Domínguez-García, S.; Ruiz, F.A.; Iglesias-Lozano, I.; Carrascal, L.; Pardillo-Díaz, R.; Gil-Salú, J.L.; Nunez-Abades, P.; et al. Targeting Protein Kinase C in Glioblastoma Treatment. Biomedicines 2021, 9, 381. [Google Scholar] [CrossRef]
- Berriz, G.F.; Beaver, J.E.; Cenik, C.; Tasan, M.; Roth, F.P. Next Generation Software for Functional Trend Analysis. Bioinformatics 2009, 25, 3043–3044. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1alpha-Responsive Genes Involved in Oxidative Phosphorylation Are Coordinately Downregulated in Human Diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef] [PubMed]
- Altman, D.G.; Bland, J.M. Standard Deviations and Standard Errors. BMJ 2005, 331, 903. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Id | median_tau_score | Name | Belongs to |
|---|---|---|---|
| BRD-K91145395 | 98.86 | prostratin | PKC activator |
| BRD-K12539581 | 98.48 | nocodazole | tubulin inhibitor |
| BRD-K86003836 | 98.31 | flubendazole | tubulin inhibitor |
| BRD-K10916986 | 98.31 | vinorelbine | tubulin inhibitor |
| BRD-A76528577 | 98.10 | vincristine | tubulin inhibitor |
| BRD-K77987382 | 97.63 | mebendazole | tubulin inhibitor |
| BRD-K37456065 | 97.37 | VU-0365114-2 | M5 modulator |
| BRD-K91623615 | 97.33 | ABT-751 | tubulin inhibitor |
| BRD-K94325918 | 96.28 | kinetin-riboside | antiproliferative and apoptosis inhibitor |
| BRD-A54927599 | 96.17 | KF-38789 | P-selectin-mediated cell adhesion inhibitor |
| BRD-K35687265 | 95.98 | ON-01910 | PLK inhibitor |
| BRD-A55594068 | 95.91 | vinblastine | tubulin inhibitor |
| BRD-K59753975 | 95.71 | vindesine | tubulin inhibitor |
| BRD-K36055864 | 94.89 | cycloheximide | protein synthesis inhibitor |
| BRD-K01976263 | 94.67 | emetine | Wnt/β-catenin inhibitor |
| BRD-A52650764 | 94.47 | ingenol | PKC activator |
| BRD-K99498722 | 91.02 | NPI-2358 | tubulin inhibitor |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirtz, A.; Bailly, Y.; Rech, F.; Pierson, J.; Dumond, H.; Dubois-Pot-Schneider, H. Molecular Characterization of the Dual Effect of the GPER Agonist G-1 in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 14309. https://doi.org/10.3390/ijms232214309
Hirtz A, Bailly Y, Rech F, Pierson J, Dumond H, Dubois-Pot-Schneider H. Molecular Characterization of the Dual Effect of the GPER Agonist G-1 in Glioblastoma. International Journal of Molecular Sciences. 2022; 23(22):14309. https://doi.org/10.3390/ijms232214309
Chicago/Turabian StyleHirtz, Alex, Yann Bailly, Fabien Rech, Julien Pierson, Hélène Dumond, and Hélène Dubois-Pot-Schneider. 2022. "Molecular Characterization of the Dual Effect of the GPER Agonist G-1 in Glioblastoma" International Journal of Molecular Sciences 23, no. 22: 14309. https://doi.org/10.3390/ijms232214309
APA StyleHirtz, A., Bailly, Y., Rech, F., Pierson, J., Dumond, H., & Dubois-Pot-Schneider, H. (2022). Molecular Characterization of the Dual Effect of the GPER Agonist G-1 in Glioblastoma. International Journal of Molecular Sciences, 23(22), 14309. https://doi.org/10.3390/ijms232214309