

Identification of Species-Specific MicroRNAs Provides Insights into Dynamic Evolution of MicroRNAs in Plants



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
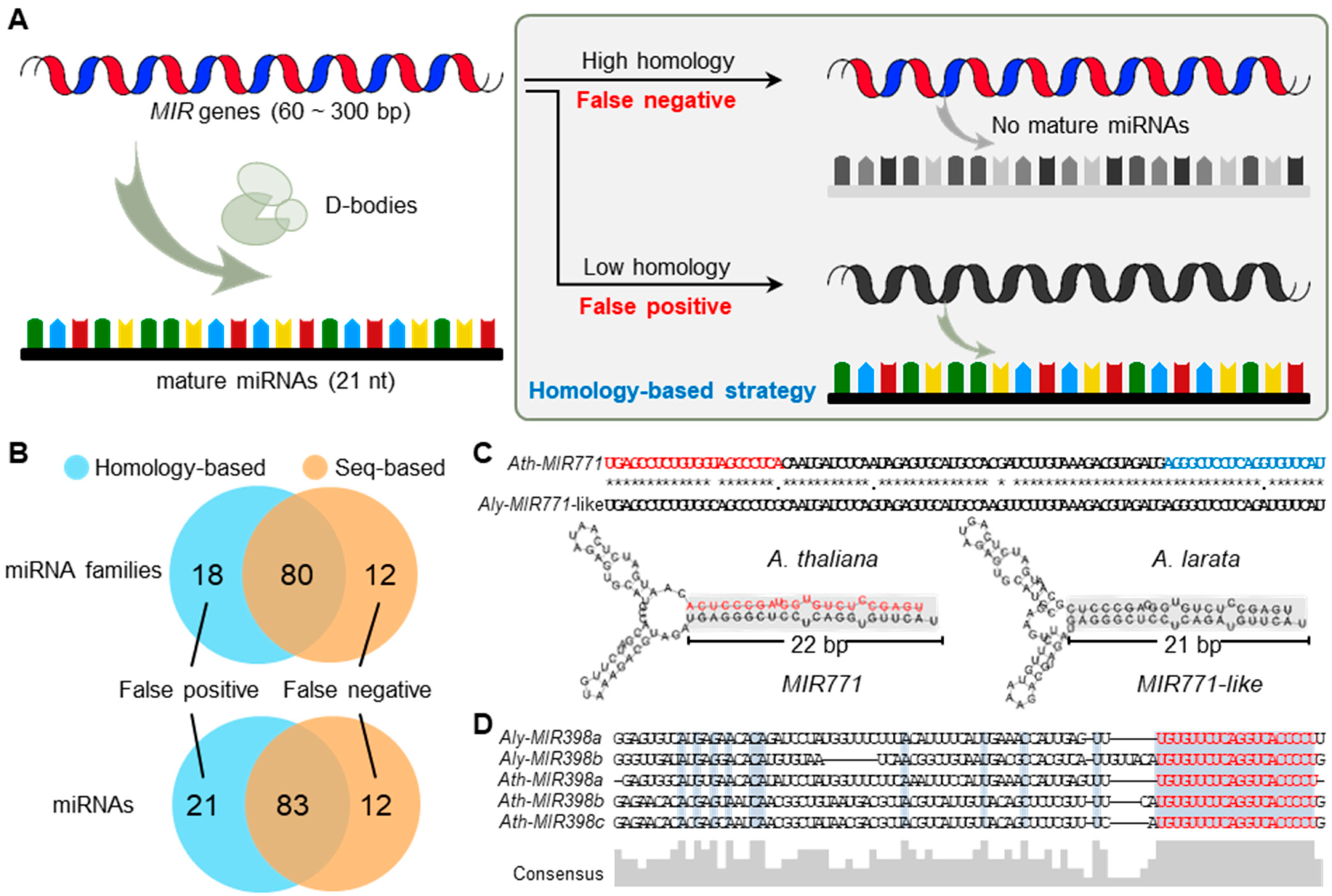
2.1. Comprehensive Comparison of Homology-Based and Seq-Based Strategies
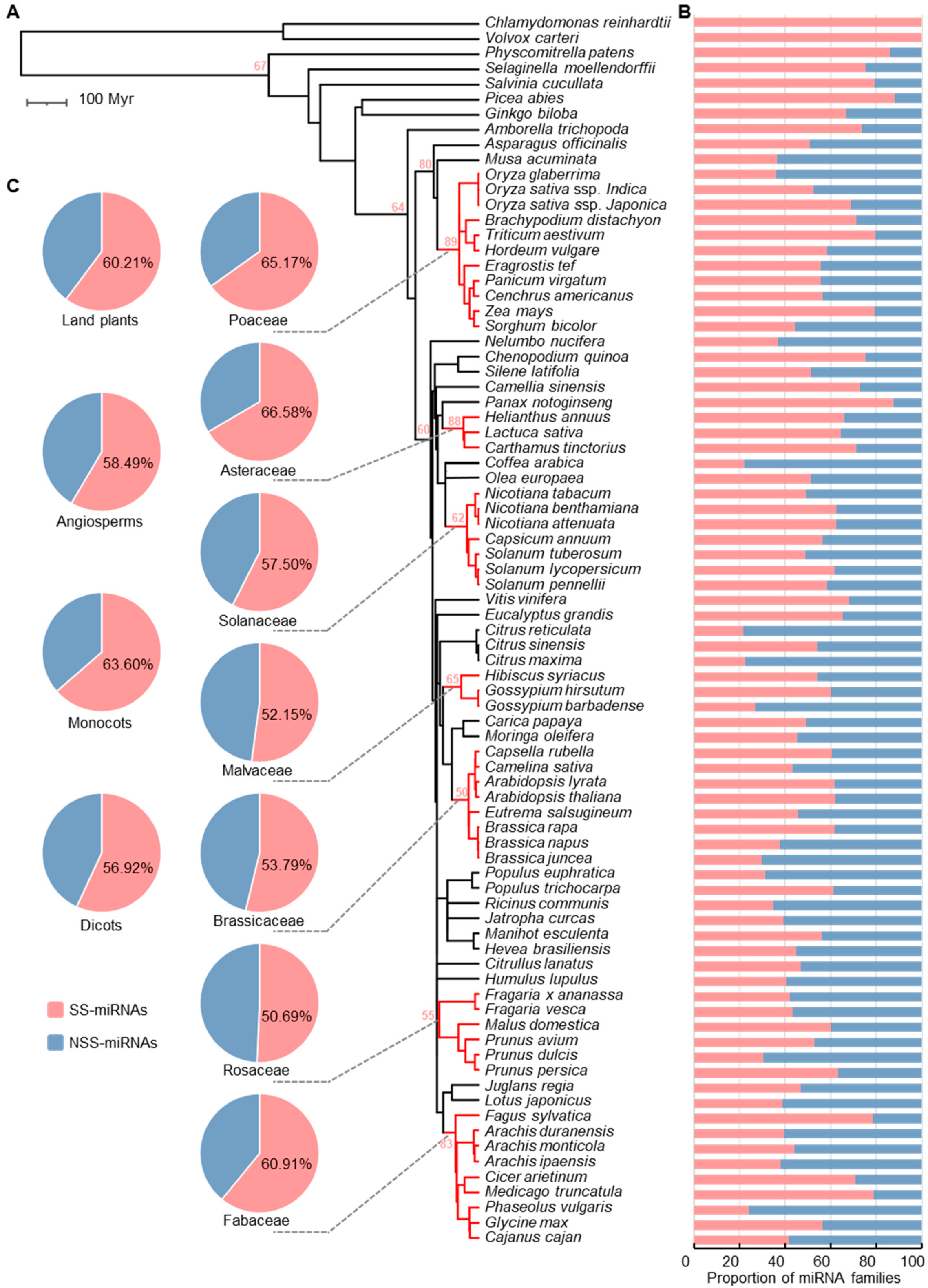
2.2. Identification of SS-miRNAs in 81 Plants
2.3. Diversification of SS-miRNAs Suggests High Turnover of miRNAs

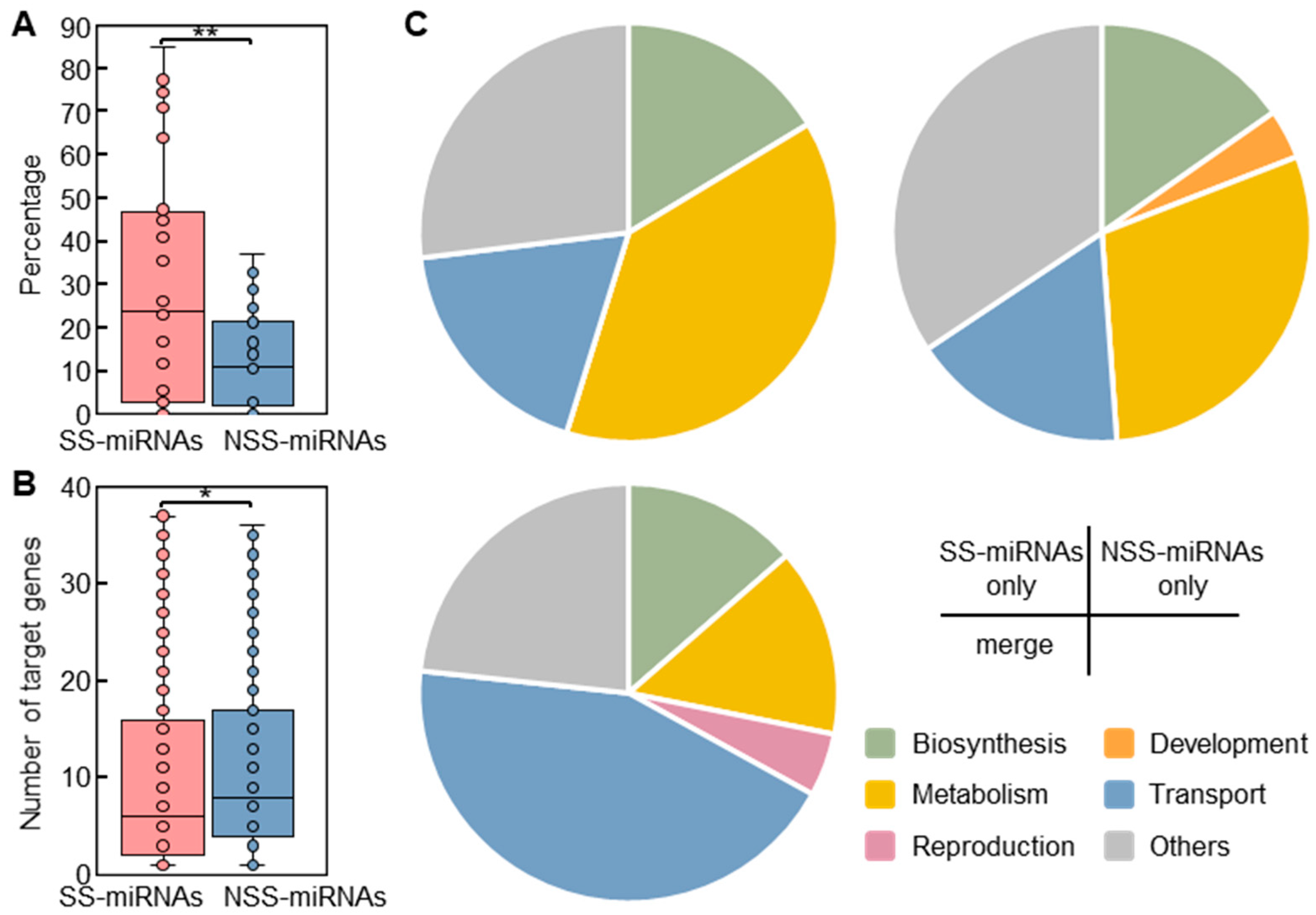
2.4. Intrinsic Features of SS-miRNAs

2.5. SS-miRNAs Lie Close to or within Genes

2.6. Association of SS-miRNA Target Genes with Metabolism

3. Discussion
4. Materials and Methods
4.1. SS-miRNA Identification Using Seq-Based Strategy
4.2. SS-miRNA Identification in A. thaliana and A. lyrata Using Homology-Based Strategy
4.3. XGBoost-Based Machine Learning Model
4.4. Target Gene Prediction
4.5. Relative Distribution of miRNAs
4.6. GO Enrichment Analysis
4.7. Other Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Voinnet, O. Origin, Biogenesis, and Activity of Plant MicroRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef]
- Bologna, N.G.; Voinnet, O. The diversity, biogenesis, and activities of endogenous silencing small RNAs in Arabidopsis. Annu. Rev. Plant Biol. 2014, 65, 473–503. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar] [CrossRef]
- Wu, H.; Li, B.; Iwakawa, H.-O.; Pan, Y.; Tang, X.; Ling-Hu, Q.; Liu, Y.; Sheng, S.; Feng, L.; Zhang, H.; et al. Plant 22-nt siRNAs mediate translational repression and stress adaptation. Nature 2020, 581, 89–93. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan micrornas. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Rogers, K.; Chen, X. Biogenesis, Turnover, and Mode of Action of Plant MicroRNAs. Plant Cell 2013, 25, 2383–2399. [Google Scholar] [CrossRef]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef]
- Baldrich, P.; Beric, A.; Meyers, B.C. Despacito: The slow evolutionary changes in plant microRNAs. Curr. Opin. Plant Biol. 2018, 42, 16–22. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, H.; Li, L. Identification and analysis of the proximal promoters of microRNA genes in Arabidopsis. Genomics 2013, 101, 187–194. [Google Scholar] [CrossRef]
- Jin, X. Regulatory Network of Serine/Arginine-Rich (SR) Proteins: The Molecular Mechanism and Physiological Function in Plants. Int. J. Mol. Sci. 2022, 23, 10147. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, Z.; Minkov, G.; Gisel, A.; Yahubyan, G.; Minkov, I.; Toneva, V.; Baev, V. The Multiverse of Plant Small RNAs: How Can We Explore It? Int. J. Mol. Sci. 2022, 23, 3979. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Moran, Y.; Agron, M.; Praher, D.; Technau, U. The evolutionary origin of plant and animal microRNAs. Nat. Ecol. Evol. 2017, 1, 27. [Google Scholar] [CrossRef]
- Ma, X.; He, K.; Shi, Z.; Li, M.; Li, F.; Chen, X.-X. Large-Scale Annotation and Evolution Analysis of MiRNA in Insects. Genome Biol. Evol. 2021, 13, evab083. [Google Scholar] [CrossRef]
- Guo, Z.; Kuang, Z.; Wang, Y.; Zhao, Y.; Tao, Y.; Cheng, C.; Yang, J.; Lu, X.; Hao, C.; Wang, T.; et al. PmiREN: A comprehensive encyclopedia of plant miRNAs. Nucleic Acids Res. 2019, 48, D1114–D1121. [Google Scholar] [CrossRef]
- Piriyapongsa, J.; Jordan, I.K. A Family of Human MicroRNA Genes from Miniature Inverted-Repeat Transposable Elements. PLoS ONE 2007, 2, e203. [Google Scholar] [CrossRef]
- Kuang, H.; Padmanabhan, C.; Li, F.; Kamei, A.; Bhaskar, P.B.; Ouyang, S.; Jiang, J.; Buell, C.R.; Baker, B. Identification of miniature inverted-repeat transposable elements (MITEs) and biogenesis of their siRNAs in the Solanaceae: New functional implications for MITEs. Genome Res. 2008, 19, 42–56. [Google Scholar] [CrossRef]
- Lu, C.; Chen, J.; Zhang, Y.; Hu, Q.; Su, W.; Kuang, H. Miniature Inverted-Repeat Transposable Elements (MITEs) Have Been Accumulated through Amplification Bursts and Play Important Roles in Gene Expression and Species Diversity in Oryza sativa. Mol. Biol. Evol. 2011, 29, 1005–1017. [Google Scholar] [CrossRef]
- Cui, J.; You, C.; Chen, X. The evolution of microRNAs in plants. Curr. Opin. Plant Biol. 2017, 35, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Piriyapongsa, J.; Jordan, I.K. Dual coding of siRNAs and miRNAs by plant transposable elements. RNA 2008, 14, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Sung, G.-H.; Spatafora, J.W.; Carrington, J. Evolution of microRNA genes by inverted duplication of target gene sequences in Arabidopsis thaliana. Nat. Genet. 2004, 36, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Fridrich, A.; Modepalli, V.; Lewandowska, M.; Aharoni, R.; Moran, Y. Unravelling the developmental and functional significance of an ancient Argonaute duplication. Nat. Commun. 2020, 11, 6187. [Google Scholar] [CrossRef]
- Prodromidou, K.; Matsas, R. Species-Specific miRNAs in Human Brain Development and Disease. Front. Cell. Neurosci. 2019, 13, 559. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Chen, Y.; Zhang, Z.; Zhao, S.; Xie, L.; Zhang, R. A species-specific miRNA participates in biomineralization by targeting CDS regions of Prisilkin-39 and ACCBP in Pinctada fucata. Sci. Rep. 2020, 10, 8971. [Google Scholar] [CrossRef]
- Chen, H.; Wang, H.; Jiang, S.; Xu, J.; Wang, L.; Qiu, L.; Song, L. An oyster species-specific miRNA scaffold42648_5080 modulates haemocyte migration by targeting integrin pathway. Fish Shellfish Immunol. 2016, 57, 160–169. [Google Scholar] [CrossRef]
- Tang, R.; Li, L.; Zhu, D.; Hou, D.; Cao, T.; Gu, H.; Zhang, J.; Chen, J.; Zhang, C.-Y.; Zen, K. Mouse miRNA-709 directly regulates miRNA-15a/16-1 biogenesis at the posttranscriptional level in the nucleus: Evidence for a microRNA hierarchy system. Cell Res. 2011, 22, 504–515. [Google Scholar] [CrossRef]
- Zhang, J.-F.; He, M.-L.; Fu, W.-M.; Wang, H.; Chen, L.-Z.; Zhu, X.; Chen, Y.; Xie, D.; Lai, P.; Chen, G.; et al. Primate-specific microRNA-637 inhibits tumorigenesis in hepatocellular carcinoma by disrupting signal transducer and activator of transcription 3 signaling. Hepatology 2011, 54, 2137–2148. [Google Scholar] [CrossRef]
- Druz, A.; Chu, C.; Majors, B.; Santuary, R.; Betenbaugh, M.; Shiloach, J. A novel microRNA mmu-miR-466h affects apoptosis regulation in mammalian cells. Biotechnol. Bioeng. 2011, 108, 1651–1661. [Google Scholar] [CrossRef]
- Mor, E.; Shomron, N. Species-specific microRNA regulation influences phenotypic variability: Perspectives on species-specific microRNA regulation. Bioessays 2013, 35, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J.; Meyers, B.C. Revisiting Criteria for Plant MicroRNA Annotation in the Era of Big Data. Plant Cell 2018, 30, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Z.; Wang, Y.; Li, L.; Yang, X. miRDeep-P2: Accurate and fast analysis of the microRNA transcriptome in plants. Bioinformatics 2018, 35, 2521–2522. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-M.; Chen, L.-T.; Patel, K.; Li, Y.-H.; Baulcombe, D.C.; Wu, S.-H. 22-nucleotide RNAs trigger secondary siRNA biogenesis in plants. Proc. Natl. Acad. Sci. USA 2010, 107, 15269–15274. [Google Scholar] [CrossRef]
- Li, J.; Song, Q.; Zuo, Z.-F.; Liu, L. MicroRNA398: A Master Regulator of Plant Development and Stress Responses. Int. J. Mol. Sci. 2022, 23, 10803. [Google Scholar] [CrossRef]
- Yang, X.; Li, L. miRDeep-P: A computational tool for analyzing the microRNA transcriptome in plants. Bioinformatics 2011, 27, 2614–2615. [Google Scholar] [CrossRef]
- Guo, Z.; Kuang, Z.; Zhao, Y.; Deng, Y.; He, H.; Wan, M.; Tao, Y.; Wang, D.; Wei, J.; Li, L.; et al. PmiREN2.0: From data annotation to functional exploration of plant microRNAs. Nucleic Acids Res. 2021, 50, D1475–D1482. [Google Scholar] [CrossRef]
- Herron, M.D.; Hackett, J.D.; Aylward, F.O.; Michod, R.E. Triassic origin and early radiation of multicellular volvocine algae. Proc. Natl. Acad. Sci. USA 2009, 106, 3254–3258. [Google Scholar] [CrossRef]
- Zhang, B.; Pan, X.; Cannon, C.H.; Cobb, G.P.; Anderson, T.A. Conservation and divergence of plant microRNA genes. Plant J. 2006, 46, 243–259. [Google Scholar] [CrossRef]
- Rodriguez, A.; Griffiths-Jones, S.; Ashurst, J.L.; Bradley, A. Identification of Mammalian microRNA Host Genes and Transcription Units. Genome Res. 2004, 14, 1902–1910. [Google Scholar] [CrossRef]
- Baskerville, S.; Bartel, D.P. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 2005, 11, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lu, M.; Miao, J.; Li, T.; Wang, E.; Cui, Q. Cepred: Predicting the Co-Expression Patterns of the Human Intronic microRNAs with Their Host Genes. PLoS ONE 2009, 4, e4421. [Google Scholar] [CrossRef]
- Radfar, M.H.; Wong, W.; Morris, Q. Computational Prediction of Intronic microRNA Targets using Host Gene Expression Reveals Novel Regulatory Mechanisms. PLoS ONE 2011, 6, e19312. [Google Scholar] [CrossRef]
- Ramalingam, P.; Palanichamy, J.K.; Singh, A.; Das, P.; Bhagat, M.; Kassab, M.A.; Sinha, S.; Chattopadhyay, P. Biogenesis of intronic miRNAs located in clusters by independent transcription and alternative splicing. RNA 2013, 20, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Qin, Y.; Yang, P.; Du, J.; Kuang, Z.; Zhao, Y.; Wang, Y.; Li, D.; Wei, J.; Guo, X.; et al. Comprehensive Annotation and Functional Exploration of MicroRNAs in Lettuce. Front. Plant Sci. 2021, 12, 781836. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Rajewsky, N. The evolution of gene regulation by transcription factors and microRNAs. Nat. Rev. Genet. 2007, 8, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Fernie, A.R.; Luo, J. Exploring the Diversity of Plant Metabolism. Trends Plant Sci. 2018, 24, 83–98. [Google Scholar] [CrossRef]
- Cronk, Q.C.B. Plant evolution and development in a post-genomic context. Nat. Rev. Genet. 2001, 2, 607–619. [Google Scholar] [CrossRef]
- Wissler, L.; Gadau, J.; Simola, D.F.; Helmkampf, M.; Bornberg-Bauer, E. Mechanisms and Dynamics of Orphan Gene Emergence in Insect Genomes. Genome Biol. Evol. 2013, 5, 439–455. [Google Scholar] [CrossRef]
- Koide, Y.; Ogino, A.; Yoshikawa, T.; Kitashima, Y.; Saito, N.; Kanaoka, Y.; Onishi, K.; Yoshitake, Y.; Tsukiyama, T.; Saito, H.; et al. Lineage-specific gene acquisition or loss is involved in interspecific hybrid sterility in rice. Proc. Natl. Acad. Sci. USA 2018, 115, E1955–E1962. [Google Scholar] [CrossRef]
- Zhang, H.; Guo, Z.; Zhuang, Y.; Suo, Y.; Du, J.; Gao, Z.; Pan, J.; Li, L.; Wang, T.; Xiao, L.; et al. MicroRNA775 regulates intrinsic leaf size and reduces cell wall pectin levels by targeting a galactosyltransferase gene in Arabidopsis. Plant Cell 2021, 33, 581–602. [Google Scholar] [CrossRef] [PubMed]
- de Felippes, F.F.; Schneeberger, K.; Dezulian, T.; Huson, D.H.; Weigel, D. Evolution of Arabidopsis thaliana microRNAs from random sequences. RNA 2008, 14, 2455–2459. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Kuang, Z.; Tao, Y.; Wang, H.; Wan, M.; Hao, C.; Shen, F.; Yang, X.; Li, L. Miniature inverted-repeat transposable elements drive rapid microRNA diversification in angiosperms. Mol. Biol. Evol. 2022, 39, msac224. [Google Scholar] [CrossRef] [PubMed]
- Fattash, I.; Rooke, R.; Wong, A.; Hui, C.; Luu, T.; Bhardwaj, P.; Yang, G. Miniature inverted-repeat transposable elements: Discovery, distribution, and activity. Genome 2013, 56, 475–486. [Google Scholar] [CrossRef]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Guestrin, C. Xgboost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Z.; Kuang, Z.; Deng, Y.; Li, L.; Yang, X. Identification of Species-Specific MicroRNAs Provides Insights into Dynamic Evolution of MicroRNAs in Plants. Int. J. Mol. Sci. 2022, 23, 14273. https://doi.org/10.3390/ijms232214273
Guo Z, Kuang Z, Deng Y, Li L, Yang X. Identification of Species-Specific MicroRNAs Provides Insights into Dynamic Evolution of MicroRNAs in Plants. International Journal of Molecular Sciences. 2022; 23(22):14273. https://doi.org/10.3390/ijms232214273
Chicago/Turabian StyleGuo, Zhonglong, Zheng Kuang, Yang Deng, Lei Li, and Xiaozeng Yang. 2022. "Identification of Species-Specific MicroRNAs Provides Insights into Dynamic Evolution of MicroRNAs in Plants" International Journal of Molecular Sciences 23, no. 22: 14273. https://doi.org/10.3390/ijms232214273
APA StyleGuo, Z., Kuang, Z., Deng, Y., Li, L., & Yang, X. (2022). Identification of Species-Specific MicroRNAs Provides Insights into Dynamic Evolution of MicroRNAs in Plants. International Journal of Molecular Sciences, 23(22), 14273. https://doi.org/10.3390/ijms232214273