
Predicting Dihydropyrimidine Dehydrogenase Deficiency and Related 5-Fluorouracil Toxicity: Opportunities and Challenges of DPYD Exon Sequencing and the Role of Phenotyping Assays

 , , and
, , and
Abstract
1. Introduction
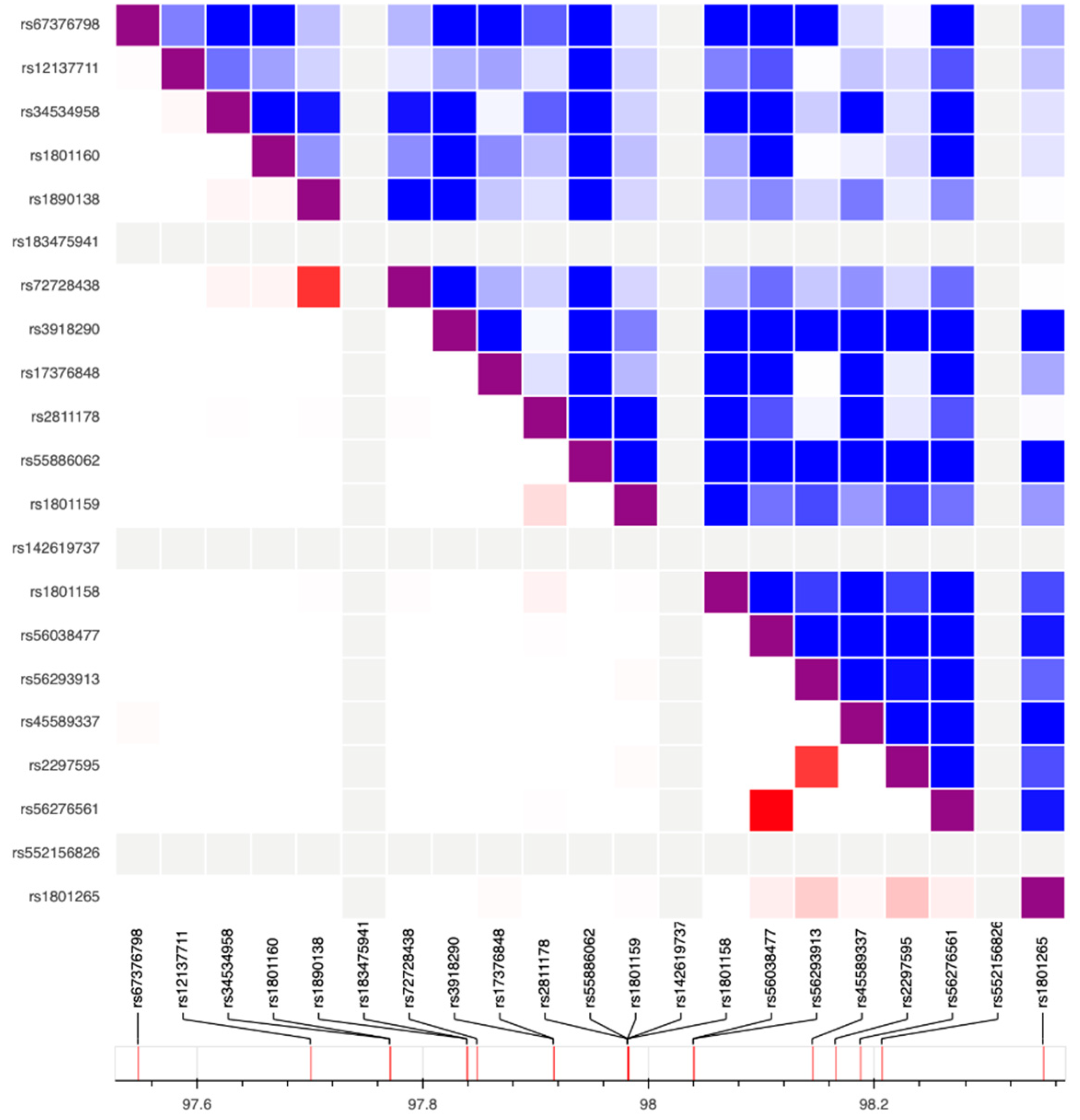
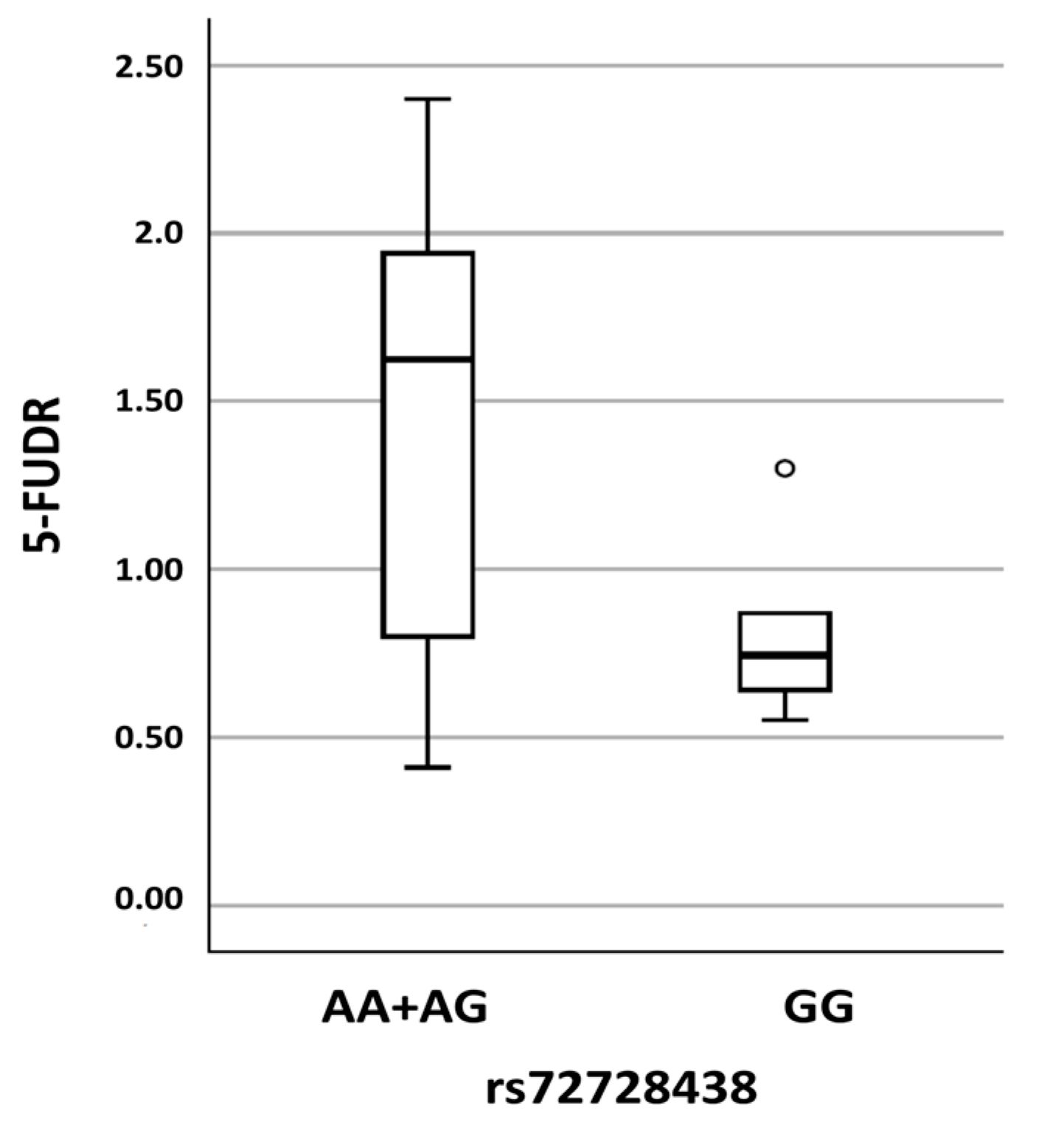
2. Results
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. 5-FU Degradation Rate
4.3. DPYD Exon Sequencing
4.4. In-Silico Prediction of Variants’ Effect
4.5. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meta-Analysis Group In Cancer; Lévy, E.; Piedbois, P.; Buyse, M.; Pignon, J.P.; Rougier, P.; Ryan, L.; Hansen, R.; Zee, B.; Weinerman, B.; et al. Toxicity of fluorouracil in patients with advanced colorectal cancer: Effect of administration schedule and prognostic factors. J. Clin. Oncol. 1998, 16, 3537–3541. [Google Scholar] [PubMed]
- Mikhail, S.E.; Sun, J.F.; Marshall, J.L. Safety of capecitabine: A review. Expert Opin. Drug Saf. 2010, 9, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Gmeiner, W.H. A narrative review of genetic factors affecting fluoropyrimidine toxicity. Precis. Cancer Med. 2021, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Barin-Le Guellec, C.; Lafay-Chebassier, C.; Ingrand, I.; Tournamille, J.F.; Boudet, A.; Lanoue, M.C.; Defossez, G.; Ingrand, P.; Perault-Pochat, M.C.; Etienne-Grimaldi, M.C. Toxicities associated with chemotherapy regimens containing a fluoropyrimidine: A real-life evaluation in France. Eur. J. Cancer 2020, 124, 37–46. [Google Scholar] [CrossRef]
- ClinCalc.com. Evidence-Based Clinical Decision Support Tools and Calculators for Medical Professionals. Available online: https://clincalc.com/DrugStats/Drugs/Fluorouracil (accessed on 3 November 2022).
- Diasio, R.B.; Beavers, T.L.; Carpenter, J.T. Familial deficiency of dihydropyrimidine dehydrogenase. Biochemical basis for familial pyrimidinemia and severe 5- fluorouracil-induced toxicity. J. Clin. Investig. 1988, 81, 47–51. [Google Scholar] [CrossRef]
- Harris, B.E.; Carpenter, J.T.; Diasio, R.B. Severe 5-fluorouracil toxicity secondary to dihydropyrimidine dehydrogenase deficiency. A potentially more common pharmacogenetic syndrome. Cancer 1991, 68, 499–501. [Google Scholar] [CrossRef]
- Takimoto, C.H.; Lu, Z.H.; Zhang, R.; Liang, M.D.; Larson, L.V.; Cantilena, L.R., Jr.; Grem, J.L.; Allegra, C.J.; Diasio, R.B.; Chu, E. Severe neurotoxicity following 5-fluorouracil-based chemotherapy in a patient with dihydropyrimidine dehydrogenase deficiency. Clin. Cancer Res. 1996, 2, 477–481. [Google Scholar]
- Diasio, R.B.; Johnson, M.R. Dihydropyrimidine dehydrogenase: Its role in 5-fluorouracil clinical toxicity and tumor resistance. Clin. Cancer Res. 1999, 5, 2672–2673. [Google Scholar]
- van den Wildenberg, S.; Streng, A.S.; van den Broek, R.; Broeren, M.; Deenen, M.J.; van Dongen, J.; Hanrath, M.A.; Lapré, C.; Brunsveld, L.; Scharnhorst, V.; et al. Quantification of uracil, dihydrouracil, thymine and dihydrothymine for reliable dihydropyrimidine dehydrogenase (DPD) phenotyping critically depend on blood and plasma storage conditions. J. Pharm. Biomed. Anal. 2022, 221, 115027. [Google Scholar] [CrossRef]
- Lostia, A.M.; Lionetto, L.; Ialongo, C.; Gentile, G.; Viterbo, A.; Malaguti, P.; Paris, I.; Marchetti, L.; Marchetti, P.; De Blasi, A.; et al. A liquid chromatography-tandem mass spectrometry method for the determination of 5-Fluorouracil degradation rate by intact peripheral blood mononuclear cells. Drug Monit. 2009, 4, 482–488. [Google Scholar] [CrossRef]
- Hodroj, K.; Barthelemy, D.; Lega, J.C.; Grenet, G.; Gagnieu, M.C.; Walter, T.; Guitton, J.; Payen-Gay, L. Issues and limitations of available biomarkers for fluoropyrimidine-based chemotherapy toxicity, a narrative review of the literature. ESMO Open 2021, 6, 100125. [Google Scholar] [CrossRef] [PubMed]
- Laures, N.; Konecki, C.; Brugel, M.; Giffard, A.L.; Abdelli, N.; Botsen, D.; Carlier, C.; Gozalo, C.; Feliu, C.; Slimano, F.; et al. Impact of Guidelines Regarding Dihydropyrimidine Dehydrogenase (DPD) Deficiency Screening Using Uracil-Based Phenotyping on the Reduction of Severe Side Effect of 5-Fluorouracil-Based Chemotherapy: A Propension Score Analysis. Pharmaceutics 2022, 14, 2119. [Google Scholar] [CrossRef] [PubMed]
- Diasio, R.B.; Offer, S.M. Testing for Dihydropyrimidine Dehydrogenase Deficiency to Individualize 5-Fluorouracil Therapy. Cancers 2022, 14, 3207. [Google Scholar] [CrossRef] [PubMed]
- Vreken, P.; Van Kuilenburg, A.B.; Meinsma, R.; Smit, G.P.; Bakker, H.D.; De Abreu, R.A.; van Gennip, A.H. A point mutation in an invariant splice donor site leads to exon skipping in two unrelated Dutch patients with dihydropyrimidine dehydrogenase deficiency. J. Inherit. Metab. Dis. 1996, 19, 645–654. [Google Scholar] [CrossRef]
- Wei, X.; McLeod, H.L.; McMurrough, J.; Gonzalez, F.J.; Fernandez-Salguero, P. Molecular basis of the human dihydropyrimidine dehydrogenase deficiency and 5-fluorouracil toxicity. J. Clin. Investig. 1996, 98, 610–615. [Google Scholar] [CrossRef]
- Lazar, A.; Mau-Holzmann, U.A.; Kolb, H.; Reichenmiller, H.E.; Riess, O.; Schömig, E. Multiple organ failure due to 5-fluorouracil chemotherapy in a patient with a rare dihydropyrimidine dehydrogenase gene variant. Onkologie 2004, 27, 559–562. [Google Scholar] [CrossRef]
- White, C.; Scott, R.J.; Paul, C.; Ziolkowski, A.; Mossman, D.; Fox, S.B.; Michael, M.; Ackland, S. Dihydropyrimidine Dehydrogenase Deficiency and Implementation of Upfront DPYD Genotyping. Clin. Pharmacol. Ther. 2022, 112, 791–802. [Google Scholar] [CrossRef]
- Sharma, B.B.; Rai, K.; Blunt, H.; Zhao, W.; Tosteson, T.D.; Brooks, G.A. Pathogenic DPYD Variants and Treatment-Related Mortality in Patients Receiving Fluoropyrimidine Chemotherapy: A Systematic Review and Meta-Analysis. Oncologist 2021, 26, 1008–1016. [Google Scholar] [CrossRef]
- Glewis, S.; Alexander, M.; Khabib, M.; Brennan, A.; Lazarakis, S.; Martin, J.; Tie, J.; Lingaratnam, S.; Michael, M. A systematic review and meta-analysis of toxicity and treatment outcomes with pharmacogenetic-guided dosing compared to standard of care BSA-based fluoropyrimidine dosing. Br. J. Cancer 2022, 127, 126–136. [Google Scholar] [CrossRef]
- Global Variome Shared LOVD DPYD (Dihydropyrimidine Dehydrogenase). Available online: https://databases.lovd.nl/shared/variants/DPYD?search_var_status=%3D%22Marked%22%7C%3D%22Public%22 (accessed on 3 November 2022).
- Johnson, M.R.; Wang, K.; Diasio, R.B. Profound dihydropyrimidine dehydrogenase deficiency resulting from a novel compound heterozygote genotype. Clin. Cancer Res. 2002, 8, 768–774. [Google Scholar]
- Offer, S.M.; Wegner, N.J.; Fossum, C.; Wang, K.; Diasio, R.B. Phenotypic profiling of DPYD variations relevant to 5-fluorouracil sensitivity using real-time cellular analysis and in vitro measurement of enzyme activity. Cancer Res. 2013, 73, 1958–1968. [Google Scholar] [CrossRef]
- Lee, A.; Shi, Q.; Pavey, E.S.; Sargent, D.J.; Alberts, S.R.; Sinicrope, F.A.; Berenberg, J.; Goldberg, R.M.; Diasio, R.B. Validation of DPYD variants DPYD*2A, I560S, and D949V as predictors of 5-fluorouracil (5-FU)-related toxicity in stage III colon cancer (CC) patients from adjuvant trial NCCTG N0147. J. Clin. Oncol. 2013, 31, abstr3510. [Google Scholar] [CrossRef]
- van Kuilenburg, A.B.; Meijer, J.; Mul, A.N.; Meinsma, R.; Schmid, V.; Dobritzsch, D.; Hennekam, R.C.; Mannens, M.M.; Kiechle, M.; Etienne-Grimaldi, M.C.; et al. Intragenic deletions and a deep intronic mutation affecting pre-mRNA splicing in the dihydropyrimidine dehydrogenase gene as novel mechanisms causing 5-fluorouracil toxicity. Hum. Genet. 2010, 128, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Meulendijks, D.; Henricks, L.M.; Sonke, G.S.; Deenen, M.J.; Froehlich, T.K.; Amstutz, U.; Largiadèr, C.R.; Jennings, B.A.; Marinaki, A.M.; Sanderson, J.D.; et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: A systematic review and meta-analysis of individual patient data. Lancet Oncol. 2015, 16, 1639–1650. [Google Scholar] [CrossRef]
- Amstutz, U.; Henricks, L.M.; Offer, S.M.; Barbarino, J.; Schellens, J.; Swen, J.J.; Klein, T.E.; McLeod, H.L.; Caudle, K.E.; Diasio, R.B.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing: 2017 Update. Clin. Pharmacol. Ther. 2018, 103, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Lunenburg, C.; van der Wouden, C.H.; Nijenhuis, M.; Crommentuijn-van Rhenen, M.H.; de Boer-Veger, N.J.; Buunk, A.M.; Houwink, E.; Mulder, H.; Rongen, G.A.; van Schaik, R.; et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction of DPYD and fluoropyrimidines. Eur. J. Hum. Genet. EJHG 2020, 28, 508–517. [Google Scholar] [CrossRef] [PubMed]
- European MedicinesAgency EMA. Press Release: EMA Recommendations on DPD Testing Prior to Treatment with Fluorouracil, Capecitabine, Tegafur and Flucytosine. 2020. Available online: https://www.ema.europa.eu/en/news/ema-recommendations-dpd-testingprior-treatment-luorouracil-capecitabine-tegafur-flucytosine (accessed on 24 February 2022).
- Hamzic, S.; Aebi, S.; Joerger, M.; Montemurro, M.; Ansari, M.; Amstutz, U.; Largiadèr, C. Fluoropyrimidine chemotherapy: Recommendations for DPYD genotyping and therapeutic drug monitoring of the Swiss Group of Pharmacogenomics and Personalised Therapy. Swiss Med. Wkly. 2020, 150, w20375. [Google Scholar] [CrossRef]
- Collie-Duguid, E.S.; Etienne, M.C.; Milano, G.; McLeod, H.L. Known variant DPYD alleles do not explain DPD deficiency in cancer patients. Pharmacogenetics 2000, 10, 217–223. [Google Scholar] [CrossRef]
- Borro, M.; Botticelli, A.; Mazzuca, F.; Onesti, E.C.; Gentile, G.; Romiti, A.; Cerbelli, B.; Mazzotti, E.; Marchetti, L.; Lionetto, L.; et al. Pre-treatment assay of 5-fluorouracil degradation rate (5-FUDR) to improve prediction of 5-fluorouracil toxicity in gastro-esophageal cancer. Oncotarget 2017, 8, 14050–14057. [Google Scholar] [CrossRef][Green Version]
- Mazzuca, F.; Borro, M.; Botticelli, A.; Mazzotti, E.; Marchetti, L.; Gentile, G.; La Torre, M.; Lionetto, L.; Simmaco, M.; Marchetti, P. Pre-treatment evaluation of 5-fluorouracil degradation rate: Association of poor and ultra-rapid metabolism with severe toxicity in a colorectal cancer patients cohort. Oncotarget 2016, 7, 20612–20620. [Google Scholar] [CrossRef]
- De Mattia, E.; Silvestri, M.; Polesel, J.; Ecca, F.; Mezzalira, S.; Scarabel, L.; Zhou, Y.; Roncato, R.; Lauschke, V.M.; Calza, S.; et al. Rare genetic variant burden in DPYD predicts severe fluoropyrimidine-related toxicity risk. Biomed Pharm. 2022, 154, 113644. [Google Scholar] [CrossRef] [PubMed]
- Bendl, J.; Stourac, J.; Salanda, O.; Pavelka, A.; Wieben, E.D.; Zendulka, J.; Brezovsky, J.; Damborsky, J. PredictSNP: Robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol. 2014, 10, e1003440. [Google Scholar] [CrossRef] [PubMed]
- Botticelli, A.; Onesti, C.E.; Strigari, L.; Occhipinti, M.; Di Pietro, F.R.; Cerbelli, B.; Petremolo, A.; Anselmi, E.; Macrini, S.; Roberto, M.; et al. A nomogram to predict 5-fluorouracil toxicity: When pharmacogenomics meets the patient. Anticancer Drugs 2017, 28, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Onesti, C.E.; Botticelli, A.; La Torre, M.; Borro, M.; Gentile, G.; Romiti, A.; Lionetto, L.; Petremolo, A.; Occhipinti, M.; Roberto, M.; et al. 5-Fluorouracil degradation rate could predict toxicity in stages II-III colorectal cancer patients undergoing adjuvant FOLFOX. Anticancer Drugs 2017, 28, 322–326. [Google Scholar] [CrossRef]
- Botticelli, A.; Borro, M.; Onesti, C.E.; Strigari, L.; Gentile, G.; Cerbelli, B.; Romiti, A.; Occhipinti, M.; Sebastiani, C.; Lionetto, L.; et al. Degradation Rate of 5-Fluorouracil in Metastatic Colorectal Cancer: A New Predictive Outcome Biomarker? PLoS ONE 2016, 11, e0163105. [Google Scholar] [CrossRef]
- Gentile, G.; Botticelli, A.; Lionetto, L.; Mazzuca, F.; Simmaco, M.; Marchetti, P.; Borro, M. Genotype-phenotype correlations in 5-fluorouracil metabolism: A candidate DPYD haplotype to improve toxicity prediction. Pharm. J. 2016, 16, 320–325. [Google Scholar] [CrossRef]
- Seck, K.; Riemer, S.; Kates, R.; Ullrich, T.; Lutz, V.; Harbeck, N.; Schmitt, M.; Kiechle, M.; Diasio, R.; Gross, E. Analysis of the DPYD gene implicated in 5-fluorouracil catabolism in a cohort of Caucasian individuals. Clin. Cancer Res. 2005, 11, 5886–5892. [Google Scholar] [CrossRef]
- Kuilenburg, A.B.P.V.; Meijer, J.; Tanck, M.W.T.; Dobritzsch, D.; Zoetekouw, L.; Dekkers, L.L.; Roelofsen, J.; Meinsma, R.; Wymenga, M.; Kulik, W.; et al. Phenotypic and clinical implications of variants in the dihydropyrimidine dehydrogenase gene. Biochim. Biophys. Acta 2016, 1862, 754–762. [Google Scholar] [CrossRef]
- Etheridge, A.S.; Gallins, P.J.; Jima, D.; Broadaway, K.A.; Ratain, M.J.; Schuetz, E.; Schadt, E.; Schroder, A.; Molony, C.; Zhou, Y.; et al. A New Liver Expression Quantitative Trait Locus Map From 1,183 Individuals Provides Evidence for Novel Expression Quantitative Trait Loci of Drug Response, Metabolic, and Sex-Biased Phenotypes. Clin. Pharmacol. Ther. 2020, 107, 1383–1393. [Google Scholar] [CrossRef]
- van Staveren, M.C.; van Kuilenburg, A.B.; Guchelaar, H.J.; Meijer, J.; Punt, C.J.; de Jong, R.S.; Gelderblom, H.; Maring, J.G. Evaluation of an oral uracil loading test to identify DPD-deficient patients using a limited sampling strategy. Br. J. Clin. Pharm. 2016, 81, 553–561. [Google Scholar] [CrossRef]
- van Staveren, M.C.; Theeuwes-Oonk, B.; Guchelaar, H.J.; van Kuilenburg, A.B.; Maring, J.G. Pharmacokinetics of orally administered uracil in healthy volunteers and in DPD-deficient patients, a possible tool for screening of DPD deficiency. Cancer Chemother. Pharm. 2011, 68, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- Offer, S.M.; Fossum, C.C.; Wegner, N.J.; Stuflesser, A.J.; Butterfield, G.L.; Diasio, R.B. Comparative functional analysis of DPYD variants of potential clinical relevance to dihydropyrimidine dehydrogenase activity. Cancer Res. 2014, 74, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Hishinuma, E.; Narita, Y.; Saito, S.; Maekawa, M.; Akai, F.; Nakanishi, Y.; Yasuda, J.; Nagasaki, M.; Yamamoto, M.; Yamaguchi, H.; et al. Functional Characterization of 21 Allelic Variants of Dihydropyrimidine Dehydrogenase Identified in 1070 Japanese Individuals. Drug Metab. Dispos. 2018, 6, 1083–1090. [Google Scholar] [CrossRef]
- SNPStats Software. Available online: http://bioinfo.iconcologia.net/SNPstats (accessed on 9 September 2022).
- Solé, X.; Guinó, E.; Valls, J.; Iniesta, R.; Moreno, V. SNPStats: A web tool for the analysis of association studies. Bioinformatics 2006, 22, 1928–1929. [Google Scholar] [CrossRef]
- Machiela, M.J.; Chanock, S.J. LDlink: A web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 2015, 31, 3555–3557. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
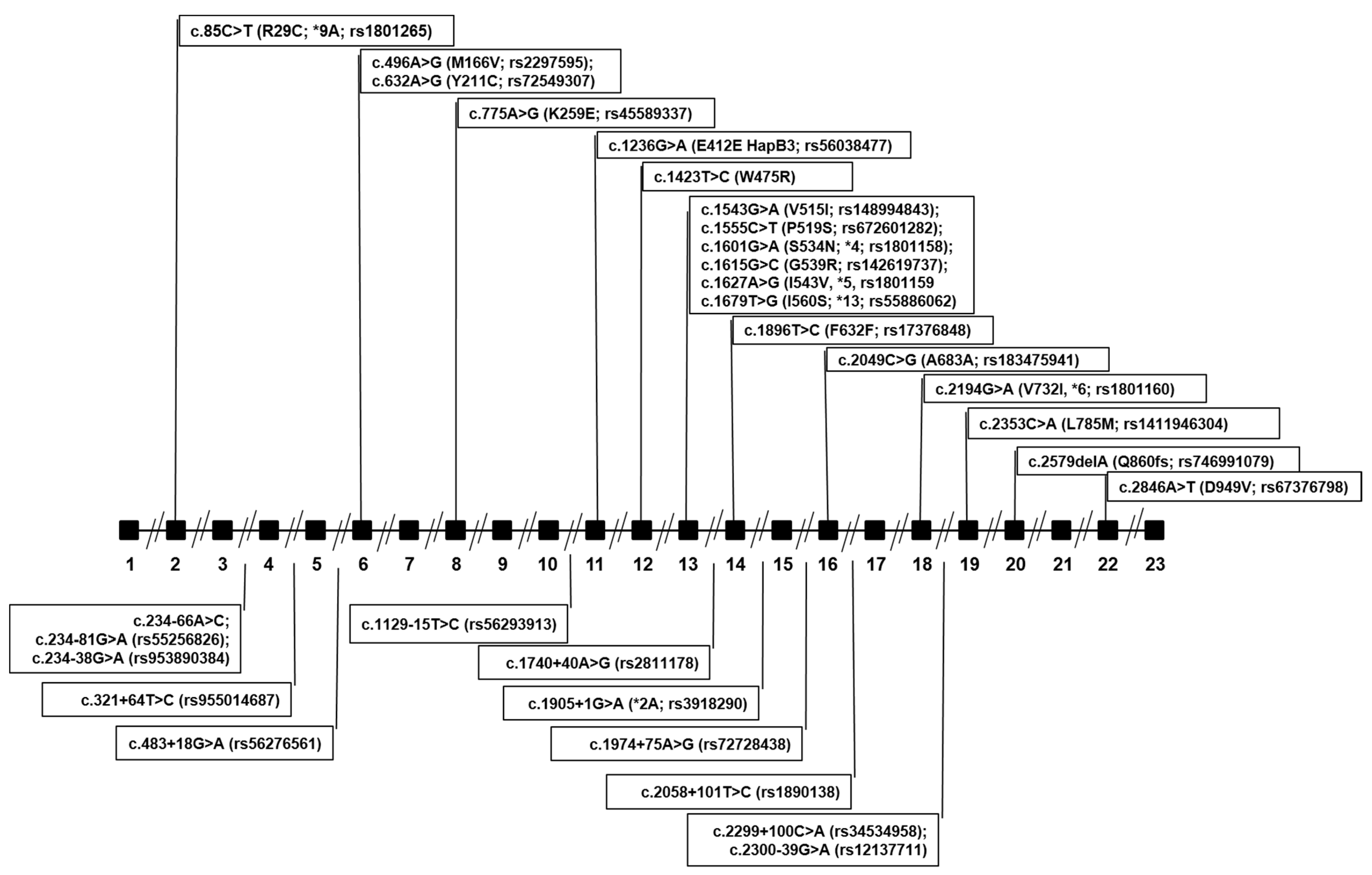
| DPYD Variants Identified in Both the NM Group and the PM Group | |||||||
|---|---|---|---|---|---|---|---|
| Locus (hg19) | Nomenclature | dbSNP rsID | Genotype | Frequencies | Notes * | ||
| NM (n, %) | PM (n, %) | Observed MAF | |||||
| chr1: 98348885 | c.85C>T, R29C, *9A | rs1801265 | TT TC CC | 30 (56) 19 (35) 5 (9) | 22 (55) 15 (38) 3 (8) | 0.266 | CPIC/DPWG: Fully functional |
| chr1: 98206101 | c.234-66A>C | Not available | AA AC | 52 (96) 2 (4) | 39 (98) 1 (2) | 0.016 | In-silico prediction: No consequences on splicing |
| chr1: 98187048 | c.483+18G>A | rs56276561 | GG GA | 53(98) 1(2) | 36 (90) 4 (10) | 0.026 | In-silico prediction: No consequences on splicing |
| chr1: 98165091 | c.496A>GM166V | rs2297595 | AA AG GG | 41 (76) 10 (19) 3 (6) | 25 (62) 14 (35) 1 (2) | 0.170 | CPIC/DPWG: normal function |
| chr1: 98039541 | c.1129-15T>C | rs56293913 | TT TC CC | 39 (72) 13 (24) 2 (4) | 26 (65) 13 (32) 1(2) | 0.170 | In-silico prediction: No consequences on splicing |
| chr1: 98039419 | c.1236G>A, E412E HapB3 | rs56038477 | GG GA | 53 (98) 1 (2) | 36(90) 4 (10) | 0.026 | CPIC/DPWG: reduced function |
| chr1: 97981421 | c.1601G>A, S534N, *4 | rs1801158 | GG GA | 52 (96) 2 (4) | 36 (90) 4 (10) | 0.032 | Insufficient evidence due to contrasting results |
| chr1: 97981395 | c.1627A>G, I543V, *5 | rs1801159 | AA AG GG | 40 (74) 14 (26) 0 (0) | 31 (78) 8 (20) 1 (2) | 0.127 | CPIC/DPWG: fully functional |
| chr1: 97981242 | c.1740+40A>G | rs2811178 | AA AG GG | 12 (22) 28 (52) 14 (26) | 8 (20) 20 (50) 12 (30) | 0.468 | In-silico prediction: No consequences on splicing |
| chr1: 97981242–97981243 | c.1740+39_1740+40 | rs796315813 (MNV) | AC/AC AC/GC GC/GC AC/GT GT/GC | 11 (20) 21 (39) 8 (15) 8 (15) 6 (11) 0 (0) | 8 (20) 17 (42) 7 (18) 3 (8) 4 (10) 1 (2) | 0.271 | In-silico prediction: No consequences on splicing |
| chr1: 97915624 | c.1896T>C, F632F | rs17376848 | T/T T/C | 50 (93) 4 (7) | 39 (98) 1 (2) | 0.026 | Synonymous |
| chr1: 97847874 | c.1974+75A>G, p.? | rs72728438 | AA AG GG | 33 (61) 20 (37) 1 (2) | 20 (50) 15 (38) 5 (12) | 0.250 | In-silico prediction: No consequences on splicing |
| chr1: 97770920 | c.2194G>A, V732I, *6 | rs1801160 | GG GA AA | 44 (81) 10 (19) 0 (0) | 31 (78) 8 (20) 1 (2) | 0.106 | CPIC/DPWG: insufficient evidence (contrasting results) |
| DPYD Variants Identified Exclusively in the PM Group | |||||||
| Locus hg19 | Nomenclature | dbSNP rsID | Genotype | Frequencies | Notes | ||
| NM (n, %) | PM (n, %) | Observed MAF | |||||
| chr1: 98206116 | c.234-81G>A | rs552156826 | G/G G/A | 54 (100) | 39 (98) 1 (2) | 0.005 | In-silico prediction: No consequences on splicing |
| chr1: 98164955 | c.632A>G, Y211C | rs72549307 | AA AG | 54 (100) | 39 (98) 1 (2) | 0.005 | In-silico prediction: Deleterious |
| chr1: 98144726 | c.775A>G, K259E | rs45589337 | AA AG | 54 (100) | 38 (95) 2 (5) | 0.011 | In-silico prediction: Deleterious |
| chr1: 98015217 | c.1423T>C, W475R | Not available | TT TC | 54 (100) | 39(98) 1(2) | 0.005 | In-silico prediction: Non-deleterious |
| chr1: 97981479 | c.1543G>A, V515I | rs148994843 | GG GA | 54 (100) | 38(95) 2(5) | 0.011 | In-silico prediction: Non-deleterious |
| chr1: 97981467 | c.1555C>T, P519S | rs672601282 | CC CT | 54 (100) | 39 (98) 1 (2) | 0.005 | In-silico prediction: Deleterious |
| chr1: 97981407 | c.1615G>C, G539R | rs142619737 | G/G G/C | 54 (100) | 39 (98) 1 (2) | 0.005 | In-silico prediction: Deleterious |
| chr1: 97981343 | c.1679T>G, I560S, *13 | rs55886062 | TT TG GG | 54 (100) 0 (0) | 38 (95) 1 (2) 1 (2) | 0.016 | CPIC/DPWG: no function |
| chr1: 97915614 | c.1905+1G>A, *2A | rs3918290 | G/G G/A | 54 (100) | 39 (98) 1 (2) | 0.005 | CPIC/DPWG: no function |
| chr1: 97658667 | c.2579delA, Q860fs | rs746991079 | A/A A/DEL | 54 (100) | 39(98) 1(2) | 0.005 | Frameshift causing stop codon and termination |
| chr1: 97547947 | c.2846A>T, D949V | rs67376798 | AA AT | 54 (100) | 38 (95) 2 (5) | 0.011 | CPIC/DPWG: reduced function |
| DPYD Variants Identified Exclusively in the NM Group | |||||||
| Locus hg19 | Nomenclature | dbSNP rsID | Genotype | Frequencies | Notes | ||
| NM (n, %) | PM (n, %) | Observed MAF | |||||
| chr1: 98206173 | c.234-138G>A | rs953890384 | G/G G/A | 53 (98) 1 (2) | 40 (100) | 0.005 | In-silico prediction: alteration of auxiliary splicing sequences |
| chr1: 98205884 | c.321+64T>C | rs955014687 | TT TC | 53 (98) 1 (2) | 40 (100) | 0.005 | In-silico prediction: No consequences on splicing |
| chr1: 97839126 | c.2049C>G, A683A | rs183475941 | G/G G/C | 52 (96) 2 (4) | 40 (100) | 0.011 | Synonymous |
| chr1: 97839016 | c.2058+101T>C | rs1890138 | T/T T/C | 50 (93) 4 (7) | 40 (100) | 0.021 | In-silico prediction: No consequences on splicing |
| chr1: 97770715 | c.2299+100C>A | rs34534958 | C/C C/A | 53 (98) 1 (2) | 40 (100) | 0.005 | In-silico prediction: No consequences on splicing |
| chr1: 97700589 | c.2300-39G>A | rs12137711 | G/G G/A | 53 (98) 1 (2) | 40 (100) | 0.005 | In-silico prediction: activation of a cryptic Donor site |
| chr1: 97700497 | c.2353C>A, L785M | rs1411946304 | C/C C/A | 53 (98) 1 (2) | 40 (100) | 0.005 | In-silico prediction: Non deleterious |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Luca, O.; Salerno, G.; De Bernardini, D.; Torre, M.S.; Simmaco, M.; Lionetto, L.; Gentile, G.; Borro, M. Predicting Dihydropyrimidine Dehydrogenase Deficiency and Related 5-Fluorouracil Toxicity: Opportunities and Challenges of DPYD Exon Sequencing and the Role of Phenotyping Assays. Int. J. Mol. Sci. 2022, 23, 13923. https://doi.org/10.3390/ijms232213923
De Luca O, Salerno G, De Bernardini D, Torre MS, Simmaco M, Lionetto L, Gentile G, Borro M. Predicting Dihydropyrimidine Dehydrogenase Deficiency and Related 5-Fluorouracil Toxicity: Opportunities and Challenges of DPYD Exon Sequencing and the Role of Phenotyping Assays. International Journal of Molecular Sciences. 2022; 23(22):13923. https://doi.org/10.3390/ijms232213923
Chicago/Turabian StyleDe Luca, Ottavia, Gerardo Salerno, Donatella De Bernardini, Maria Simona Torre, Maurizio Simmaco, Luana Lionetto, Giovanna Gentile, and Marina Borro. 2022. "Predicting Dihydropyrimidine Dehydrogenase Deficiency and Related 5-Fluorouracil Toxicity: Opportunities and Challenges of DPYD Exon Sequencing and the Role of Phenotyping Assays" International Journal of Molecular Sciences 23, no. 22: 13923. https://doi.org/10.3390/ijms232213923
APA StyleDe Luca, O., Salerno, G., De Bernardini, D., Torre, M. S., Simmaco, M., Lionetto, L., Gentile, G., & Borro, M. (2022). Predicting Dihydropyrimidine Dehydrogenase Deficiency and Related 5-Fluorouracil Toxicity: Opportunities and Challenges of DPYD Exon Sequencing and the Role of Phenotyping Assays. International Journal of Molecular Sciences, 23(22), 13923. https://doi.org/10.3390/ijms232213923