



MET Signaling Pathways, Resistance Mechanisms, and Opportunities for Target Therapies



Abstract
1. Introduction
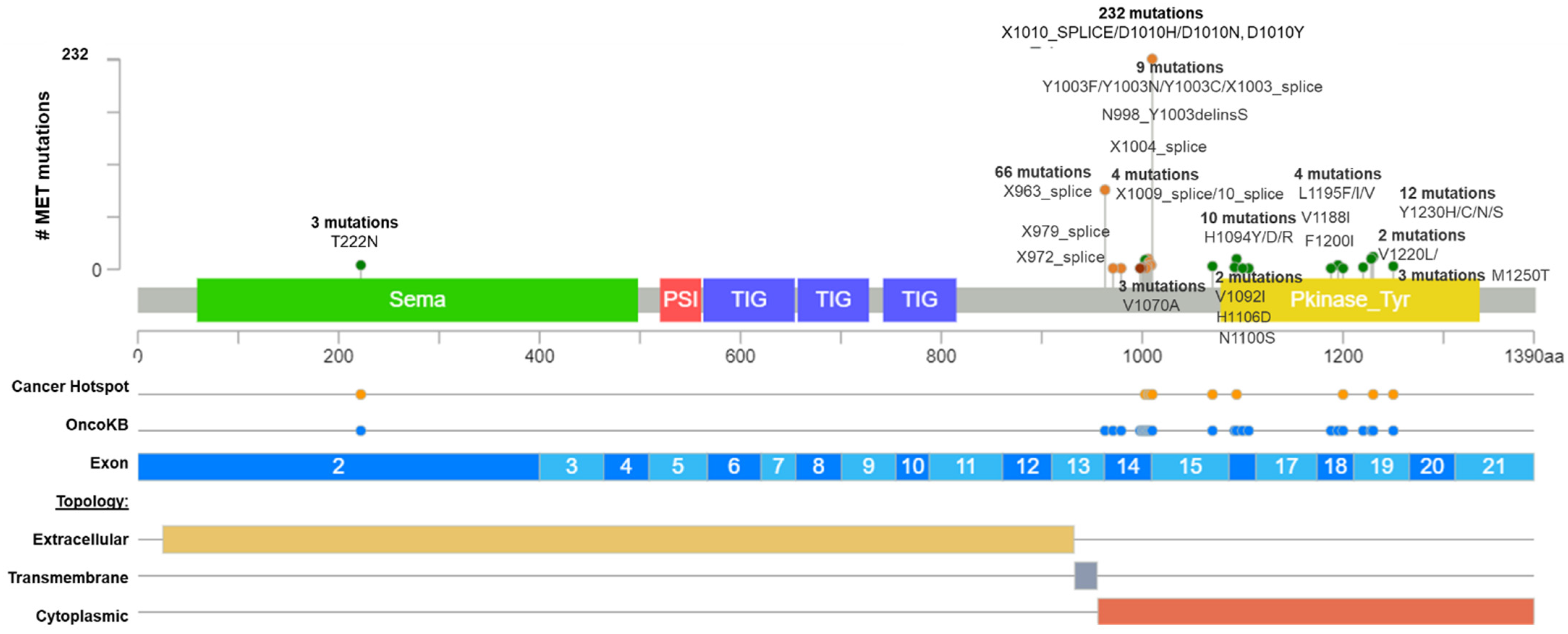
2. MET in Cancer Initiation and Driver Mutations
2.1. MET Amplification
2.2. MET Exon 14 Skipping Alterations
3. Actionable MET Mutations and MET-Target Therapies
3.1. Type I MET-Targeting Drugs Directed to Actionable Mutations
3.1.1. Crizotinib
3.1.2. Tepotinib
3.1.3. Capmatinib
3.1.4. Savolitinib
3.2. MET Alterations and Treatment in the Context of Mestastasic NSCLC
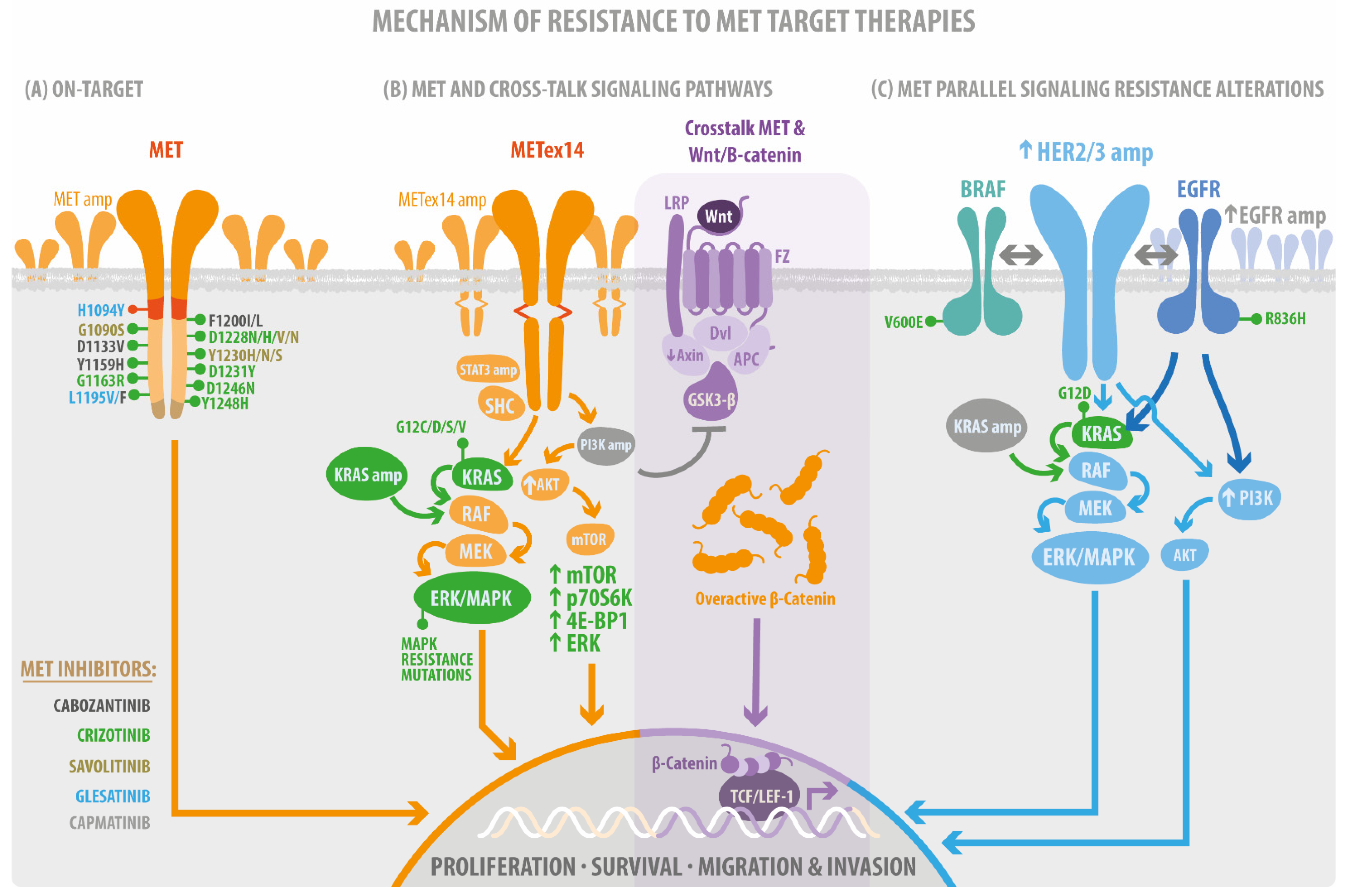
4. Resistance Mutations as a Bypass of MET-Target Therapies
4.1. Acquisition of On-Target Mutations Resistance to MET TKIs
4.2. MET Parallel Signaling Resistance Alteration
4.3. Resistance Mutation on MET Signaling Pathways
4.3.1. MET/RAS/MAPK, the Proliferative and Survival Signaling
4.3.2. MET/PI3K/AKT/mTOR Cellular Motility and Invasion Signaling
4.3.3. MET/Src/Fak Mechanosensing Signaling
4.3.4. MET/Wnt/β-Catenin
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.S.; Park, M.; Blair, D.G.; Tainsky, M.A.; Huebner, K.; Croce, C.M.; Vande Woude, G.F. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984, 311, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ferrell, L.D.; Faouzi, S.; Maher, J.J.; Bishop, J.M. Activation of the Met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J. Cell Biol. 2001, 153, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Tward, A.D.; Jones, K.D.; Yant, S.; Cheung, S.T.; Fan, S.T.; Chen, X.; Kay, M.A.; Wang, R.; Bishop, J.M. Distinct pathways of genomic progression to benign and malignant tumors of the liver. Proc. Natl. Acad. Sci. USA 2007, 104, 14771–14776. [Google Scholar] [CrossRef] [PubMed]
- Mi, J.; Hooker, E.; Balog, S.; Zeng, H.; Johnson, D.T.; He, Y.; Yu, E.J.; Wu, H.; Le, V.; Lee, D.H.; et al. Activation of hepatocyte growth factor/MET signaling initiates oncogenic transformation and enhances tumor aggressiveness in the murine prostate. J. Biol. Chem. 2018, 293, 20123–20136. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, E.; Maeshima, A.; Nakajima, T.; Nakamura, T. Expression of c-met/HGF receptor in human non-small cell lung carcinomas in vitro and in vivo and its prognostic significance. Jpn. J. Cancer Res. 1996, 87, 1063–1069. [Google Scholar] [CrossRef]
- Siegfried, J.M.; Weissfeld, L.A.; Luketich, J.D.; Weyant, R.J.; Gubish, C.T.; Landreneau, R.J. The clinical significance of hepatocyte growth factor for non-small cell lung cancer. Ann. Thorac. Surg. 1998, 66, 1915–1918. [Google Scholar] [CrossRef]
- Overbeck, T.R.; Cron, D.A.; Schmitz, K.; Rittmeyer, A.; Korber, W.; Hugo, S.; Schnalke, J.; Lukat, L.; Hugo, T.; Hinterthaner, M.; et al. Top-level MET gene copy number gain defines a subtype of poorly differentiated pulmonary adenocarcinomas with poor prognosis. Transl. Lung Cancer Res. 2020, 9, 603–616. [Google Scholar] [CrossRef]
- Yin, W.; Cheng, J.; Tang, Z.; Toruner, G.; Hu, S.; Guo, M.; Robinson, M.; Medeiros, L.J.; Tang, G. MET Amplification (MET/CEP7 Ratio >/= 1.8) Is an Independent Poor Prognostic Marker in Patients With Treatment-naive Non-Small-cell Lung Cancer. Clin. Lung Cancer 2021, 22, e512–e518. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, H.S.; Kim, B.J.; Jang, H.J.; Lee, J. Prognostic value of c-Met overexpression in hepatocellular carcinoma: A meta-analysis and review. Oncotarget 2017, 8, 90351–90357. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, C.; Dai, C.; Liu, X.; Li, W.; Huang, M.; Zhao, X.; Ji, D.; Li, J.; Guo, W. Amplification and expression of c-MET correlate with poor prognosis of patients with gastric cancer and upregulate the expression of PDL1. Acta Biochim. Biophys. Sin. (Shanghai) 2021, 53, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Zagouri, F.; Bago-Horvath, Z.; Rossler, F.; Brandstetter, A.; Bartsch, R.; Papadimitriou, C.A.; Dimitrakakis, C.; Tsigginou, A.; Papaspyrou, I.; Giannos, A.; et al. High MET expression is an adverse prognostic factor in patients with triple-negative breast cancer. Br. J. Cancer 2013, 108, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Noonan, S.A.; Berry, L.; Lu, X.; Gao, D.; Baron, A.E.; Chesnut, P.; Sheren, J.; Aisner, D.L.; Merrick, D.; Doebele, R.C.; et al. Identifying the Appropriate FISH Criteria for Defining MET Copy Number-Driven Lung Adenocarcinoma through Oncogene Overlap Analysis. J. Thorac. Oncol. 2016, 11, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Consortium, A.P.G. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Frampton, G.M.; Ali, S.M.; Rosenzweig, M.; Chmielecki, J.; Lu, X.; Bauer, T.M.; Akimov, M.; Bufill, J.A.; Lee, C.; Jentz, D.; et al. Activation of MET via Diverse Exon 14 Splicing Alterations Occurs in Multiple Tumor Types and Confers Clinical Sensitivity to MET Inhibitors. Cancer Discov. 2015, 5, 850–859. [Google Scholar] [CrossRef]
- Paik, P.K.; Drilon, A.; Fan, P.D.; Yu, H.; Rekhtman, N.; Ginsberg, M.S.; Borsu, L.; Schultz, N.; Berger, M.F.; Rudin, C.M.; et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015, 5, 842–849. [Google Scholar] [CrossRef]
- Onozato, R.; Kosaka, T.; Kuwano, H.; Sekido, Y.; Yatabe, Y.; Mitsudomi, T. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J. Thorac. Oncol. 2009, 4, 5–11. [Google Scholar] [CrossRef]
- Kong-Beltran, M.; Seshagiri, S.; Zha, J.; Zhu, W.; Bhawe, K.; Mendoza, N.; Holcomb, T.; Pujara, K.; Stinson, J.; Fu, L.; et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006, 66, 283–289. [Google Scholar] [CrossRef]
- Kron, A.; Scheffler, M.; Heydt, C.; Ruge, L.; Schaepers, C.; Eisert, A.K.; Merkelbach-Bruse, S.; Riedel, R.; Nogova, L.; Fischer, R.N.; et al. Genetic Heterogeneity of MET-Aberrant NSCLC and Its Impact on the Outcome of Immunotherapy. J. Thorac. Oncol. 2021, 16, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Sattler, M.; Salgia, R. MET in the Driver’s Seat: Exon 14 Skipping Mutations as Actionable Targets in Lung Cancer. J. Thorac. Oncol. 2016, 11, 1381–1383. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, J.; Tawfik, O. Detection of MET exon 14 skipping mutations in non-small cell lung cancer: Overview and community perspective. Expert Rev. Anticancer Ther. 2021, 21, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Pennell, N.A.; Davies, K.D. MET Exon 14 Skipping Mutations in Non-Small-Cell Lung Cancer: An Overview of Biology, Clinical Outcomes, and Testing Considerations. JCO Precis. Oncol. 2021, 5. [Google Scholar] [CrossRef]
- Awad, M.M.; Lee, J.K.; Madison, R.; Classon, A.; Kmak, J.; Frampton, G.M.; Alexander, B.M.; Venstrom, J.; Schrock, A.B. Characterization of 1,387 NSCLCs with MET exon 14 (METex14) skipping alterations (SA) and potential acquired resistance (AR) mechanisms. J. Clin. Oncol. 2020, 38, 9511. [Google Scholar] [CrossRef]
- Poirot, B.; Doucet, L.; Benhenda, S.; Champ, J.; Meignin, V.; Lehmann-Che, J. MET Exon 14 Alterations and New Resistance Mutations to Tyrosine Kinase Inhibitors: Risk of Inadequate Detection with Current Amplicon-Based NGS Panels. J. Thorac. Oncol. 2017, 12, 1582–1587. [Google Scholar] [CrossRef]
- Davies, K.D.; Lomboy, A.; Lawrence, C.A.; Yourshaw, M.; Bocsi, G.T.; Camidge, D.R.; Aisner, D.L. DNA-Based versus RNA-Based Detection of MET Exon 14 Skipping Events in Lung Cancer. J. Thorac. Oncol. 2019, 14, 737–741. [Google Scholar] [CrossRef]
- Kim, E.K.; Kim, K.A.; Lee, C.Y.; Kim, S.; Chang, S.; Cho, B.C.; Shim, H.S. Molecular Diagnostic Assays and Clinicopathologic Implications of MET Exon 14 Skipping Mutation in Non-small-cell Lung Cancer. Clin. Lung Cancer 2019, 20, e123–e132. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385 e318. [Google Scholar] [CrossRef]
- Drilon, A.; Cappuzzo, F.; Ou, S.I.; Camidge, D.R. Targeting MET in Lung Cancer: Will Expectations Finally Be MET? J. Thorac. Oncol. 2017, 12, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Le, X. Heterogeneity in MET-Aberrant NSCLC. J. Thorac. Oncol. 2021, 16, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Recondo, G.; Bahcall, M.; Spurr, L.F.; Che, J.; Ricciuti, B.; Leonardi, G.C.; Lo, Y.C.; Li, Y.Y.; Lamberti, G.; Nguyen, T.; et al. Molecular Mechanisms of Acquired Resistance to MET Tyrosine Kinase Inhibitors in Patients with MET Exon 14-Mutant NSCLC. Clin. Cancer Res. 2020, 26, 2615–2625. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.G.; Zou, H.Y.; Arango, M.E.; Li, Q.; Lee, J.H.; McDonnell, S.R.; Yamazaki, S.; Alton, G.R.; Mroczkowski, B.; Los, G. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol. Cancer Ther. 2007, 6, 3314–3322. [Google Scholar] [CrossRef] [PubMed]
- McDermott, U.; Iafrate, A.J.; Gray, N.S.; Shioda, T.; Classon, M.; Maheswaran, S.; Zhou, W.; Choi, H.G.; Smith, S.L.; Dowell, L.; et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res. 2008, 68, 3389–3395. [Google Scholar] [CrossRef]
- Yasuda, H.; de Figueiredo-Pontes, L.L.; Kobayashi, S.; Costa, D.B. Preclinical rationale for use of the clinically available multitargeted tyrosine kinase inhibitor crizotinib in ROS1-translocated lung cancer. J. Thorac. Oncol. 2012, 7, 1086–1090. [Google Scholar] [CrossRef]
- Solomon, B.J.; Mok, T.; Kim, D.W.; Wu, Y.L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef]
- Rodig, S.J.; Shapiro, G.I. Crizotinib, a small-molecule dual inhibitor of the c-Met and ALK receptor tyrosine kinases. Curr. Opin. Investig. Drugs 2010, 11, 1477–1490. [Google Scholar]
- Drilon, A.E.; Camidge, D.R.; Ou, S.-H.I.; Clark, J.W.; Socinski, M.A.; Weiss, J.; Riely, G.J.; Winter, M.; Wang, S.C.; Monti, K.; et al. Efficacy and safety of crizotinib in patients (pts) with advanced MET exon 14-altered non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2016, 34, 108. [Google Scholar] [CrossRef]
- Camidge, D.R.; Otterson, G.A.; Clark, J.W.; Ignatius Ou, S.H.; Weiss, J.; Ades, S.; Shapiro, G.I.; Socinski, M.A.; Murphy, D.A.; Conte, U.; et al. Crizotinib in Patients With MET-Amplified NSCLC. J. Thorac. Oncol. 2021, 16, 1017–1029. [Google Scholar] [CrossRef]
- Drilon, A.; Clark, J.W.; Weiss, J.; Ou, S.H.I.; Camidge, D.R.; Solomon, B.J.; Otterson, G.A.; Villaruz, L.C.; Riely, G.J.; Heist, R.S. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat. Med. 2020, 26, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in Non-Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N. Engl. J. Med. 2020, 383, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Seto, T.; Han, J.Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.W.; Hida, T.; de Jonge, M.; Orlov, S.V.; et al. Capmatinib in MET Exon 14-Mutated or MET-Amplified Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Gavine, P.R.; Ren, Y.; Han, L.; Lv, J.; Fan, S.; Zhang, W.; Xu, W.; Liu, Y.J.; Zhang, T.; Fu, H.; et al. Volitinib, a potent and highly selective c-Met inhibitor, effectively blocks c-Met signaling and growth in c-MET amplified gastric cancer patient-derived tumor xenograft models. Mol. Oncol. 2015, 9, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Fang, J.; Li, X.; Cao, L.; Zhou, J.; Guo, Q.; Liang, Z.; Cheng, Y.; Jiang, L.; Yang, N.; et al. Once-daily savolitinib in Chinese patients with pulmonary sarcomatoid carcinomas and other non-small-cell lung cancers harbouring MET exon 14 skipping alterations: A multicentre, single-arm, open-label, phase 2 study. Lancet Respir. Med. 2021, 9, 1154–1164. [Google Scholar] [CrossRef]
- Sequist, L.V.; Han, J.Y.; Ahn, M.J.; Cho, B.C.; Yu, H.; Kim, S.W.; Yang, J.C.; Lee, J.S.; Su, W.C.; Kowalski, D.; et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: Interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. 2020, 21, 373–386. [Google Scholar] [CrossRef]
- Tamura, T.; Kurishima, K.; Nakazawa, K.; Kagohashi, K.; Ishikawa, H.; Satoh, H.; Hizawa, N. Specific organ metastases and survival in metastatic non-small-cell lung cancer. Mol. Clin. Oncol. 2015, 3, 217–221. [Google Scholar] [CrossRef]
- Offin, M.; Luo, J.; Guo, R.; Lyo, J.K.; Falcon, C.; Dienstag, J.; Wilkins, O.; Chang, J.; Rudin, C.M.; Riely, G.; et al. CNS Metastases in Patients With MET Exon 14-Altered Lung Cancers and Outcomes With Crizotinib. JCO Precis. Oncol. 2020, 4, 871–876. [Google Scholar] [CrossRef]
- Klempner, S.J.; Borghei, A.; Hakimian, B.; Ali, S.M.; Ou, S.I. Intracranial Activity of Cabozantinib in MET Exon 14-Positive NSCLC with Brain Metastases. J. Thorac. Oncol. 2017, 12, 152–156. [Google Scholar] [CrossRef]
- Li, E.; Huang, X.; Zhang, G.; Liang, T. Combinational blockade of MET and PD-L1 improves pancreatic cancer immunotherapeutic efficacy. J. Exp. Clin. Cancer Res. 2021, 40, 279. [Google Scholar] [CrossRef]
- Domenech, M.; Munoz Marmol, A.M.; Mate, J.L.; Estival, A.; Moran, T.; Cucurull, M.; Saigi, M.; Hernandez, A.; Sanz, C.; Hernandez-Gallego, A.; et al. Correlation between PD-L1 expression and MET gene amplification in patients with advanced non-small cell lung cancer and no other actionable oncogenic driver. Oncotarget 2021, 12, 1802–1810. [Google Scholar] [CrossRef] [PubMed]
- Roth, K.G.; Mambetsariev, I.; Salgia, R. Prolonged survival and response to tepotinib in a non-small-cell lung cancer patient with brain metastases harboring MET exon 14 mutation: A research report. Cold Spring Harb. Mol. Case Stud. 2020, 6, a005785. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Forjaz, G.; Mooradian, M.J.; Meza, R.; Kong, C.Y.; Cronin, K.A.; Mariotto, A.B.; Lowy, D.R.; Feuer, E.J. The Effect of Advances in Lung-Cancer Treatment on Population Mortality. N. Engl. J. Med. 2020, 383, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Vander Velde, R.; Yoon, N.; Marusyk, V.; Durmaz, A.; Dhawan, A.; Miroshnychenko, D.; Lozano-Peral, D.; Desai, B.; Balynska, O.; Poleszhuk, J.; et al. Resistance to targeted therapies as a multifactorial, gradual adaptation to inhibitor specific selective pressures. Nat. Commun. 2020, 11, 2393. [Google Scholar] [CrossRef] [PubMed]
- Meador, C.B.; Hata, A.N. Acquired resistance to targeted therapies in NSCLC: Updates and evolving insights. Pharmacol. Ther. 2020, 210, 107522. [Google Scholar] [CrossRef] [PubMed]
- Wood, K.C. Mapping the Pathways of Resistance to Targeted Therapies. Cancer Res. 2015, 75, 4247–4251. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, S.; Swanton, C.; Taylor, B.S.; Costello, J.F. Treatment-Induced Mutagenesis and Selective Pressures Sculpt Cancer Evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a026617. [Google Scholar] [CrossRef]
- Awad, M.M.; Bahcall, M.; Sholl, L.M.; Wilson, F.H.; Paweletz, C.; Capelletti, M.; Leonardi, G.C.; Watanabe, M.; Baba, H.; Chambers, E.S.; et al. Mechanisms of acquired resistance to MET tyrosine kinase inhibitors (TKIs) in MET exon 14 (METex14) mutant non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2018, 36, 9069. [Google Scholar] [CrossRef]
- Fujino, T.; Kobayashi, Y.; Suda, K.; Koga, T.; Nishino, M.; Ohara, S.; Chiba, M.; Shimoji, M.; Tomizawa, K.; Takemoto, T.; et al. Sensitivity and Resistance of MET Exon 14 Mutations in Lung Cancer to Eight MET Tyrosine Kinase Inhibitors In Vitro. J. Thorac. Oncol. 2019, 14, 1753–1765. [Google Scholar] [CrossRef]
- Ou, S.I.; Young, L.; Schrock, A.B.; Johnson, A.; Klempner, S.J.; Zhu, V.W.; Miller, V.A.; Ali, S.M. Emergence of Preexisting MET Y1230C Mutation as a Resistance Mechanism to Crizotinib in NSCLC with MET Exon 14 Skipping. J. Thorac. Oncol. 2017, 12, 137–140. [Google Scholar] [CrossRef]
- Li, A.; Yang, J.J.; Zhang, X.C.; Zhang, Z.; Su, J.; Gou, L.Y.; Bai, Y.; Zhou, Q.; Yang, Z.; Han-Zhang, H.; et al. Acquired MET Y1248H and D1246N Mutations Mediate Resistance to MET Inhibitors in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 4929–4937. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.; Jamme, P.; Cortot, A.B.; Kherrouche, Z.; Tulasne, D. When the MET receptor kicks in to resist targeted therapies. Oncogene 2021, 40, 4061–4078. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Kiedrowski, L.A.; Ravera, E.; Cheng, H.; Halmos, B. Response to Dual Crizotinib and Osimertinib Treatment in a Lung Cancer Patient with MET Amplification Detected by Liquid Biopsy Who Acquired Secondary Resistance to EGFR Tyrosine Kinase Inhibition. J. Thorac. Oncol. 2018, 13, e169–e172. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yang, S.; Wang, K.; Sun, S.Y. MET inhibitors for targeted therapy of EGFR TKI-resistant lung cancer. J. Hematol. Oncol. 2019, 12, 63. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Kim, S.; Kim, T.M.; Kim, D.W.; Kim, S.; Kim, M.; Ahn, Y.O.; Keam, B.; Heo, D.S. Acquired Resistance of MET-Amplified Non-small Cell Lung Cancer Cells to the MET Inhibitor Capmatinib. Cancer Res. Treat. 2019, 51, 951–962. [Google Scholar] [CrossRef]
- Wu, Y.L.; Zhang, L.; Kim, D.W.; Liu, X.; Lee, D.H.; Yang, J.C.; Ahn, M.J.; Vansteenkiste, J.F.; Su, W.C.; Felip, E.; et al. Phase Ib/II Study of Capmatinib (INC280) Plus Gefitinib After Failure of Epidermal Growth Factor Receptor (EGFR) Inhibitor Therapy in Patients With EGFR-Mutated, MET Factor-Dysregulated Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 3101–3109. [Google Scholar] [CrossRef]
- Vogel, W.; Ullrich, A. Multiple in vivo phosphorylated tyrosine phosphatase SHP-2 engages binding to Grb2 via tyrosine 584. Cell Growth Differ. 1996, 7, 1589–1597. [Google Scholar]
- Graziani, A.; Gramaglia, D.; dalla Zonca, P.; Comoglio, P.M. Hepatocyte growth factor/scatter factor stimulates the Ras-guanine nucleotide exchanger. J. Biol. Chem. 1993, 268, 9165–9168. [Google Scholar] [CrossRef]
- Paumelle, R.; Tulasne, D.; Kherrouche, Z.; Plaza, S.; Leroy, C.; Reveneau, S.; Vandenbunder, B.; Fafeur, V. Hepatocyte growth factor/scatter factor activates the ETS1 transcription factor by a RAS-RAF-MEK-ERK signaling pathway. Oncogene 2002, 21, 2309–2319. [Google Scholar] [CrossRef]
- Li, Y.; Guessous, F.; Johnson, E.B.; Eberhart, C.G.; Li, X.N.; Shu, Q.; Fan, S.; Lal, B.; Laterra, J.; Schiff, D.; et al. Functional and molecular interactions between the HGF/c-Met pathway and c-Myc in large-cell medulloblastoma. Lab. Invest. 2008, 88, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Fujita-Sato, S.; Galeas, J.; Truitt, M.; Pitt, C.; Urisman, A.; Bandyopadhyay, S.; Ruggero, D.; McCormick, F. Enhanced MET Translation and Signaling Sustains K-Ras-Driven Proliferation under Anchorage-Independent Growth Conditions. Cancer Res. 2015, 75, 2851–2862. [Google Scholar] [CrossRef] [PubMed]
- Scorilas, A.; Karameris, A.; Arnogiannaki, N.; Ardavanis, A.; Bassilopoulos, P.; Trangas, T.; Talieri, M. Overexpression of matrix-metalloproteinase-9 in human breast cancer: A potential favourable indicator in node-negative patients. Br. J. Cancer 2001, 84, 1488–1496. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.B.; Nabha, S.M.; Atanaskova, N. Role of MAP kinase in tumor progression and invasion. Cancer Metastasis Rev. 2003, 22, 395–403. [Google Scholar] [CrossRef]
- Rotow, J.K.; Gui, P.; Wu, W.; Raymond, V.M.; Lanman, R.B.; Kaye, F.J.; Peled, N.; Fece de la Cruz, F.; Nadres, B.; Corcoran, R.B.; et al. Co-occurring Alterations in the RAS-MAPK Pathway Limit Response to MET Inhibitor Treatment in MET Exon 14 Skipping Mutation-Positive Lung Cancer. Clin. Cancer Res. 2020, 26, 439–449. [Google Scholar] [CrossRef]
- Suzawa, K.; Offin, M.; Lu, D.; Kurzatkowski, C.; Vojnic, M.; Smith, R.S.; Sabari, J.K.; Tai, H.; Mattar, M.; Khodos, I.; et al. Activation of KRAS Mediates Resistance to Targeted Therapy in MET Exon 14-mutant Non-small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 1248–1260. [Google Scholar] [CrossRef]
- Bahcall, M.; Awad, M.M.; Sholl, L.M.; Wilson, F.H.; Xu, M.; Wang, S.; Palakurthi, S.; Choi, J.; Ivanova, E.V.; Leonardi, G.C.; et al. Amplification of Wild-type KRAS Imparts Resistance to Crizotinib in MET Exon 14 Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 5963–5976. [Google Scholar] [CrossRef]
- Graziani, A.; Gramaglia, D.; Cantley, L.C.; Comoglio, P.M. The tyrosine-phosphorylated hepatocyte growth factor/scatter factor receptor associates with phosphatidylinositol 3-kinase. J. Biol. Chem. 1991, 266, 22087–22090. [Google Scholar] [CrossRef]
- Jamme, P.; Fernandes, M.; Copin, M.C.; Descarpentries, C.; Escande, F.; Morabito, A.; Gregoire, V.; Jamme, M.; Baldacci, S.; Tulasne, D.; et al. Alterations in the PI3K Pathway Drive Resistance to MET Inhibitors in NSCLC Harboring MET Exon 14 Skipping Mutations. J. Thorac. Oncol. 2020, 15, 741–751. [Google Scholar] [CrossRef]
- Que, W.; Chen, J.; Chuang, M.; Jiang, D. Knockdown of c-Met enhances sensitivity to bortezomib in human multiple myeloma U266 cells via inhibiting Akt/mTOR activity. APMIS 2012, 120, 195–203. [Google Scholar] [CrossRef]
- Shang, R.; Song, X.; Wang, P.; Zhou, Y.; Lu, X.; Wang, J.; Xu, M.; Chen, X.; Utpatel, K.; Che, L.; et al. Cabozantinib-based combination therapy for the treatment of hepatocellular carcinoma. Gut 2021, 70, 1746–1757. [Google Scholar] [CrossRef] [PubMed]
- Guessous, F.; Yang, Y.; Johnson, E.; Marcinkiewicz, L.; Smith, M.; Zhang, Y.; Kofman, A.; Schiff, D.; Christensen, J.; Abounader, R. Cooperation between c-Met and focal adhesion kinase family members in medulloblastoma and implications for therapy. Mol. Cancer Ther. 2012, 11, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Chen, H.C. Direct interaction of focal adhesion kinase (FAK) with Met is required for FAK to promote hepatocyte growth factor-induced cell invasion. Mol. Cell. Biol. 2006, 26, 5155–5167. [Google Scholar] [CrossRef] [PubMed]
- Hui, A.Y.; Meens, J.A.; Schick, C.; Organ, S.L.; Qiao, H.; Tremblay, E.A.; Schaeffer, E.; Uniyal, S.; Chan, B.M.; Elliott, B.E. Src and FAK mediate cell-matrix adhesion-dependent activation of Met during transformation of breast epithelial cells. J. Cell. Biochem. 2009, 107, 1168–1181. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Qu, X.; Liu, S.; Zhang, S.; Liu, J.; Qu, J.; Zheng, H.; Liu, Y.; Che, X. Dual inhibition of MET and SRC kinase activity as a combined targeting strategy for colon cancer. Exp. Ther. Med. 2017, 14, 1357–1366. [Google Scholar] [CrossRef][Green Version]
- Lu, J.; Li, X.; Tu, K.; Guan, Y.; Fung, K.P.; Liu, F. Verticillin A suppresses HGF-induced migration and invasion via repression of the c-Met/FAK/Src pathway in human gastric and cervical cancer cells. OncoTargets Ther. 2019, 12, 5823–5833. [Google Scholar] [CrossRef]
- Kaposi-Novak, P.; Lee, J.S.; Gomez-Quiroz, L.; Coulouarn, C.; Factor, V.M.; Thorgeirsson, S.S. Met-regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J. Clin. Investig. 2006, 116, 1582–1595. [Google Scholar] [CrossRef]
- Kim, K.H.; Seol, H.J.; Kim, E.H.; Rheey, J.; Jin, H.J.; Lee, Y.; Joo, K.M.; Lee, J.; Nam, D.H. Wnt/beta-catenin signaling is a key downstream mediator of MET signaling in glioblastoma stem cells. NeuroOncology 2013, 15, 161–171. [Google Scholar] [CrossRef]
- Zhan, N.; Michael, A.A.; Wu, K.; Zeng, G.; Bell, A.; Tao, J.; Monga, S.P. The Effect of Selective c-MET Inhibitor on Hepatocellular Carcinoma in the MET-Active, beta-Catenin-Mutated Mouse Model. Gene Expr. 2018, 18, 135–147. [Google Scholar] [CrossRef]
- Fang, D.; Hawke, D.; Zheng, Y.; Xia, Y.; Meisenhelder, J.; Nika, H.; Mills, G.B.; Kobayashi, R.; Hunter, T.; Lu, Z. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J. Biol. Chem. 2007, 282, 11221–11229. [Google Scholar] [CrossRef]
- Etnyre, D.; Stone, A.L.; Fong, J.T.; Jacobs, R.J.; Uppada, S.B.; Botting, G.M.; Rajanna, S.; Moravec, D.N.; Shambannagari, M.R.; Crees, Z.; et al. Targeting c-Met in melanoma: Mechanism of resistance and efficacy of novel combinatorial inhibitor therapy. Cancer Biol. Ther. 2014, 15, 1129–1141. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Davies, K.D. MET Copy Number as a Secondary Driver of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Resistance in EGFR-Mutant Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2019, 37, 855–857. [Google Scholar] [CrossRef] [PubMed]
- Rivas, S.; Armisén, R. El cáncer de pulmón de células no pequeñas en la era de la medicina de precisión (Non-small cell lung cancer in the era of precision medicine). Rev. Médica Clínica Condes 2022, 33, 25–35. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cancer Type | N° GENIE Samples | N° Samples after Filters | METex14 | MET Amp |
|---|---|---|---|---|
| NSCLC | 17,137 | 10,231 | 4% | 2% |
| Renal | 1986 | 1556 | 1.2% | 1.2% |
| Hepatobiliary | 2517 | 1854 | 0.4% | 1.2% |
| Colorectal | 11,893 | 7370 | 0% | 0.4% |
| Ovarian | 4606 | 4481 | 0.1% | 0.2% |
| Breast | 13,388 | 8365 | 0% | 0.2% |
| Prostate | 4379 | 3530 | 0.1% | 0.1% |
| Total analysis | 56,682 | 36,095 | 5.8% | 5.3% |
| Drug’s Type | Drug Name | Clinical Trial | Alterations | ORR | DOR | PFS | Disease Control Rate |
|---|---|---|---|---|---|---|---|
| (Months) | (Months) | ||||||
| Selective type Ia | Crizotinib | PROFILE-1001 | METex14 | 32% | 9.1 | 7.3 | |
| Multitarget TKIs | (NCT00585195) | GCN ≥1.8–≤ 2.2 | 33% | 12.1 | 1.8 | ||
| GCN ≥2.2–≤ 4 | 14.3% | 3.7 | 1.9 | ||||
| GCN > 4 | 40% | 5.5 | 6.7 | ||||
| Selective type Ib monotarget MET TKIs | Capmatinib | GEOMETRY mono-1 trial (NCT02414139) | METex14 | 40.6% previously treated | 9.7 | 5.4 | 78% |
| 67.9% treatment-naive | 12.6 | 12.4 | 96% | ||||
| NCT01324479 | MET GCN > 6 | 47% | N.S | 9.3 | 80% | ||
| Tepotinib | VISION trial (NCT02864992) | METex14 | 46% | 11.1 | 8.5 | 65.70% | |
| Savolitinib | (NCT02897479) | METex14 | 49.2% | - | 6.9 months | 93.40% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivas, S.; Marín, A.; Samtani, S.; González-Feliú, E.; Armisén, R. MET Signaling Pathways, Resistance Mechanisms, and Opportunities for Target Therapies. Int. J. Mol. Sci. 2022, 23, 13898. https://doi.org/10.3390/ijms232213898
Rivas S, Marín A, Samtani S, González-Feliú E, Armisén R. MET Signaling Pathways, Resistance Mechanisms, and Opportunities for Target Therapies. International Journal of Molecular Sciences. 2022; 23(22):13898. https://doi.org/10.3390/ijms232213898
Chicago/Turabian StyleRivas, Solange, Arnaldo Marín, Suraj Samtani, Evelin González-Feliú, and Ricardo Armisén. 2022. "MET Signaling Pathways, Resistance Mechanisms, and Opportunities for Target Therapies" International Journal of Molecular Sciences 23, no. 22: 13898. https://doi.org/10.3390/ijms232213898
APA StyleRivas, S., Marín, A., Samtani, S., González-Feliú, E., & Armisén, R. (2022). MET Signaling Pathways, Resistance Mechanisms, and Opportunities for Target Therapies. International Journal of Molecular Sciences, 23(22), 13898. https://doi.org/10.3390/ijms232213898