
The Role of Copper Homeostasis in Brain Disease


Abstract
1. Introduction
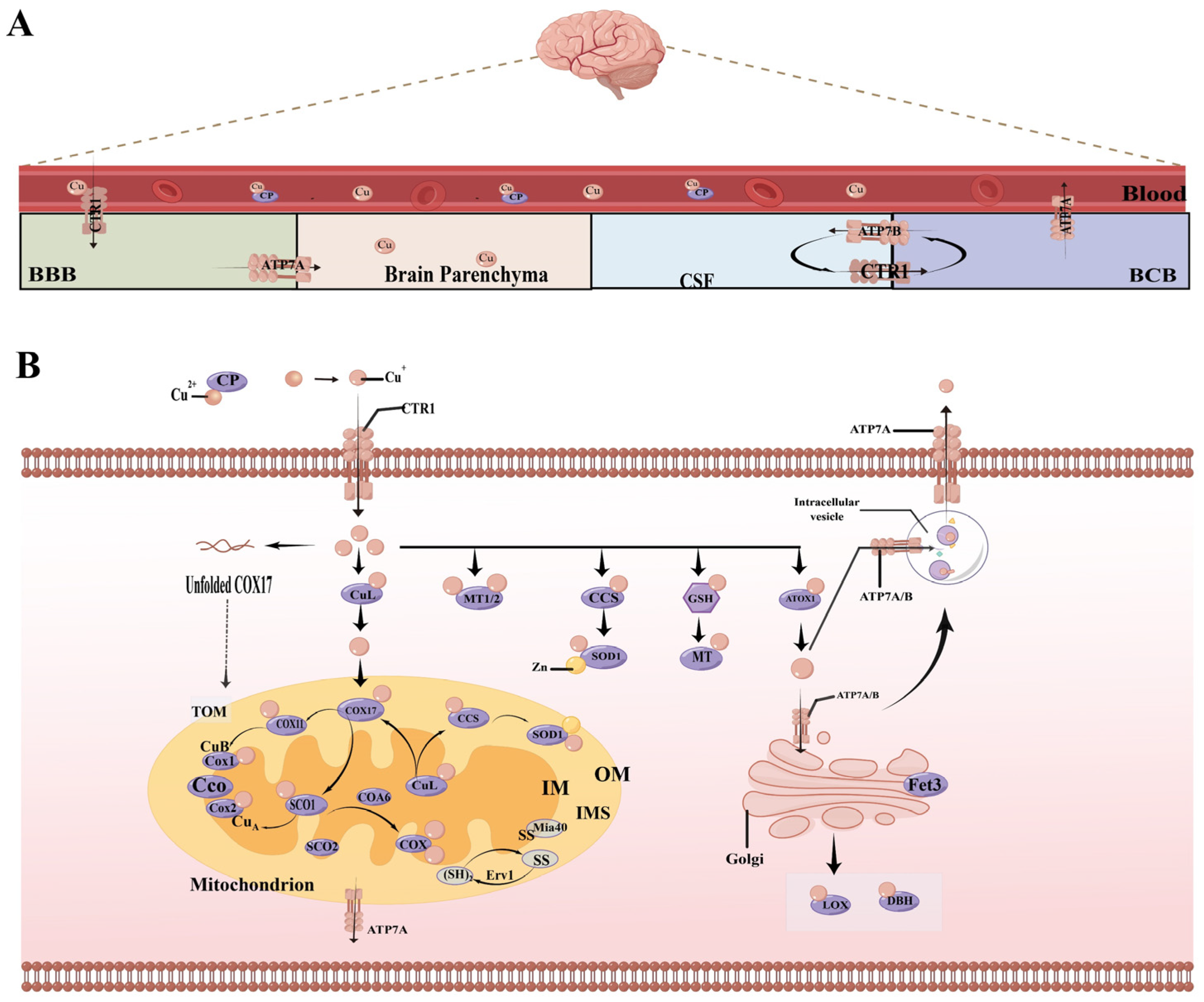
2. Copper Metabolism in the Brain
3. The Physiological and Pathological Role of Copper in the Brain
3.1. Copper and Inflammation
3.2. Copper and Immunity
3.3. Copper and Oxidative Stress
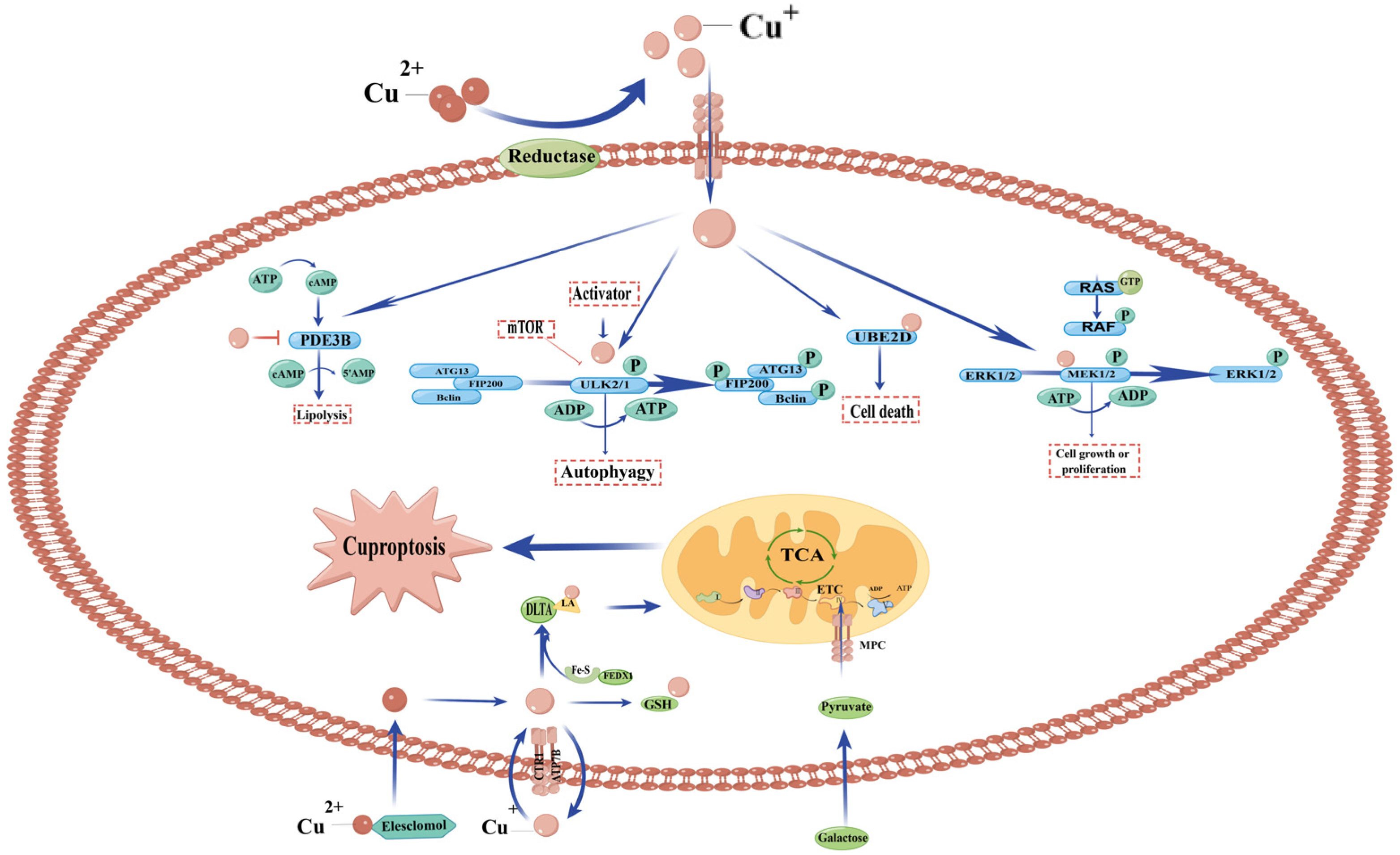
3.4. Copper and Cell Death (Cuproptosis)
4. The Copper Signal Pathway in Brain Diseases
4.1. Alzheimer’s Disease (AD)
4.2. Menkes Disease (MD)
4.3. Wilson’s Disease (WD)
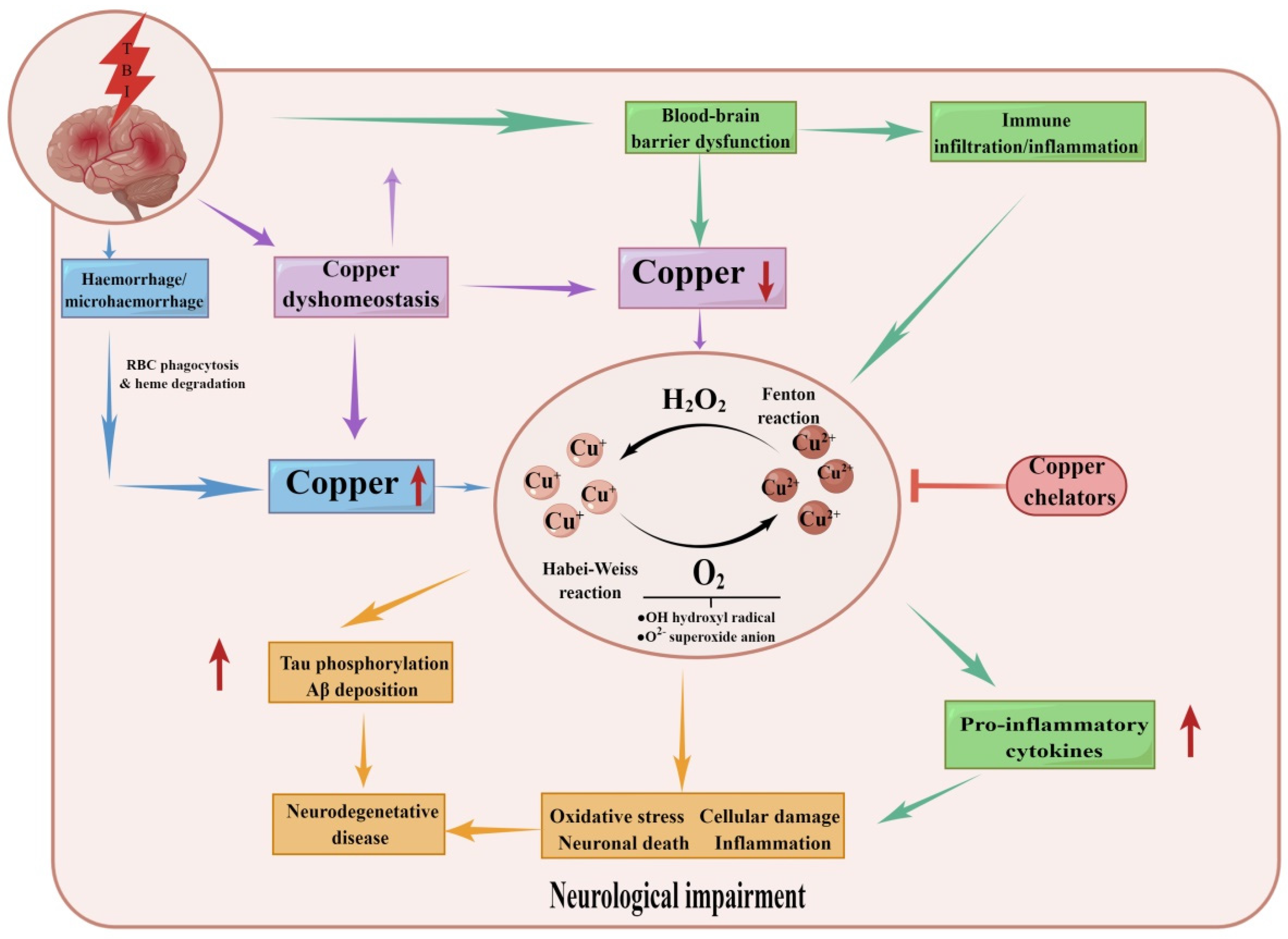
4.4. Traumatic Brain Injury
4.5. Intracerebral Hemorrhage (ICH)
4.6. Ischemic Stroke
4.7. Spinal Cord Injury (SCI)
4.8. Glioma
4.9. Other Diseases
5. The Drugs for Copper
5.1. The Increase of Copper
5.2. The Decrease in Copper
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CP | Ceruloplasmin |
| CTR1 | Copper transporter 1 |
| CuL | Copper ligand |
| COX1/2/11/17 | Copper chaperone for cytochrome c oxidase 1/2/11/17 |
| CCS | Copper chaperone for superoxide dismutase |
| CCO | Cytochrome c oxidase |
| OM | Outer membrane of mitochondria |
| IM | Inner membrane |
| IMS | Intermembrane space |
| SOD | Cu-Zn superoxide dismutase |
| MT1 and MT2 | Metallothionein |
| ATP7A/B | ATPase 7A/B |
| GSH | Glutathione |
| LOX | Lysyl oxidase |
| DBH | Dopamine β-hydroxylase |
| SCO1/2 | Synthesis of cytochrome oxidase 1/2 |
| COA6 | Cytochrome c oxidase assembly factor 6 |
| ATOX1 | Antioxidant protein 1 |
References
- Rihel, J. Copper on the brain. Nat. Chem. Biol. 2018, 14, 638–639. [Google Scholar] [CrossRef] [PubMed]
- Burkhead, J.L.; Gogolin Reynolds, K.A.; Abdel-Ghany, S.E.; Cohu, C.M.; Pilon, M. Copper homeostasis. New Phytol. 2009, 182, 799–816. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.E.; Turski, M.L.; Nose, Y.; Casad, M.; Rockman, H.A.; Thiele, D.J. Cardiac copper deficiency activates a systemic signaling mechanism that communicates with the copper acquisition and storage organs. Cell Metab. 2010, 11, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.E.; Nevitt, T.; Thiele, D.J. Mechanisms for copper acquisition, distribution and regulation. Nat. Chem. Biol. 2008, 4, 176–185. [Google Scholar] [CrossRef]
- Ge, E.J.; Bush, A.I.; Casini, A.; Cobine, P.A.; Cross, J.R.; DeNicola, G.M.; Dou, Q.P.; Franz, K.J.; Gohil, V.M.; Gupta, S.; et al. Connecting copper and cancer: From transition metal signalling to metalloplasia. Nat. Rev. Cancer 2022, 22, 102–113. [Google Scholar] [CrossRef]
- Pierson, H.; Yang, H.; Lutsenko, S. Copper Transport and Disease: What Can We Learn from Organoids? Annu. Rev. Nutr. 2019, 39, 75–94. [Google Scholar] [CrossRef]
- Lutsenko, S. Human copper homeostasis: A network of interconnected pathways. Curr. Opin. Chem. Biol. 2010, 14, 211–217. [Google Scholar] [CrossRef]
- Barrow, L.; Tanner, M.S. Copper distribution among serum proteins in paediatric liver disorders and malignancies. Eur. J. Clin. Investig. 1988, 18, 555–560. [Google Scholar] [CrossRef]
- Han, M.; Lin, Z.; Zhang, Y. The alteration of copper homeostasis in inflammation induced by lipopolysaccharides. Biol. Trace Elem. Res. 2013, 154, 268–274. [Google Scholar] [CrossRef]
- Menkes, J.H.; Alter, M.; Steigleder, G.K.; Weakley, D.R.; Sung, J.H. A sex-linked recessive disorder with retardation of growth, peculiar hair, and focal cerebral and cerebellar degeneration. Pediatrics 1962, 29, 764–779. [Google Scholar]
- Compston, A. Progressive lenticular degeneration: A familial nervous disease associated with cirrhosis of the liver, by S. A. Kinnier Wilson, (From the National Hospital, and the Laboratory of the National Hospital, Queen Square, London). Brain J. Neurol. 2009, 132, 1997–2001. [Google Scholar] [CrossRef] [PubMed]
- Que, E.L.; Domaille, D.W.; Chang, C.J. Metals in neurobiology: Probing their chemistry and biology with molecular imaging. Chem. Rev. 2008, 108, 1517–1549. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Kaler, S.G. ATP7A-related copper transport diseases-emerging concepts and future trends. Nat. Rev. Neurol. 2011, 7, 15–29. [Google Scholar] [CrossRef]
- Madsen, E.; Gitlin, J.D. Copper and iron disorders of the brain. Annu. Rev. Neurosci. 2007, 30, 317–337. [Google Scholar] [CrossRef] [PubMed]
- Zlatic, S.; Comstra, H.S.; Gokhale, A.; Petris, M.J.; Faundez, V. Molecular basis of neurodegeneration and neurodevelopmental defects in Menkes disease. Neurobiol. Dis. 2015, 81, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.; White, A.R. Copper complexes as therapeutic agents. Metallomics 2012, 4, 127–138. [Google Scholar] [CrossRef]
- Gaier, E.D.; Eipper, B.A.; Mains, R.E. Copper signaling in the mammalian nervous system: Synaptic effects. J. Neurosci. Res. 2013, 91, 2–19. [Google Scholar] [CrossRef]
- Zheng, W.; Monnot, A.D. Regulation of brain iron and copper homeostasis by brain barrier systems: Implication in neurodegenerative diseases. Pharmacol. Ther. 2012, 133, 177–188. [Google Scholar] [CrossRef]
- Choi, B.-S.; Zheng, W. Copper transport to the brain by the blood-brain barrier and blood-CSF barrier. Brain Res. 2009, 1248, 14–21. [Google Scholar] [CrossRef]
- Lee, J.; Petris, M.J.; Thiele, D.J. Characterization of mouse embryonic cells deficient in the Ctr1 high affinity copper transporter. Identification of a Ctr1-independent copper transport system. J. Biol. Chem. 2002, 277, 40253–40259. [Google Scholar] [CrossRef] [PubMed]
- Scheiber, I.F.; Mercer, J.F.B.; Dringen, R. Metabolism and functions of copper in brain. Prog. Neurobiol. 2014, 116, 33–57. [Google Scholar] [CrossRef] [PubMed]
- Ridge, P.G.; Zhang, Y.; Gladyshev, V.N. Comparative genomic analyses of copper transporters and cuproproteomes reveal evolutionary dynamics of copper utilization and its link to oxygen. PLoS ONE 2008, 3, e1378. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Occurrence of copper proteins through the three domains of life: A bioinformatic approach. J. Proteome Res. 2008, 7, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.J.; Winge, D.R. Copper metallochaperones. Annu. Rev. Biochem. 2010, 79, 537–562. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.T.; Bourassa, D.; Harankhedkar, S.; McCallum, A.M.; Zlatic, S.A.; Calvo, J.S.; Meloni, G.; Faundez, V.; Fahrni, C.J. Ratiometric two-photon microscopy reveals attomolar copper buffering in normal and Menkes mutant cells. Proc. Natl. Acad. Sci. USA 2019, 116, 12167–12172. [Google Scholar] [CrossRef]
- Cotruvo, J.A., Jr.; Aron, A.T.; Ramos-Torres, K.M.; Chang, C.J. Synthetic fluorescent probes for studying copper in biological systems. Chem. Soc. Rev. 2015, 44, 4400–4414. [Google Scholar] [CrossRef]
- White, C.; Kambe, T.; Fulcher, Y.G.; Sachdev, S.W.; Bush, A.I.; Fritsche, K.; Lee, J.; Quinn, T.P.; Petris, M.J. Copper transport into the secretory pathway is regulated by oxygen in macrophages. J. Cell. Sci. 2009, 122, 1315–1321. [Google Scholar] [CrossRef]
- White, C.; Lee, J.; Kambe, T.; Fritsche, K.; Petris, M.J. A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J. Biol. Chem. 2009, 284, 33949–33956. [Google Scholar] [CrossRef]
- Hernandez, S.; Tsuchiya, Y.; García-Ruiz, J.P.; Lalioti, V.; Nielsen, S.; Cassio, D.; Sandoval, I.V. ATP7B copper-regulated traffic and association with the tight junctions: Copper excretion into the bile. Gastroenterology 2008, 134, 1215–1223. [Google Scholar] [CrossRef]
- Faller, P.; Hureau, C. A bioinorganic view of Alzheimer’s disease: When misplaced metal ions (re)direct the electrons to the wrong target. Chemistry 2012, 18, 15910–15920. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.; Oikonomou, G.; Prober, D.A. Norepinephrine is required to promote wakefulness and for hypocretin-induced arousal in zebrafish. ELife 2015, 4, e07000. [Google Scholar] [CrossRef] [PubMed]
- Szerdahelyi, P.; Kása, P. Histochemical demonstration of copper in normal rat brain and spinal cord. Evidence of localization in glial cells. Histochemistry 1986, 85, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Kodama, H.; Meguro, Y.; Abe, T.; Rayner, M.H.; Suzuki, K.T.; Kobayashi, S.; Nishimura, M. Genetic expression of Menkes disease in cultured astrocytes of the macular mouse. J. Inherit. Metab. Dis. 1991, 14, 896–901. [Google Scholar] [CrossRef]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Smith, Z.M.; Francis, P.S.; Denoyer, D.; Meggyesy, P.M.; Fontaine, S.; Cater, M.A. Copper accumulation in senescent cells: Interplay between copper transporters and impaired autophagy. Redox Biol. 2018, 16, 322–331. [Google Scholar] [CrossRef]
- Wang, L.-M.; Becker, J.S.; Wu, Q.; Oliveira, M.F.; Bozza, F.A.; Schwager, A.L.; Hoffman, J.M.; Morton, K.A. Bioimaging of copper alterations in the aging mouse brain by autoradiography, laser ablation inductively coupled plasma mass spectrometry and immunohistochemistry. Metallomics 2010, 2, 348–353. [Google Scholar] [CrossRef]
- Serpa, R.F.B.; de Jesus, E.F.O.; Anjos, M.J.; de Oliveira, L.F.; Marins, L.A.; do Carmo, M.G.T.; Corrêa Junior, J.D.; Rocha, M.S.; Lopes, R.T.; Martinez, A.M.B. Topographic trace-elemental analysis in the brain of Wistar rats by X-ray microfluorescence with synchrotron radiation. Anal. Sci. 2008, 24, 839–842. [Google Scholar] [CrossRef][Green Version]
- Schlief, M.L.; Craig, A.M.; Gitlin, J.D. NMDA receptor activation mediates copper homeostasis in hippocampal neurons. J. Neurosci. 2005, 25, 239–246. [Google Scholar] [CrossRef]
- Gaggelli, E.; Kozlowski, H.; Valensin, D.; Valensin, G. Copper homeostasis and neurodegenerative disorders (Alzheimer’s, prion, and Parkinson’s diseases and amyotrophic lateral sclerosis). Chem. Rev. 2006, 106, 1995–2044. [Google Scholar] [CrossRef]
- Barnham, K.J.; Bush, A.I. Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chem. Soc. Rev. 2014, 43, 6727–6749. [Google Scholar] [CrossRef]
- Dodani, S.C.; Firl, A.; Chan, J.; Nam, C.I.; Aron, A.T.; Onak, C.S.; Ramos-Torres, K.M.; Paek, J.; Webster, C.M.; Feller, M.B.; et al. Copper is an endogenous modulator of neural circuit spontaneous activity. Proc. Natl. Acad. Sci. USA 2014, 111, 16280–16285. [Google Scholar] [CrossRef] [PubMed]
- Conforti, A.; Franco, L.; Milanino, R.; Totorizzo, A.; Velo, G.P. Copper metabolism during acute inflammation: Studies on liver and serum copper concentrations in normal and inflamed rats. Br. J. Pharmacol. 1983, 79, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Milanino, R.; Velo, G.P. Multiple actions of copper in control of inflammation: Studies in copper-deficient rats. Agents Actions Suppl. 1981, 8, 209–230. [Google Scholar] [PubMed]
- Tapiero, H.; Townsend, D.M.; Tew, K.D. Trace elements in human physiology and pathology. Copper. Biomed Pharmacother. 2003, 57, 386–398. [Google Scholar] [CrossRef]
- Milanino, R.; Mazzoli, S.; Passarella, E.; Tarter, G.; Velo, G.P. Carrageenan oedema in copper-deficient rats. Agents Actions 1978, 8, 618–622. [Google Scholar] [CrossRef]
- Milanino, R.; Conforti, A.; Fracasso, M.E.; Franco, L.; Leone, R.; Passarella, E.; Tarter, G.; Velo, G.P. Concerning the role of endogenous copper in the acute inflammatory process. Agents Actions 1979, 9, 581–588. [Google Scholar] [CrossRef]
- Wei, H.; Frei, B.; Beckman, J.S.; Zhang, W.-J. Copper chelation by tetrathiomolybdate inhibits lipopolysaccharide-induced inflammatory responses in vivo. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H712–H720. [Google Scholar] [CrossRef]
- Wei, H.; Zhang, W.-J.; McMillen, T.S.; Leboeuf, R.C.; Frei, B. Copper chelation by tetrathiomolybdate inhibits vascular inflammation and atherosclerotic lesion development in apolipoprotein E-deficient mice. Atherosclerosis 2012, 223, 306–313. [Google Scholar] [CrossRef]
- Rao, K.M. MAP kinase activation in macrophages. J. Leukoc. Biol. 2001, 69, 3–10. [Google Scholar] [CrossRef]
- Qin, L.; Yang, Y.-B.; Yang, Y.-X.; Zhu, N.; Li, S.-X.; Liao, D.-F.; Zheng, X.-L. Anti-inflammatory activity of ezetimibe by regulating NF-κB/MAPK pathway in THP-1 macrophages. Pharmacology 2014, 93, 69–75. [Google Scholar] [CrossRef]
- Chesi, G.; Hegde, R.N.; Iacobacci, S.; Concilli, M.; Parashuraman, S.; Festa, B.P.; Polishchuk, E.V.; Di Tullio, G.; Carissimo, A.; Montefusco, S.; et al. Identification of p38 MAPK and JNK as new targets for correction of Wilson disease-causing ATP7B mutants. Hepatology 2016, 63, 1842–1859. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, F.; Davinelli, S.; Krishnan, S.; Gallo, R.C.; Scapagnini, G.; Zella, D.; Curreli, S. Sulfur compounds block MCP-1 production by Mycoplasma fermentans-infected macrophages through NF-κB inhibition. J. Transl. Med. 2014, 12, 145. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Anderson, D.E.; Barnitz, R.A.; Snow, A.; Bidere, N.; Zheng, L.; Hegde, V.; Lam, L.T.; Staudt, L.M.; Levens, D.; et al. Ribosomal protein S3: A KH domain subunit in NF-kappaB complexes that mediates selective gene regulation. Cell 2007, 131, 927–939. [Google Scholar] [CrossRef]
- Chen, L.; Kuang, P.; Liu, H.; Wei, Q.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; et al. Sodium Fluoride (NaF) Induces Inflammatory Responses Via Activating MAPKs/NF-κB Signaling Pathway and Reducing Anti-inflammatory Cytokine Expression in the Mouse Liver. Biol. Trace Elem. Res. 2019, 189, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tang, J.; Zuo, Y.; Yu, Y.; Luo, P.; Yao, X.; Dong, Y.; Wang, P.; Liu, L.; Zhou, H. Stauntoside B inhibits macrophage activation by inhibiting NF-κB and ERK MAPK signalling. Pharmacol. Res. 2016, 111, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K. Innate recognition of lipopolysaccharide by Toll-like receptor 4-MD-2. Trends Microbiol. 2004, 12, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.-C.; Jee, S.Y.; Lee, S.G.; Park, S.J.; Lee, J.R.; Kim, S.C. Anti-inflammatory activity of the methanol extract of moutan cortex in LPS-activated Raw264.7 cells. Evid. Based Complement Altern. Med. 2007, 4, 327–333. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Wu, D.-C.; Huang, F.-P.; Yang, G.-Y. Inhibition of MEK/ERK 1/2 pathway reduces pro-inflammatory cytokine interleukin-1 expression in focal cerebral ischemia. Brain Res. 2004, 996, 55–66. [Google Scholar] [CrossRef]
- Koksal, C.; Ercan, M.; Bozkurt, A.K.; Cortelekoglu, T.; Konukoglu, D. Abdominal aortic aneurysm or aortic occlusive disease: Role of trace element imbalance. Angiology 2007, 58, 191–195. [Google Scholar] [CrossRef]
- Stadler, N.; Lindner, R.A.; Davies, M.J. Direct detection and quantification of transition metal ions in human atherosclerotic plaques: Evidence for the presence of elevated levels of iron and copper. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 949–954. [Google Scholar] [CrossRef]
- Sudhahar, V.; Das, A.; Horimatsu, T.; Ash, D.; Leanhart, S.; Antipova, O.; Vogt, S.; Singla, B.; Csanyi, G.; White, J.; et al. Copper Transporter ATP7A (Copper-Transporting P-Type ATPase/Menkes ATPase) Limits Vascular Inflammation and Aortic Aneurysm Development: Role of MicroRNA-125b. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2320–2337. [Google Scholar] [CrossRef] [PubMed]
- Veldhuis, N.A.; Gaeth, A.P.; Pearson, R.B.; Gabriel, K.; Camakaris, J. The multi-layered regulation of copper translocating P-type ATPases. Biometals Int. J. Role Met. Ions Biol. Biochem. Med. 2009, 22, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Rivest, S. Molecular insights on the cerebral innate immune system. Brain Behav. Immun. 2003, 17, 13–19. [Google Scholar] [CrossRef]
- Rivest, S. Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 2009, 9, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; White, C.; Lee, J.; Peterson, T.S.; Bush, A.I.; Sun, G.Y.; Weisman, G.A.; Petris, M.J. Altered microglial copper homeostasis in a mouse model of Alzheimer’s disease. J. Neurochem. 2010, 114, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Niciu, M.J.; Ma, X.M.; El Meskini, R.; Ronnett, G.V.; Mains, R.E.; Eipper, B.A. Developmental changes in the expression of ATP7A during a critical period in postnatal neurodevelopment. Neuroscience 2006, 139, 947–964. [Google Scholar] [CrossRef] [PubMed]
- Telianidis, J.; Hung, Y.H.; Materia, S.; Fontaine, S.L. Role of the P-Type ATPases, ATP7A and ATP7B in brain copper homeostasis. Front. Aging Neurosci. 2013, 5, 44. [Google Scholar] [CrossRef]
- Huuskonen, M.T.; Tuo, Q.Z.; Loppi, S.; Dhungana, H.; Korhonen, P.; McInnes, L.E.; Donnelly, P.S.; Grubman, A.; Wojciechowski, S.; Lejavova, K.; et al. The Copper bis(thiosemicarbazone) Complex Cu(II)(atsm) Is Protective Against Cerebral Ischemia through Modulation of the Inflammatory Milieu. Neurotherapeutics 2017, 14, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Percival, S.S. Copper and immunity. Am. J. Clin. Nutr. 1998, 67, 1064S–1068S. [Google Scholar] [CrossRef]
- Prohaska, J.R.; Downing, S.W.; Lukasewycz, O.A. Chronic dietary copper deficiency alters biochemical and morphological properties of mouse lymphoid tissues. J. Nutr. 1983, 113, 1583–1590. [Google Scholar] [CrossRef]
- Lukasewycz, O.A.; Prohaska, J.R. The immune response in copper deficiency. Ann. N. Y. Acad. Sci. 1990, 587, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Koller, L.D.; Mulhern, S.A.; Frankel, N.C.; Steven, M.G.; Williams, J.R. Immune dysfunction in rats fed a diet deficient in copper. Am. J. Clin. Nutr. 1987, 45, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Bala, S.; Failla, M.L. Copper deficiency reversibly impairs DNA synthesis in activated T lymphocytes by limiting interleukin 2 activity. Proc. Natl. Acad. Sci. USA 1992, 89, 6794–6797. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.M. Copper deficiency in humans. Semin. Hematol. 1983, 20, 118–128. [Google Scholar]
- Higuchi, S.; Higashi, A.; Nakamura, T.; Yanabe, Y.; Matsuda, I. Anti-neutrophil antibodies in patients with nutritional copper deficiency. Eur. J. Pediatr. 1991, 150, 327–330. [Google Scholar] [CrossRef]
- Boyne, R.; Arthur, J.R. Effects of selenium and copper deficiency on neutrophil function in cattle. J. Comp. Pathol. 1981, 91, 271–276. [Google Scholar] [CrossRef]
- Babu, U.; Failla, M.L. Copper status and function of neutrophils are reversibly depressed in marginally and severely copper-deficient rats. J. Nutr. 1990, 120, 1700–1709. [Google Scholar] [CrossRef]
- Ciriolo, M.R.; Marasco, M.R.; Iannone, M.; Nisticò, G.; Rotilio, G. Decrease of immunoreactive catalase protein in specific areas of ageing rat brain. Neurosci. Lett. 1997, 228, 21–24. [Google Scholar] [CrossRef]
- Uriu-Adams, J.Y.; Keen, C.L. Copper, oxidative stress, and human health. Mol. Asp. Med. 2005, 26, 268–298. [Google Scholar] [CrossRef]
- Chen, Y.; Saari, J.T.; Kang, Y.J. Weak antioxidant defenses make the heart a target for damage in copper-deficient rats. Free Radic. Biol. Med. 1994, 17, 529–536. [Google Scholar] [CrossRef]
- Olin, K.L.; Walter, R.M.; Keen, C.L. Copper deficiency affects selenoglutathione peroxidase and selenodeiodinase activities and antioxidant defense in weanling rats. Am. J. Clin. Nutr. 1994, 59, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Strain, J.J. Newer aspects of micronutrients in chronic disease: Copper. Proc. Nutr. Soc. 1994, 53, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Uriu-Adams, J.Y.; Rucker, R.B.; Commisso, J.F.; Keen, C.L. Diabetes and dietary copper alter 67Cu metabolism and oxidant defense in the rat. J. Nutr. Biochem. 2005, 16, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Prohaska, J.R.; Brokate, B. Lower copper, zinc-superoxide dismutase protein but not mRNA in organs of copper-deficient rats. Arch. Biochem. Biophys. 2001, 393, 170–176. [Google Scholar] [CrossRef]
- West, E.C.; Prohaska, J.R. Cu,Zn-superoxide dismutase is lower and copper chaperone CCS is higher in erythrocytes of copper-deficient rats and mice. Exp. Biol. Med. 2004, 229, 756–764. [Google Scholar] [CrossRef]
- Chung, K.; Romero, N.; Tinker, D.; Keen, C.L.; Amemiya, K.; Rucker, R. Role of copper in the regulation and accumulation of superoxide dismutase and metallothionein in rat liver. J. Nutr. 1988, 118, 859–864. [Google Scholar] [CrossRef]
- Perera, C.S.; St Clair, D.K.; McClain, C.J. Differential regulation of manganese superoxide dismutase activity by alcohol and TNF in human hepatoma cells. Arch. Biochem. Biophys. 1995, 323, 471–476. [Google Scholar] [CrossRef]
- Yen, T.C.; King, K.L.; Lee, H.C.; Yeh, S.H.; Wei, Y.H. Age-dependent increase of mitochondrial DNA deletions together with lipid peroxides and superoxide dismutase in human liver mitochondria. Free Radic. Biol. Med. 1994, 16, 207–214. [Google Scholar] [CrossRef]
- Hellman, N.E.; Gitlin, J.D. Ceruloplasmin metabolism and function. Annu. Rev. Nutr. 2002, 22, 439–458. [Google Scholar] [CrossRef]
- Prohaska, J.R.; Brokate, B. The timing of perinatal copper deficiency in mice influences offspring survival. J. Nutr. 2002, 132, 3142–3145. [Google Scholar] [CrossRef]
- Chao, P.Y.; Allen, K.G. Glutathione production in copper-deficient isolated rat hepatocytes. Free Radic. Biol. Med. 1992, 12, 145–150. [Google Scholar] [PubMed]
- Fabisiak, J.P.; Tyurin, V.A.; Tyurina, Y.Y.; Borisenko, G.G.; Korotaeva, A.; Pitt, B.R.; Lazo, J.S.; Kagan, V.E. Redox regulation of copper-metallothionein. Arch. Biochem. Biophys. 1999, 363, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.-D.; Liu, Y.; Hu, K.; Jiang, J.; Li, S.-H.; Feng, L.; Zhou, X.-Q. Copper exposure induces oxidative injury, disturbs the antioxidant system and changes the Nrf2/ARE (CuZnSOD) signaling in the fish brain: Protective effects of myo-inositol. Aquat. Toxicol. 2014, 155, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, S.M.; dos Santos, N.M.S.; Calejo, M.; Fontainhas-Fernandes, A.; Sousa, M. Copper toxicity in gills of the teleost fish, Oreochromis niloticus: Effects in apoptosis induction and cell proliferation. Aquat. Toxicol. 2009, 94, 219–228. [Google Scholar] [CrossRef]
- Sahin, E.; Gümüşlü, S. Alterations in brain antioxidant status, protein oxidation and lipid peroxidation in response to different stress models. Behav. Brain Res. 2004, 155, 241–248. [Google Scholar] [CrossRef]
- Upadhyay, R.; Panda, S.K. Zinc reduces copper toxicity induced oxidative stress by promoting antioxidant defense in freshly grown aquatic duckweed Spirodela polyrhiza L. J. Hazard Mater. 2010, 175, 1081–1084. [Google Scholar] [CrossRef]
- Halliwell, B. The role of oxygen radicals in human disease, with particular reference to the vascular system. Haemostasis 1993, 23 (Suppl. S1), 118–126. [Google Scholar] [CrossRef]
- Chang, H.-H.; Guo, M.-K.; Kasten, F.H.; Chang, M.-C.; Huang, G.-F.; Wang, Y.-L.; Wang, R.-S.; Jeng, J.-H. Stimulation of glutathione depletion, ROS production and cell cycle arrest of dental pulp cells and gingival epithelial cells by HEMA. Biomaterials 2005, 26, 745–753. [Google Scholar] [CrossRef]
- Wu, G.; Fang, Y.-Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef]
- Opazo, C.M.; Lotan, A.; Xiao, Z.; Zhang, B.; Greenough, M.A.; Lim, C.M.; Trytell, H.; Ramírez, A.; Ukuwela, A.A.; Mawal, C.H.; et al. Nutrient copper signaling promotes protein turnover by allosteric activation of ubiquitin E2D conjugases. bioRxiv 2021. [Google Scholar] [CrossRef]
- Turski, M.L.; Brady, D.C.; Kim, H.J.; Kim, B.-E.; Nose, Y.; Counter, C.M.; Winge, D.R.; Thiele, D.J. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol. Cell. Biol. 2012, 32, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Kahlson, M.A.; Dixon, S.J. Copper-induced cell death. Science 2022, 375, 1231–1232. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kroemer, G. Cuproptosis: A copper-triggered modality of mitochondrial cell death. Cell. Res. 2022, 32, 417–418. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-R.; Bu, L.-L.; Cai, L. Cuproptosis: Lipoylated TCA cycle proteins-mediated novel cell death pathway. Signal Transduct Target Ther. 2022, 7, 158. [Google Scholar] [CrossRef] [PubMed]
- Kirshner, J.R.; He, S.; Balasubramanyam, V.; Kepros, J.; Yang, C.-Y.; Zhang, M.; Du, Z.; Barsoum, J.; Bertin, J. Elesclomol induces cancer cell apoptosis through oxidative stress. Mol. Cancer Ther. 2008, 7, 2319–2327. [Google Scholar] [CrossRef]
- Cai, K.; Tonelli, M.; Frederick, R.O.; Markley, J.L. Human Mitochondrial Ferredoxin 1 (FDX1) and Ferredoxin 2 (FDX2) Both Bind Cysteine Desulfurase and Donate Electrons for Iron-Sulfur Cluster Biosynthesis. Biochemistry 2017, 56, 487–499. [Google Scholar] [CrossRef]
- Sheftel, A.D.; Stehling, O.; Pierik, A.J.; Elsässer, H.-P.; Mühlenhoff, U.; Webert, H.; Hobler, A.; Hannemann, F.; Bernhardt, R.; Lill, R. Humans possess two mitochondrial ferredoxins, Fdx1 and Fdx2, with distinct roles in steroidogenesis, heme, and Fe/S cluster biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 11775–11780. [Google Scholar] [CrossRef]
- Shi, Y.; Ghosh, M.; Kovtunovych, G.; Crooks, D.R.; Rouault, T.A. Both human ferredoxins 1 and 2 and ferredoxin reductase are important for iron-sulfur cluster biogenesis. Biochim. Biophys. Acta 2012, 1823, 484–492. [Google Scholar] [CrossRef]
- Arroyo, J.D.; Jourdain, A.A.; Calvo, S.E.; Ballarano, C.A.; Doench, J.G.; Root, D.E.; Mootha, V.K. A Genome-wide CRISPR Death Screen Identifies Genes Essential for Oxidative Phosphorylation. Cell Metab. 2016, 24, 875–885. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Detappe, A.; Cai, K.; Keys, H.R.; Brune, Z.; Ying, W.; Thiru, P.; Reidy, M.; Kugener, G.; Rossen, J.; et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat. Chem. Biol. 2019, 15, 681–689. [Google Scholar] [CrossRef]
- Brookmeyer, R.; Gray, S.; Kawas, C. Projections of Alzheimer’s disease in the United States and the public health impact of delaying disease onset. Am. J. Public Health 1998, 88, 1337–1342. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Korshavn, K.J.; Kochi, A.; Derrick, J.S.; Lim, M.H. Cholesterol and metal ions in Alzheimer’s disease. Chem. Soc. Rev. 2014, 43, 6672–6682. [Google Scholar] [CrossRef]
- The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. Neurobiol. Aging 1997, 18, S1–S2. [Google Scholar] [CrossRef]
- Arnold, S.E.; Hyman, B.T.; Flory, J.; Damasio, A.R.; Van Hoesen, G.W. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb. Cortex 1991, 1, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Vinters, H.V.; Cole, G.M.; Khachaturian, Z.S. Alzheimer’s disease: Etiologies, pathophysiology, cognitive reserve, and treatment opportunities. Neurology 1998, 51, S2–S17. [Google Scholar] [CrossRef]
- Selkoe, D.J. Presenilins, β-amyloid precursor protein and the molecular basis of Alzheimer’s disease. Clin. Neurosci. Res. 2001, 1, 91–103. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, Y.H.; Zhang, W.; Gao, H.L.; Zhong, M.L.; Huang, T.T.; Guo, R.F.; Liu, N.N.; Li, D.D.; Li, Y.; et al. Copper chelators promote nonamyloidogenic processing of AbetaPP via MT1/2/CREB-dependent signaling pathways in AbetaPP/PS1 transgenic mice. J. Pineal Res. 2018, 65, e12502. [Google Scholar] [CrossRef]
- Kepp, K.P. Bioinorganic chemistry of Alzheimer’s disease. Chem. Rev. 2012, 112, 5193–5239. [Google Scholar] [CrossRef]
- Savelieff, M.G.; Lee, S.; Liu, Y.; Lim, M.H. Untangling amyloid-β, tau, and metals in Alzheimer’s disease. ACS Chem. Biol. 2013, 8, 856–865. [Google Scholar] [CrossRef]
- Bush, A.I.; Tanzi, R.E. Therapeutics for Alzheimer’s disease based on the metal hypothesis. Neurother. J. Am. Soc. Exp. Neurother. 2008, 5, 421–432. [Google Scholar] [CrossRef]
- Chelly, J.; Tümer, Z.; Tønnesen, T.; Petterson, A.; Ishikawa-Brush, Y.; Tommerup, N.; Horn, N.; Monaco, A.P. Isolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding protein. Nat. Genet. 1993, 3, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.F.; Livingston, J.; Hall, B.; Paynter, J.A.; Begy, C.; Chandrasekharappa, S.; Lockhart, P.; Grimes, A.; Bhave, M.; Siemieniak, D. Isolation of a partial candidate gene for Menkes disease by positional cloning. Nat. Genet. 1993, 3, 20–25. [Google Scholar] [CrossRef]
- Vulpe, C.; Levinson, B.; Whitney, S.; Packman, S.; Gitschier, J. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat. Genet. 1993, 3, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Menkes, J.H. Kinky hair disease: Twenty five years later. Brain Dev. 1988, 10, 77–79. [Google Scholar] [CrossRef]
- Petris, M.J.; Mercer, J.F.; Culvenor, J.G.; Lockhart, P.; Gleeson, P.A.; Camakaris, J. Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: A novel mechanism of regulated trafficking. EMBO J. 1996, 15, 6084–6095. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, L.M.; Soma, S.; Yuan, S.; Silva, A.; Zulkifli, M.; Snavely, T.C.; Greene, H.F.; Nunez, E.; Lynch, B.; De Ville, C.; et al. Elesclomol alleviates Menkes pathology and mortality by escorting Cu to cuproenzymes in mice. Science 2020, 368, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Camakaris, J.; Mann, J.R.; Danks, D.M. Copper metabolism in mottled mouse mutants: Copper concentrations in tissues during development. Biochem. J. 1979, 180, 597–604. [Google Scholar] [CrossRef]
- Phillips, M.; Camakaris, J.; Danks, D.M. Comparisons of copper deficiency states in the murine mutants blotchy and brindled. Changes in copper-dependent enzyme activity in 13-day-old mice. Biochem. J. 1986, 238, 177–183. [Google Scholar] [CrossRef]
- Prohaska, J.R.; Geissler, J.; Brokate, B.; Broderius, M. Copper, zinc-superoxide dismutase protein but not mRNA is lower in copper-deficient mice and mice lacking the copper chaperone for superoxide dismutase. Exp. Biol. Med. 2003, 228, 959–966. [Google Scholar] [CrossRef]
- Qin, Z.; Itoh, S.; Jeney, V.; Ushio-Fukai, M.; Fukai, T. Essential role for the Menkes ATPase in activation of extracellular superoxide dismutase: Implication for vascular oxidative stress. FASEB J. 2006, 20, 334–336. [Google Scholar] [CrossRef]
- Iwase, T.; Nishimura, M.; Sugimura, H.; Igarashi, H.; Ozawa, F.; Shinmura, K.; Suzuki, M.; Tanaka, M.; Kino, I. Localization of Menkes gene expression in the mouse brain; its association with neurological manifestations in Menkes model mice. Acta Neuropathol. 1996, 91, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Niciu, M.J.; Ma, X.-M.; El Meskini, R.; Pachter, J.S.; Mains, R.E.; Eipper, B.A. Altered ATP7A expression and other compensatory responses in a murine model of Menkes disease. Neurobiol. Dis. 2007, 27, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, N. Histochemical localization of copper in various organs of brindled mice. Pathol. Int. 1994, 44, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Murata, Y.; Kodama, H.; Mori, Y.; Kobayashi, M.; Abe, T. Mottled gene expression and copper distribution in the macular mouse, an animal model for Menkes disease. J. Inherit. Metab. Dis. 1998, 21, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Ito, M.; Tsuda, H.; Mikawa, H. Biochemical study on the critical period for treatment of the mottled brindled mouse. J. Neurochem. 1990, 55, 885–889. [Google Scholar] [CrossRef]
- Cumings, J.N. The copper and iron content of brain and liver in the normal and in hepato-lenticular degeneration. Brain J. Neurol. 1948, 71, 410–415. [Google Scholar] [CrossRef]
- Coffey, A.J.; Durkie, M.; Hague, S.; McLay, K.; Emmerson, J.; Lo, C.; Klaffke, S.; Joyce, C.J.; Dhawan, A.; Hadzic, N.; et al. A genetic study of Wilson’s disease in the United Kingdom. Brain 2013, 136, 1476–1487. [Google Scholar] [CrossRef]
- Czlonkowska, A.; Litwin, T.; Dusek, P.; Ferenci, P.; Lutsenko, S.; Medici, V.; Rybakowski, J.K.; Weiss, K.H.; Schilsky, M.L. Wilson disease. Nat. Rev. Dis. Prim. 2018, 4, 21. [Google Scholar] [CrossRef]
- Lutsenko, S.; Barnes, N.L.; Bartee, M.Y.; Dmitriev, O.Y. Function and regulation of human copper-transporting ATPases. Physiol. Rev. 2007, 87, 1011–1046. [Google Scholar] [CrossRef]
- Payne, A.S.; Gitlin, J.D. Functional expression of the menkes disease protein reveals common biochemical mechanisms among the copper-transporting P-type ATPases. J. Biol. Chem. 1998, 273, 3765–3770. [Google Scholar] [CrossRef]
- Walshe, J.M. Monitoring copper in Wilson’s disease. Adv. Clin. Chem. 2010, 50, 151–163. [Google Scholar] [PubMed]
- Gitlin, J.D. Wilson disease. Gastroenterology 2003, 125, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.A.; Schilsky, M.L.; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: An update. Hepatology 2008, 47, 2089–2111. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, D.A.; Medici, V. Wilson disease: At the crossroads between genetics and epigenetics—A review of the evidence. Liver Res. 2017, 1, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; Kim, H.W.; Nakagawa, O.; Ozumi, K.; Lessner, S.M.; Aoki, H.; Akram, K.; McKinney, R.D.; Ushio-Fukai, M.; Fukai, T. Novel role of antioxidant-1 (Atox1) as a copper-dependent transcription factor involved in cell proliferation. J. Biol. Chem. 2008, 283, 9157–9167. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Yuzbasiyan-Gurkan, V. Wilson disease. Medicine 1992, 71, 139–164. [Google Scholar] [CrossRef] [PubMed]
- Hyder, A.A.; Wunderlich, C.A.; Puvanachandra, P.; Gururaj, G.; Kobusingye, O.C. The impact of traumatic brain injuries: A global perspective. NeuroRehabilitation 2007, 22, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, J.L.; Grimes, J.; Ling, G.S.F. Pathophysiology of battlefield associated traumatic brain injury. Pathophysiology 2013, 20, 23–30. [Google Scholar] [CrossRef]
- Jones, P.W.; Taylor, D.M.; Williams, D.R. Analysis and chemical speciation of copper and zinc in wound fluid. J. Inorg. Biochem. 2000, 81, 1–10. [Google Scholar] [CrossRef]
- Mirastschijski, U.; Martin, A.; Jorgensen, L.N.; Sampson, B.; Ågren, M.S. Zinc, copper, and selenium tissue levels and their relation to subcutaneous abscess, minor surgery, and wound healing in humans. Biol. Trace Elem. Res. 2013, 153, 76–83. [Google Scholar] [CrossRef]
- Peng, F.; Muzik, O.; Gatson, J.; Kernie, S.G.; Diaz-Arrastia, R. Assessment of Traumatic Brain Injury by Increased 64Cu Uptake on 64CuCl2 PET/CT. J. Nucl. Med. 2015, 56, 1252–1257. [Google Scholar] [CrossRef] [PubMed]
- Mikawa, S.; Kinouchi, H.; Kamii, H.; Gobbel, G.T.; Chen, S.F.; Carlson, E.; Epstein, C.J.; Chan, P.H. Attenuation of acute and chronic damage following traumatic brain injury in copper, zinc-superoxide dismutase transgenic mice. J. Neurosurg. 1996, 85, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Kinouchi, H.; Epstein, C.J.; Mizui, T.; Carlson, E.; Chen, S.F.; Chan, P.H. Attenuation of focal cerebral ischemic injury in transgenic mice overexpressing CuZn superoxide dismutase. Proc. Natl. Acad. Sci. USA 1991, 88, 11158–11162. [Google Scholar] [CrossRef] [PubMed]
- Yunoki, M.; Kawauchi, M.; Ukita, N.; Noguchi, Y.; Nishio, S.; Ono, Y.; Asari, S.; Ohmoto, T.; Asanuma, M.; Ogawa, N. Effects of lecithinized superoxide dismutase on traumatic brain injury in rats. J. Neurotrauma 1997, 14, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Shie, F.-S.; Zhang, J.; Lee, C.-P.; Ho, Y.-S. Prevention of mitochondrial dysfunction in post-traumatic mouse brain by superoxide dismutase. J. Neurochem. 2005, 95, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Dash, P.K.; Redell, J.B.; Hergenroeder, G.; Zhao, J.; Clifton, G.L.; Moore, A. Serum ceruloplasmin and copper are early biomarkers for traumatic brain injury-associated elevated intracranial pressure. J. Neurosci. Res. 2010, 88, 1719–1726. [Google Scholar] [CrossRef]
- Penkowa, M.; Giralt, M.; Thomsen, P.S.; Carrasco, J.; Hidalgo, J. Zinc or copper deficiency-induced impaired inflammatory response to brain trauma may be caused by the concomitant metallothionein changes. J. Neurotrauma 2001, 18, 447–463. [Google Scholar] [CrossRef]
- Isaev, N.K.; Stelmashook, E.V.; Genrikhs, E.E. Role of zinc and copper ions in the pathogenetic mechanisms of traumatic brain injury and Alzheimer’s disease. Rev. Neurosci. 2020, 31, 233–243. [Google Scholar] [CrossRef]
- Han, M.; Ding, S.; Zhang, Y.; Lin, Z.; Li, K. Serum Copper Homeostasis in Hypertensive Intracerebral Hemorrhage and its Clinical Significance. Biol. Trace Elem. Res. 2018, 185, 56–62. [Google Scholar] [CrossRef]
- Noshita, N.; Sugawara, T.; Hayashi, T.; Lewén, A.; Omar, G.; Chan, P.H. Copper/zinc superoxide dismutase attenuates neuronal cell death by preventing extracellular signal-regulated kinase activation after transient focal cerebral ischemia in mice. J. Neurosci. 2002, 22, 7923–7930. [Google Scholar] [CrossRef]
- Sen, C.K.; Khanna, S.; Venojarvi, M.; Trikha, P.; Ellison, E.C.; Hunt, T.K.; Roy, S. Copper-induced vascular endothelial growth factor expression and wound healing. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1821–H1827. [Google Scholar] [CrossRef] [PubMed]
- Sakata, H.; Niizuma, K.; Wakai, T.; Narasimhan, P.; Maier, C.M.; Chan, P.H. Neural stem cells genetically modified to overexpress cu/zn-superoxide dismutase enhance amelioration of ischemic stroke in mice. Stroke 2012, 43, 2423–2429. [Google Scholar] [CrossRef] [PubMed]
- Wakai, T.; Sakata, H.; Narasimhan, P.; Yoshioka, H.; Kinouchi, H.; Chan, P.H. Transplantation of neural stem cells that overexpress SOD1 enhances amelioration of intracerebral hemorrhage in mice. J. Cereb. Blood Flow Metab. 2014, 34, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Keep, R.F.; Hoff, J.T.; Xi, G. Brain injury after intracerebral hemorrhage: The role of thrombin and iron. Stroke 2007, 38, 759–762. [Google Scholar] [CrossRef]
- Cherukuri, S.; Potla, R.; Sarkar, J.; Nurko, S.; Harris, Z.L.; Fox, P.L. Unexpected role of ceruloplasmin in intestinal iron absorption. Cell Metab. 2005, 2, 309–319. [Google Scholar] [CrossRef]
- Heuser, R.R. The Role for Cardiologists in Stroke Intervention. Prog. Cardiovasc. Dis. 2017, 59, 549–554. [Google Scholar] [CrossRef]
- Lai, M.; Wang, D.; Lin, Z.; Zhang, Y. Small Molecule Copper and Its Relative Metabolites in Serum of Cerebral Ischemic Stroke Patients. J. Stroke Cerebrovasc. Dis. 2016, 25, 214–219. [Google Scholar] [CrossRef]
- Xiao, Y.; Yuan, Y.; Liu, Y.; Yu, Y.; Jia, N.; Zhou, L.; Wang, H.; Huang, S.; Zhang, Y.; Yang, H.; et al. Circulating Multiple Metals and Incident Stroke in Chinese Adults. Stroke 2019, 50, 1661–1668. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, L.-P.; Dong, X.-H.; Cai, J.; Jiang, G.-J.; Zhang, C.; Xie, H.-H. Trace Amounts of Copper in Drinking Water Aggravate Cerebral Ischemic Injury via Impairing Endothelial Progenitor Cells in Mice. CNS Neurosci. Ther. 2015, 21, 677–680. [Google Scholar] [CrossRef]
- Yang, L.; Chen, X.; Cheng, H.; Zhang, L. Dietary Copper Intake and Risk of Stroke in Adults: A Case-Control Study Based on National Health and Nutrition Examination Survey 2013–2018. Nutrients 2022, 14, 409. [Google Scholar] [CrossRef]
- Zhang, J.; Cao, J.; Zhang, H.; Jiang, C.; Lin, T.; Zhou, Z.; Song, Y.; Li, Y.; Liu, C.; Liu, L.; et al. Plasma copper and the risk of first stroke in hypertensive patients: A nested case-control study. Am. J. Clin. Nutr. 2019, 110, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Bi, C.; Lin, T.; Liu, L.; Song, Y.; Wang, P.; Wang, B.; Fang, C.; Ma, H.; Huang, X.; et al. Association between plasma copper levels and first stroke: A community-based nested case-control study. Nutr. Neurosci. 2021, 34, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Mirończuk, A.; Kapica-Topczewska, K.; Socha, K.; Soroczyńska, J.; Jamiołkowski, J.; Kułakowska, A.; Kochanowicz, J. Selenium, Copper, Zinc Concentrations and Cu/Zn, Cu/Se Molar Ratios in the Serum of Patients with Acute Ischemic Stroke in Northeastern Poland-A New Insight into Stroke Pathophysiology. Nutrients 2021, 13, 2139. [Google Scholar] [CrossRef]
- Stenudd, M.; Sabelström, H.; Frisén, J. Role of endogenous neural stem cells in spinal cord injury and repair. JAMA Neurol. 2015, 72, 235–237. [Google Scholar] [CrossRef]
- Silva, N.A.; Sousa, N.; Reis, R.L.; Salgado, A.J. From basics to clinical: A comprehensive review on spinal cord injury. Prog. Neurobiol. 2014, 114, 25–57. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Narasimhan, P.; Saito, A.; Liu, J.; Chan, P.H. Increased expression of a proline-rich Akt substrate (PRAS40) in human copper/zinc-superoxide dismutase transgenic rats protects motor neurons from death after spinal cord injury. J. Cereb. Blood Flow Metab. 2008, 28, 44–52. [Google Scholar] [CrossRef]
- Siklós, L.; Engelhardt, J.; Harati, Y.; Smith, R.G.; Joó, F.; Appel, S.H. Ultrastructural evidence for altered calcium in motor nerve terminals in amyotropic lateral sclerosis. Ann. Neurol. 1996, 39, 203–216. [Google Scholar] [CrossRef]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar] [CrossRef]
- Kong, J.; Xu, Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 1998, 18, 3241–3250. [Google Scholar] [CrossRef]
- Menzies, F.M.; Cookson, M.R.; Taylor, R.W.; Turnbull, D.M.; Chrzanowska-Lightowlers, Z.M.A.; Dong, L.; Figlewicz, D.A.; Shaw, P.J. Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain J. Neurol. 2002, 125, 1522–1533. [Google Scholar] [CrossRef]
- Fujita, K.; Yamauchi, M.; Shibayama, K.; Ando, M.; Honda, M.; Nagata, Y. Decreased cytochrome c oxidase activity but unchanged superoxide dismutase and glutathione peroxidase activities in the spinal cords of patients with amyotrophic lateral sclerosis. J. Neurosci. Res. 1996, 45, 276–281. [Google Scholar] [CrossRef]
- Borthwick, G.M.; Johnson, M.A.; Ince, P.G.; Shaw, P.J.; Turnbull, D.M. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: Implications for the role of mitochondria in neuronal cell death. Ann. Neurol. 1999, 46, 787–790. [Google Scholar] [CrossRef]
- Mattiazzi, M.; D’Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Beal, M.F.; Manfredi, G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar] [CrossRef] [PubMed]
- Curti, D.; Malaspina, A.; Facchetti, G.; Camana, C.; Mazzini, L.; Tosca, P.; Zerbi, F.; Ceroni, M. Amyotrophic lateral sclerosis: Oxidative energy metabolism and calcium homeostasis in peripheral blood lymphocytes. Neurology 1996, 47, 1060–1064. [Google Scholar] [CrossRef]
- Vijayvergiya, C.; Beal, M.F.; Buck, J.; Manfredi, G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J. Neurosci. 2005, 25, 2463–2470. [Google Scholar] [CrossRef]
- Chan, P.H.; Yang, G.Y.; Chen, S.F.; Carlson, E.; Epstein, C.J. Cold-induced brain edema and infarction are reduced in transgenic mice overexpressing CuZn-superoxide dismutase. Ann. Neurol. 1991, 29, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.-L.; Zhang, N.; Xie, X.-M.; Chen, Y.-J.; Wang, R.; Shen, L.; Zhou, J.-S.; Hu, J.-G.; Lü, H.-Z. Transcriptome profile of rat genes in injured spinal cord at different stages by RNA-sequencing. BMC Genom. 2017, 18, 173. [Google Scholar] [CrossRef] [PubMed]
- Rydén, L.; Björk, I. Reinvestigation of some physicochemical and chemical properties of human ceruloplasmin (ferroxidase). Biochemistry 1976, 15, 3411–3417. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.K.; Kim, I.H. Ceruloplasmin has a distinct active site for the catalyzing glutathione-dependent reduction of alkyl hydroperoxide. Biochemistry 1999, 38, 12104–12110. [Google Scholar] [CrossRef]
- Shi, X.; Stoj, C.; Romeo, A.; Kosman, D.J.; Zhu, Z. Fre1p Cu2+ reduction and Fet3p Cu1+ oxidation modulate copper toxicity in Saccharomyces cerevisiae. J. Biol. Chem. 2003, 278, 50309–50315. [Google Scholar] [CrossRef]
- Taylor, A.B.; Stoj, C.S.; Ziegler, L.; Kosman, D.J.; Hart, P.J. The copper-iron connection in biology: Structure of the metallo-oxidase Fet3p. Proc. Natl. Acad. Sci. USA 2005, 102, 15459–15464. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Shen, L.; Wang, R.; Tang, J.; Ding, S.-Q.; Wang, S.-N.; Guo, X.-Y.; Hu, J.-G.; Lü, H.-Z. Increased ceruloplasmin expression caused by infiltrated leukocytes, activated microglia, and astrocytes in injured female rat spinal cords. J. Neurosci. Res. 2018, 96, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Seelig, J.; Heller, R.A.; Hackler, J.; Haubruck, P.; Moghaddam, A.; Biglari, B.; Schomburg, L. Selenium and copper status—Potential signposts for neurological remission after traumatic spinal cord injury. J. Trace Elem. Med. Biol. Organ Soc. Miner. Trace Elem. 2020, 57, 126415. [Google Scholar] [CrossRef] [PubMed]
- Kaler, S.G. Inborn errors of copper metabolism. Handb. Clin. Neurol. 2013, 113, 1745–1754. [Google Scholar] [CrossRef] [PubMed]
- Rathore, K.I.; Kerr, B.J.; Redensek, A.; López-Vales, R.; Jeong, S.Y.; Ponka, P.; David, S. Ceruloplasmin protects injured spinal cord from iron-mediated oxidative damage. J. Neurosci. 2008, 28, 12736–12747. [Google Scholar] [CrossRef] [PubMed]
- Sproull, M.; Brechbiel, M.; Camphausen, K. Antiangiogenic therapy through copper chelation. Expert Opin. Targets 2003, 7, 405–409. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, W.; Zhang, H.J.; Domann, F.E.; Oberley, L.W. Overexpression of copper zinc superoxide dismutase suppresses human glioma cell growth. Cancer Res. 2002, 62, 1205–1212. [Google Scholar]
- Brem, S.; Tsanaclis, A.M.; Zagzag, D. Anticopper treatment inhibits pseudopodial protrusion and the invasive spread of 9L gliosarcoma cells in the rat brain. Neurosurgery 1990, 26, 391–396. [Google Scholar] [CrossRef]
- Brem, S.S.; Zagzag, D.; Tsanaclis, A.M.; Gately, S.; Elkouby, M.P.; Brien, S.E. Inhibition of angiogenesis and tumor growth in the brain. Suppression of endothelial cell turnover by penicillamine and the depletion of copper, an angiogenic cofactor. Am. J. Pathol. 1990, 137, 1121–1142. [Google Scholar]
- Hu, G.F. Copper stimulates proliferation of human endothelial cells under culture. J. Cell Biochem. 1998, 69, 326–335. [Google Scholar] [CrossRef]
- Ziche, M.; Jones, J.; Gullino, P.M. Role of prostaglandin E1 and copper in angiogenesis. J. Natl. Cancer Inst. 1982, 69, 475–482. [Google Scholar]
- Buccarelli, M.; D’Alessandris, Q.G.; Matarrese, P.; Mollinari, C.; Signore, M.; Cappannini, A.; Martini, M.; D’Aliberti, P.; De Luca, G.; Pedini, F.; et al. Elesclomol-induced increase of mitochondrial reactive oxygen species impairs glioblastoma stem-like cell survival and tumor growth. J. Exp. Clin. Cancer Res. CR 2021, 40, 228. [Google Scholar] [CrossRef]
- Pan, Q.; Kleer, C.G.; van Golen, K.L.; Irani, J.; Bottema, K.M.; Bias, C.; De Carvalho, M.; Mesri, E.A.; Robins, D.M.; Dick, R.D.; et al. Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis. Cancer Res. 2002, 62, 4854–4859. [Google Scholar] [PubMed]
- Nagai, M.; Vo, N.H.; Shin Ogawa, L.; Chimmanamada, D.; Inoue, T.; Chu, J.; Beaudette-Zlatanova, B.C.; Lu, R.; Blackman, R.K.; Barsoum, J.; et al. The oncology drug elesclomol selectively transports copper to the mitochondria to induce oxidative stress in cancer cells. Free Radic. Biol. Med. 2012, 52, 2142–2150. [Google Scholar] [CrossRef] [PubMed]
- Modica-Napolitano, J.S.; Bharath, L.P.; Hanlon, A.J.; Hurley, L.D. The Anticancer Agent Elesclomol Has Direct Effects on Mitochondrial Bioenergetic Function in Isolated Mammalian Mitochondria. Biomolecules 2019, 9, 298. [Google Scholar] [CrossRef] [PubMed]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [PubMed]
- Toyokuni, S.; Okamoto, K.; Yodoi, J.; Hiai, H. Persistent oxidative stress in cancer. FEBS Lett. 1995, 358, 1–3. [Google Scholar] [CrossRef]
- Rae, C.; Tesson, M.; Babich, J.W.; Boyd, M.; Sorensen, A.; Mairs, R.J. The role of copper in disulfiram-induced toxicity and radiosensitization of cancer cells. J. Nucl. Med. 2013, 54, 953–960. [Google Scholar] [CrossRef]
- Chen, S.H.; Liu, S.H.; Liang, Y.C.; Lin, J.K.; Lin-Shiau, S.Y. Oxidative stress and c-Jun-amino-terminal kinase activation involved in apoptosis of primary astrocytes induced by disulfiram-Cu(2+) complex. Eur. J. Pharmacol. 2001, 414, 177–188. [Google Scholar] [CrossRef]
- Cen, D.; Gonzalez, R.I.; Buckmeier, J.A.; Kahlon, R.S.; Tohidian, N.B.; Meyskens, F.L. Disulfiram induces apoptosis in human melanoma cells: A redox-related process. Mol. Cancer Ther. 2002, 1, 197–204. [Google Scholar]
- Daniel, K.G.; Chen, D.; Yan, B.; Dou, Q.P. Copper-binding compounds as proteasome inhibitors and apoptosis inducers in human cancer. Front. Biosci. 2007, 12, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Gupte, A.; Mumper, R.J. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat Rev. 2009, 35, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Cui, Q.C.; Yang, H.; Dou, Q.P. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 2006, 66, 10425–10433. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Zhang, Y.; Lu, L.; Zhang, H.; Zhao, C.; Pu, Y.; Yin, L. Copper induces microglia-mediated neuroinflammation through ROS/NF-κB pathway and mitophagy disorder. Food Chem. Toxicol. 2022, 168, 113369. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol. 2020, 41, 758–770. [Google Scholar] [CrossRef]
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci. 2020, 43, 144–154. [Google Scholar] [CrossRef]
- Kardos, J.; Héja, L.; Simon, Á.; Jablonkai, I.; Kovács, R.; Jemnitz, K. Copper signalling: Causes and consequences. Cell Commun. Signal. 2018, 16, 71. [Google Scholar] [CrossRef]
- Pal, A.; Badyal, R.K.; Vasishta, R.K.; Attri, S.V.; Thapa, B.R.; Prasad, R. Biochemical, histological, and memory impairment effects of chronic copper toxicity: A model for non-Wilsonian brain copper toxicosis in Wistar rat. Biol. Trace Elem. Res. 2013, 153, 257–268. [Google Scholar] [CrossRef]
- Colombo, E.; Triolo, D.; Bassani, C.; Bedogni, F.; Di Dario, M.; Dina, G.; Fredrickx, E.; Fermo, I.; Martinelli, V.; Newcombe, J.; et al. Dysregulated copper transport in multiple sclerosis may cause demyelination via astrocytes. Proc. Natl. Acad. Sci. USA 2021, 118, e2025804118. [Google Scholar] [CrossRef]
- Madsen, E.; Gitlin, J.D. Copper deficiency. Curr. Opin. Gastroenterol. 2007, 23, 187–192. [Google Scholar] [CrossRef]
- Kaler, S.G.; Holmes, C.S.; Goldstein, D.S.; Tang, J.; Godwin, S.C.; Donsante, A.; Liew, C.J.; Sato, S.; Patronas, N. Neonatal diagnosis and treatment of Menkes disease. N. Engl. J. Med. 2008, 358, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, B.H.; Kim, Y.-M.; Choi, J.-H.; Kim, G.-H.; Cheon, C.K.; Yoo, H.-W. Novel mutations and clinical outcomes of copper-histidine therapy in Menkes disease patients. Metab. Brain Dis. 2015, 30, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Duan, D.; Xu, J.; Fang, J. Redox-Dependent Copper Carrier Promotes Cellular Copper Uptake and Oxidative Stress-Mediated Apoptosis of Cancer Cells. ACS Appl. Mater. Interfaces 2018, 10, 33010–33021. [Google Scholar] [CrossRef] [PubMed]
- Aubert, L.; Nandagopal, N.; Steinhart, Z.; Lavoie, G.; Nourreddine, S.; Berman, J.; Saba-El-Leil, M.K.; Papadopoli, D.; Lin, S.; Hart, T.; et al. Copper bioavailability is a KRAS-specific vulnerability in colorectal cancer. Nat. Commun. 2020, 11, 3701. [Google Scholar] [CrossRef]
- Witt, B.; Schaumloffel, D.; Schwerdtle, T. Subcellular Localization of Copper-Cellular Bioimaging with Focus on Neurological Disorders. Int. J. Mol. Sci. 2020, 21, 2341. [Google Scholar] [CrossRef]
- Behar, A.E.; Sabater, L.; Baskin, M.; Hureau, C.; Maayan, G. A Water-Soluble Peptoid Chelator that Can Remove Cu from Amyloid-β Peptides and Stop the Formation of Reactive Oxygen Species Associated with Alzheimer’s Disease. Angew. Chem. 2021, 60, 24588–24597. [Google Scholar] [CrossRef]
- Krishnan, N.; Felice, C.; Rivera, K.; Pappin, D.J.; Tonks, N.K. DPM-1001 decreased copper levels and ameliorated deficits in a mouse model of Wilson’s disease. Genes Dev. 2018, 32, 944–952. [Google Scholar] [CrossRef]
- Telpoukhovskaia, M.A.; Orvig, C. Werner coordination chemistry and neurodegeneration. Chem. Soc. Rev. 2013, 42, 1836–1846. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| NO. | Clinical Trial Identifier | Condition/Disease | Condition/Disease | Number of Participants |
|---|---|---|---|---|
| List of the published clinical trials that highlight the application of copper chelators | ||||
| 1 | NCT04737278 | Neuralgia Myalgia | Drug: Cunermuspir Other: Placebo | 56 |
| 2 | NCT04422431 | Wilson’s Disease | Drug: Bis-choline tetrathiomolybdate | 31 |
| 3 | NCT03539952 | Wilson’s Disease | Drug: TETA 4HCL Drug: Penicillamine | 53 |
| 4 | NCT03299829 | Trientine Treatment for Wilson’s Disease | Drug: Trientine | 48 |
| 5 | NCT02273596 | Wilson’s Disease | Drug: ALXN1840 | 29 |
| 6 | NCT01472874 | Wilson’s Disease | Drug: Once a day trientine | 8 |
| 7 | NCT00325572 | Autism Pervasive Developmental Disorder | Drug: Oral zinc and vitamin C supplements Other: Oral placebo | 89 |
| 8 | NCT00113542 | Psoriasis | Drug: Tetrathiomolybdate (TM) | 10 |
| 9 | NCT00003751 | Brain and Central Nervous System Tumors | Drug: Penicillamine Radiation: Radiation therapy | 40 |
| List of the published clinical trials that highlight the application of copper supply agent | ||||
| 10 | NCT03283800 | Lipodermatosclerosis Chronic Venous Insufficiency Venous Insufficiency Varicose Veins | Other: Copper-impregnated compression stocking Other: Normal compression stocking | 16 |
| 11 | NCT03034135 | Recurrent Glioblastoma | Drug: Disulfiram/copper Drug: Temozolomide (TMZ) | 23 |
| 12 | NCT01971112 | Upper Respiratory Infections Lower Respiratory Tract Infections | Dietary Supplement: Multivitamins and minerals | 320 |
| 13 | NCT01177579 | Copper Deficiency | Dietary Supplement: Copper gluconate | 70 |
| 14 | NCT00001262 | Kinky Hair Syndrome | Drug: Copper histidine | 60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, Y.; Li, S.; Huang, X.; Chen, X.; Shan, H.; Zhang, M. The Role of Copper Homeostasis in Brain Disease. Int. J. Mol. Sci. 2022, 23, 13850. https://doi.org/10.3390/ijms232213850
An Y, Li S, Huang X, Chen X, Shan H, Zhang M. The Role of Copper Homeostasis in Brain Disease. International Journal of Molecular Sciences. 2022; 23(22):13850. https://doi.org/10.3390/ijms232213850
Chicago/Turabian StyleAn, Yumei, Sunao Li, Xinqi Huang, Xueshi Chen, Haiyan Shan, and Mingyang Zhang. 2022. "The Role of Copper Homeostasis in Brain Disease" International Journal of Molecular Sciences 23, no. 22: 13850. https://doi.org/10.3390/ijms232213850
APA StyleAn, Y., Li, S., Huang, X., Chen, X., Shan, H., & Zhang, M. (2022). The Role of Copper Homeostasis in Brain Disease. International Journal of Molecular Sciences, 23(22), 13850. https://doi.org/10.3390/ijms232213850