
In Silico Identification of Multi-Target Ligands as Promising Hit Compounds for Neurodegenerative Diseases Drug Development
 ,
,  , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
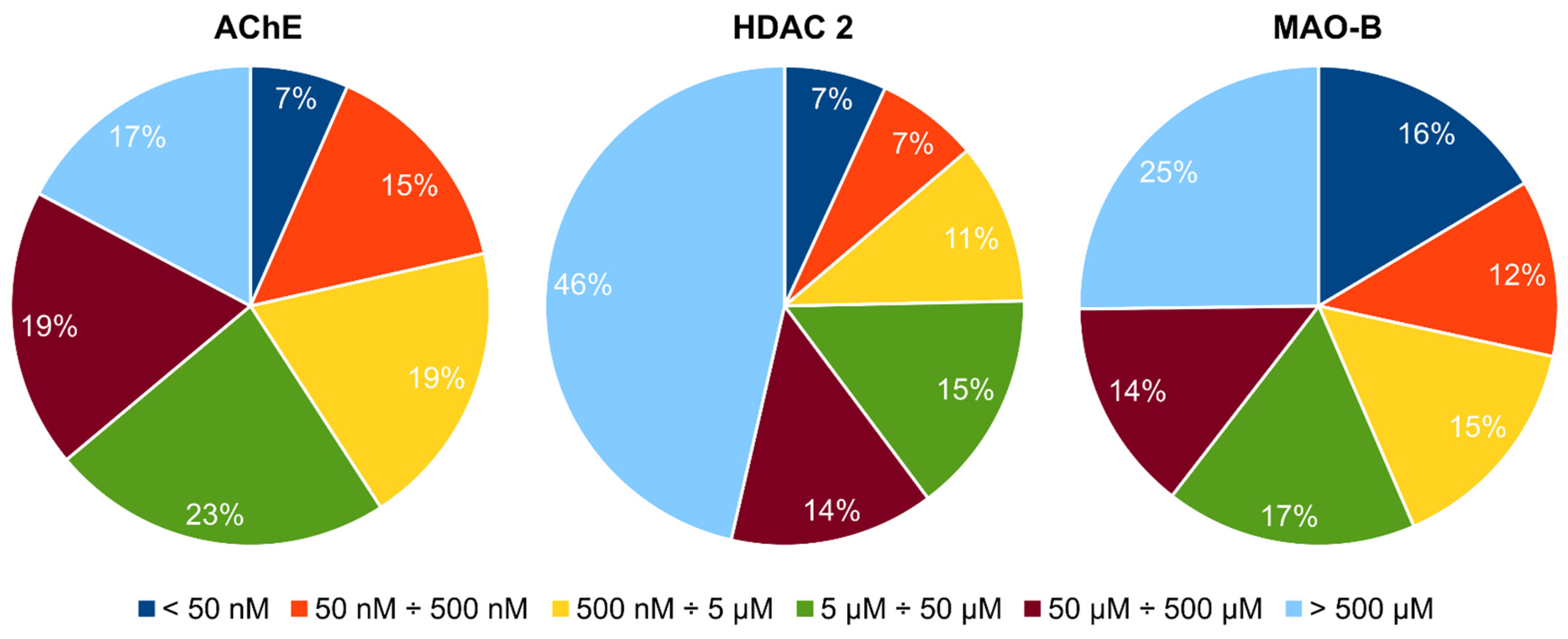
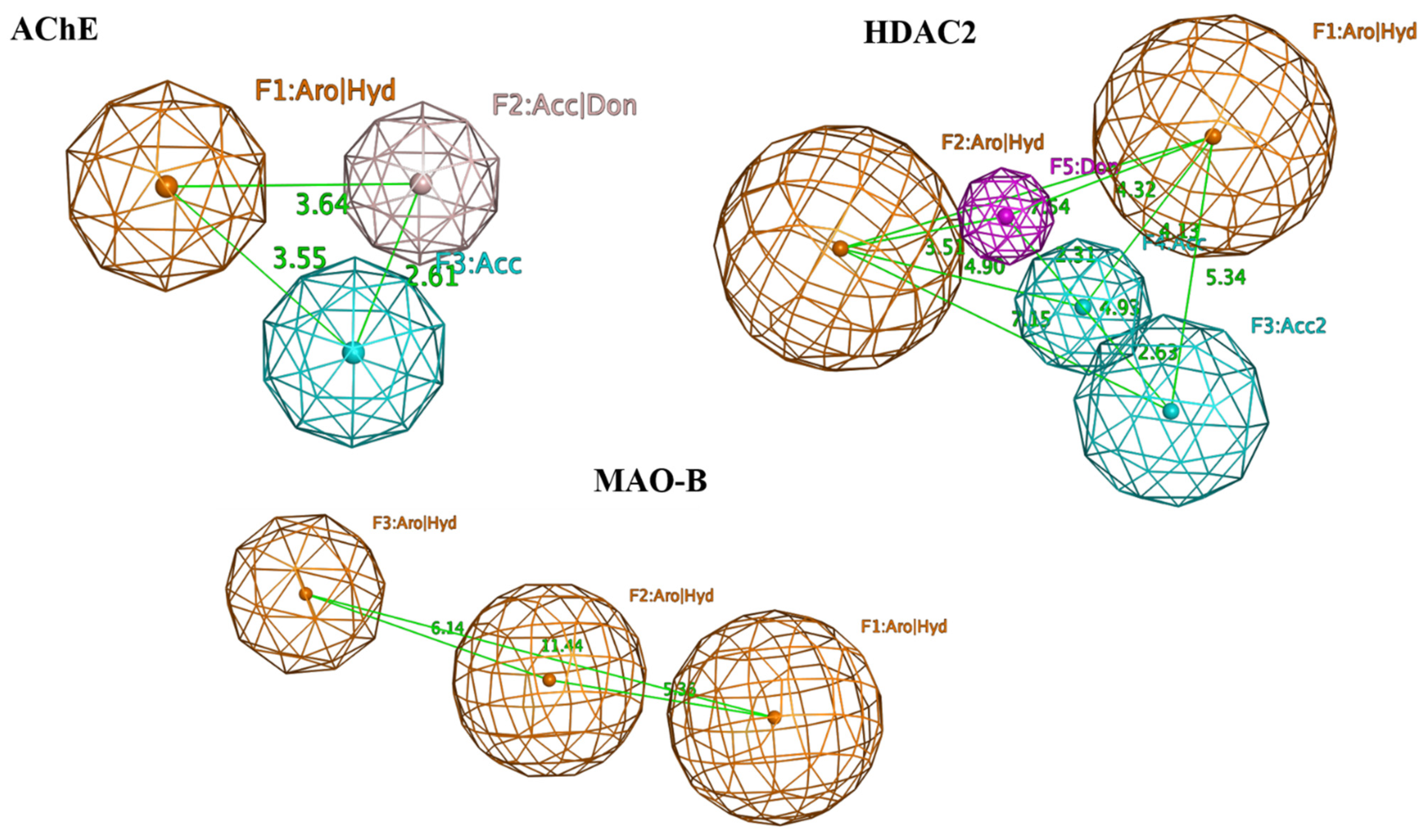
2.1. Structure-Based Virtual Screening to Identify Potential Multi-Target Ligands
2.2. BBB Penetration and Safety Profile Prediction of the Identified Hits
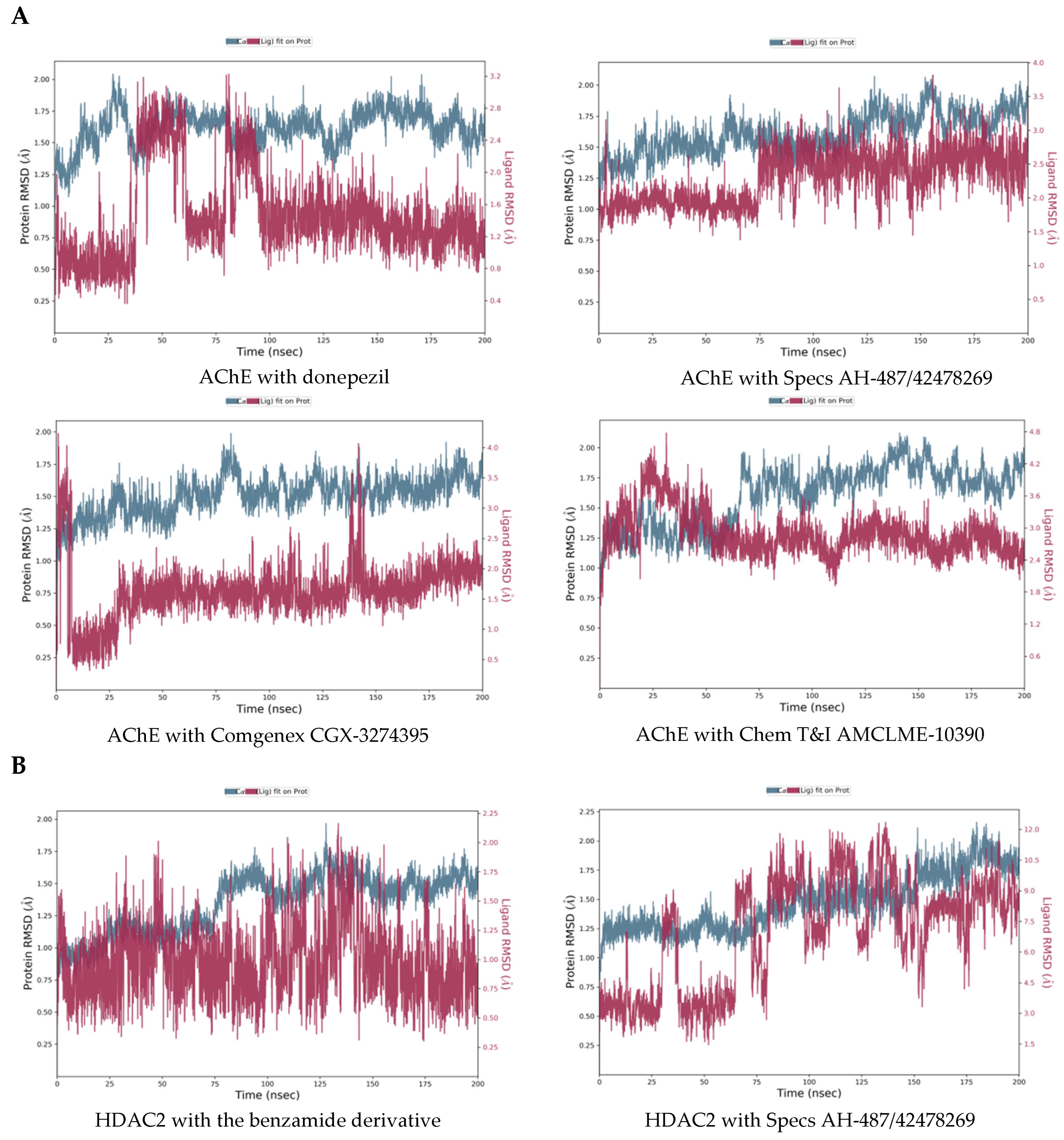
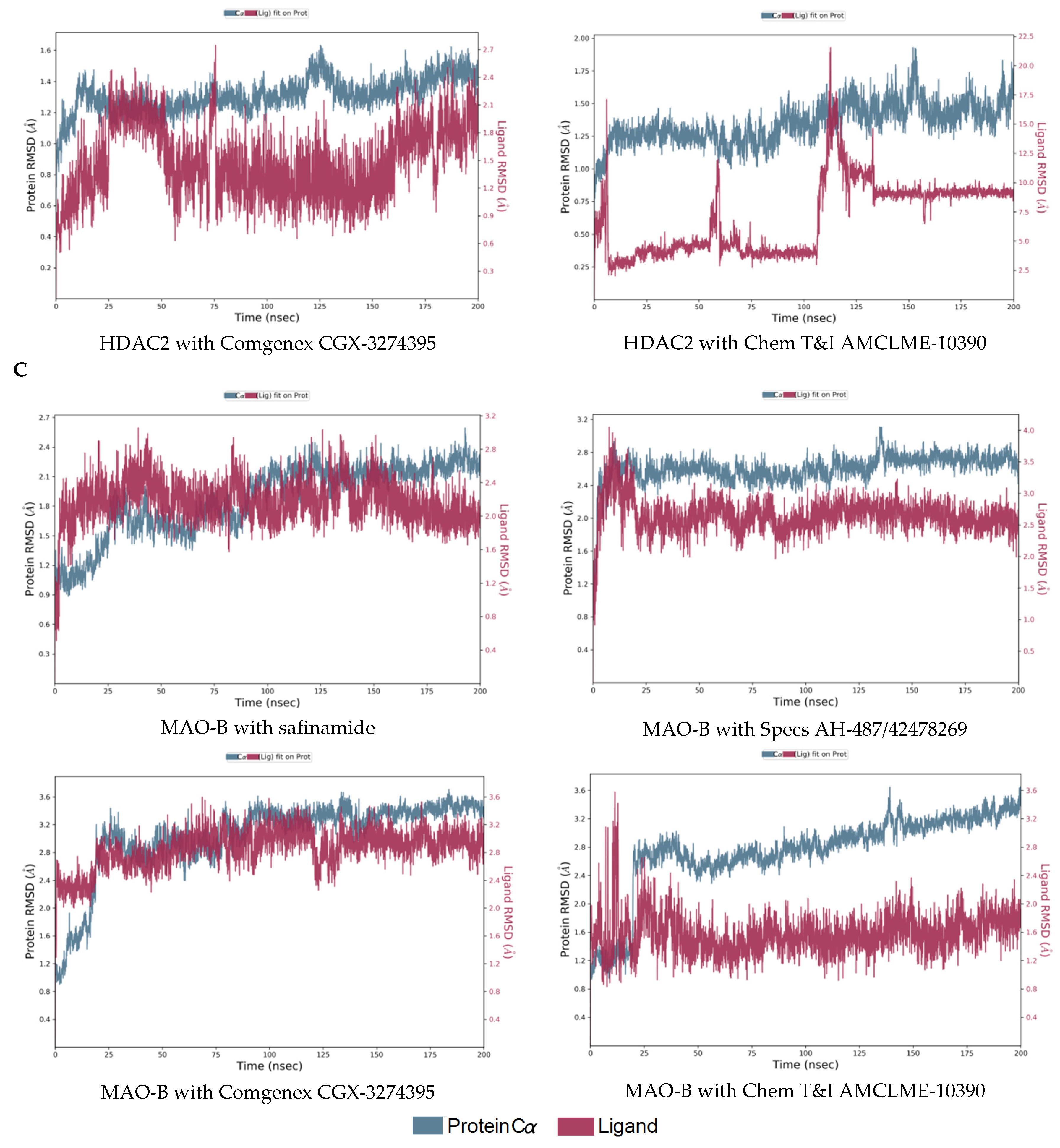
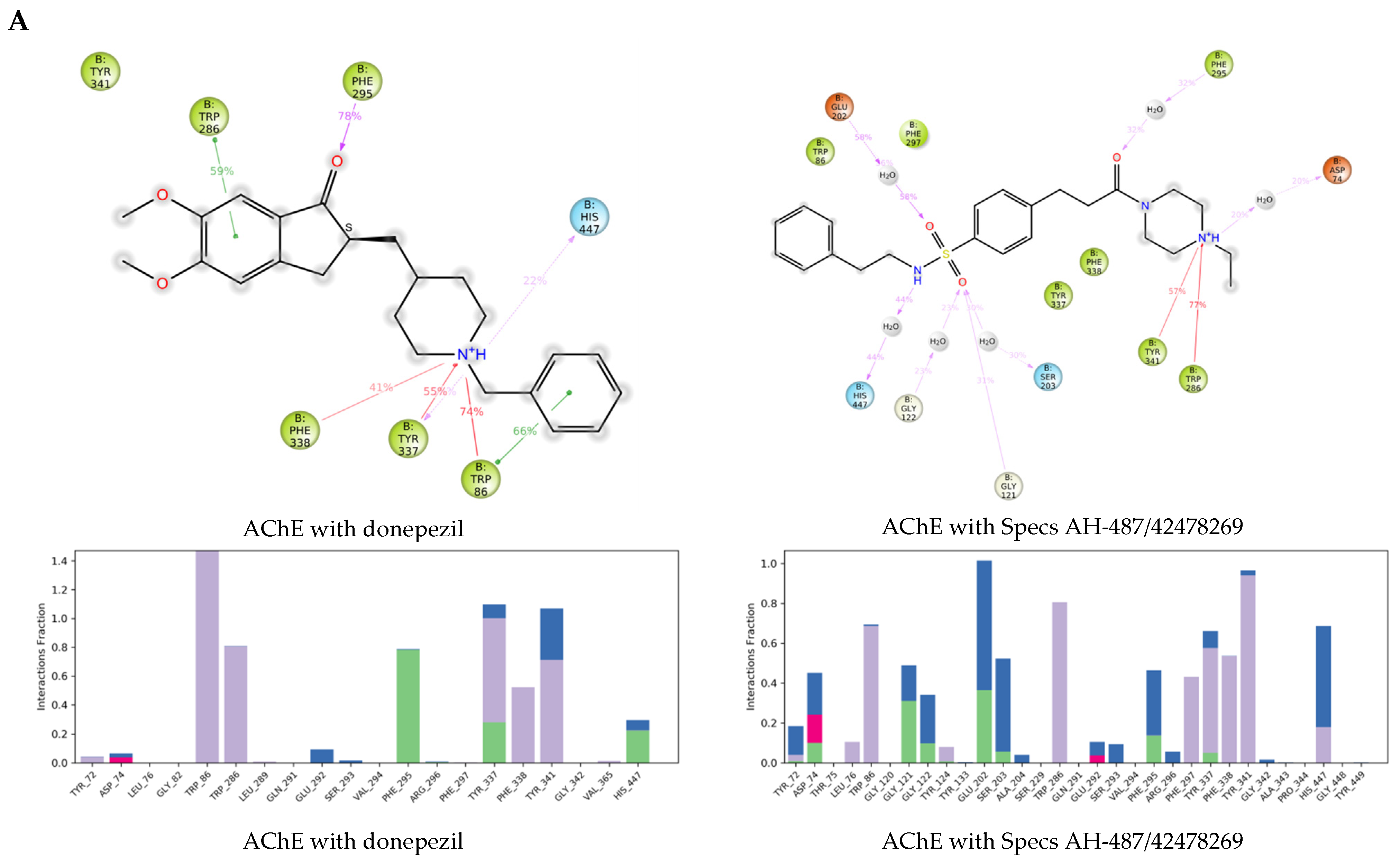
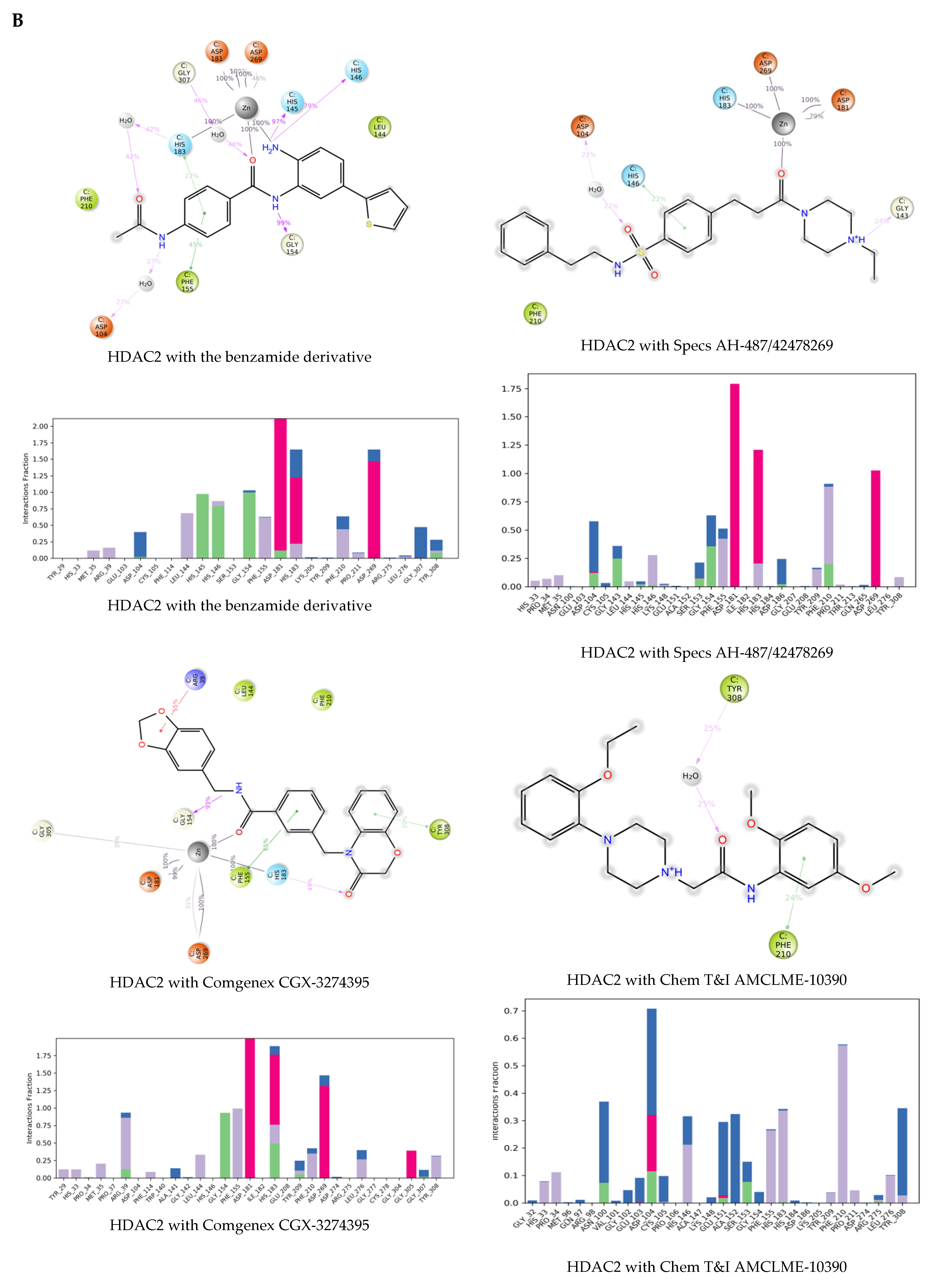
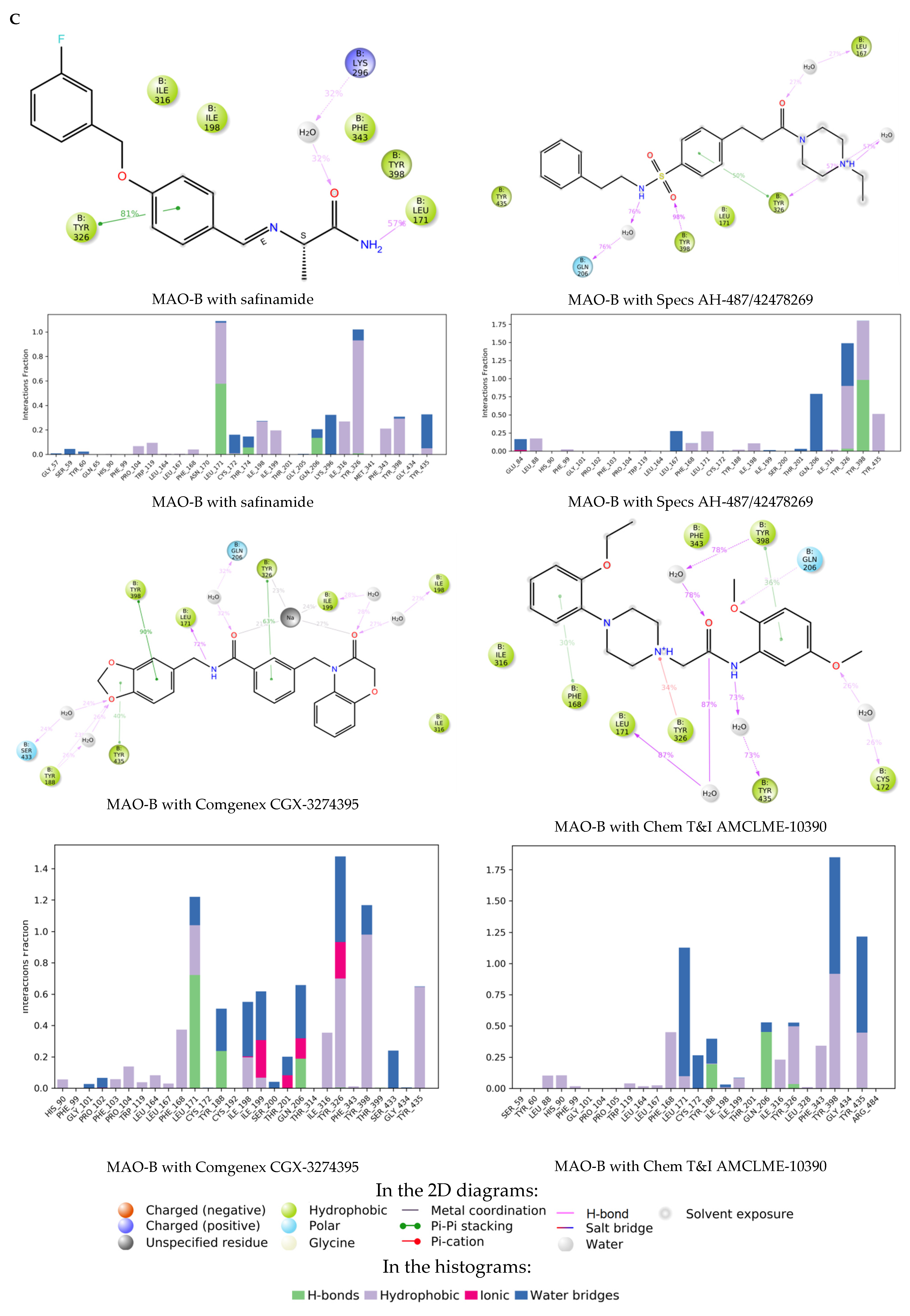
2.3. Molecular Dynamics Simulations of the Selected Hits
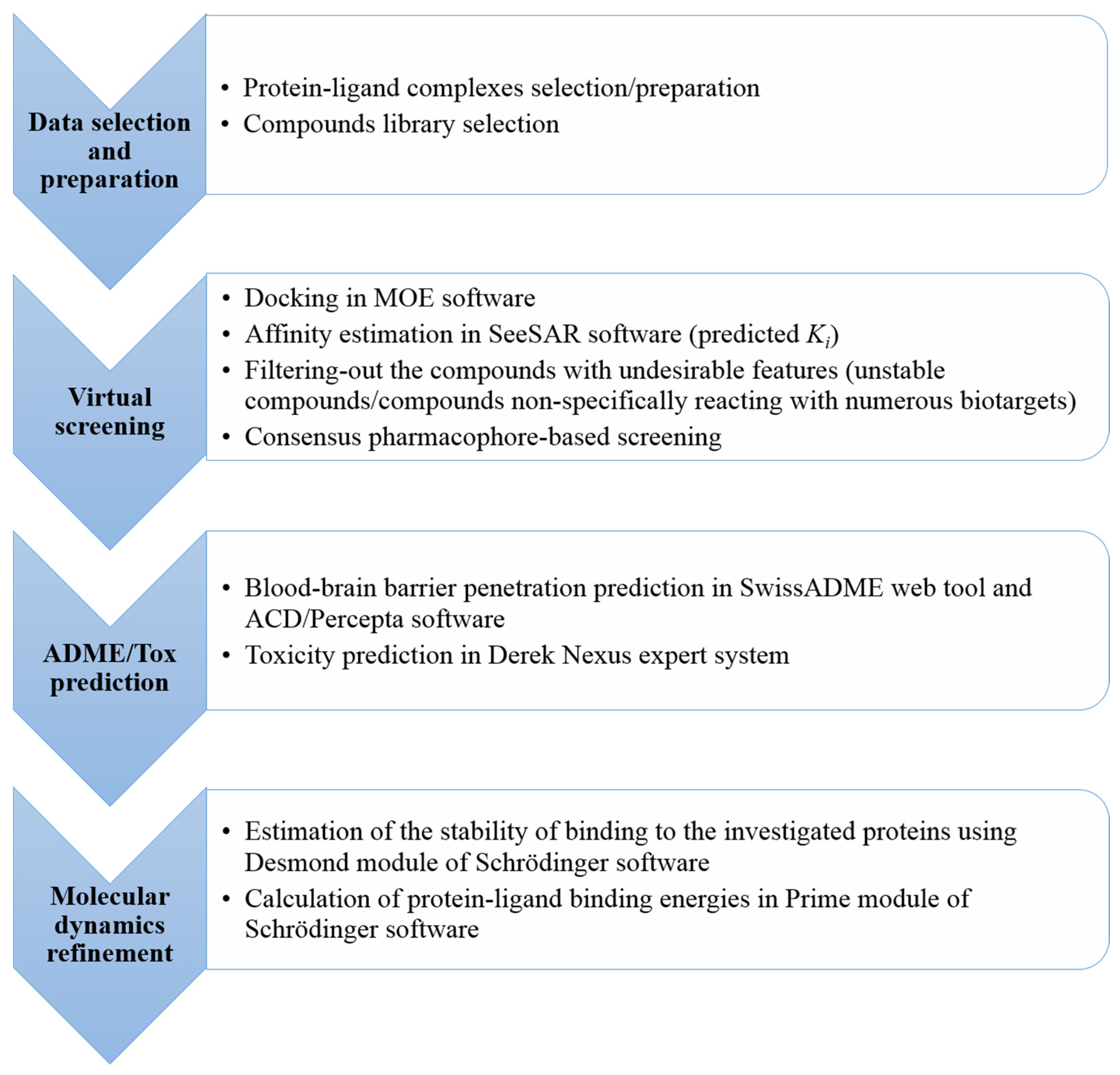
3. Materials and Methods
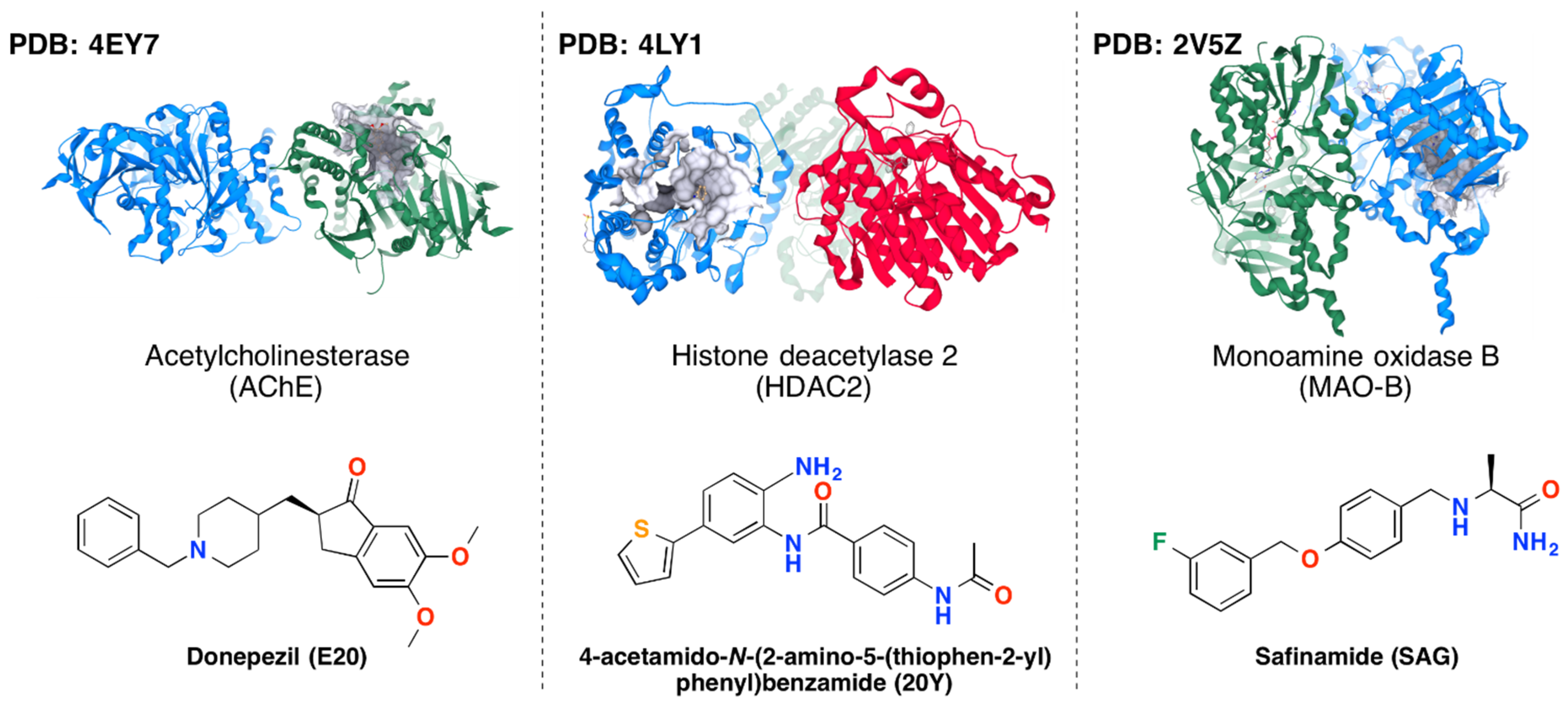
3.1. Protein-Ligand Complexes
3.2. Compounds Library
3.3. Docking and Virtual Screening
3.4. Selection of the Potential Multi-Target Ligands
3.5. Blood-Brain Barrier Penetration Prediction
3.6. Safety Profile Elucidation
3.7. Molecular Dynamics Simulations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Przedborski, S. The Two-Century Journey of Parkinson Disease Research. Nat. Rev. Neurosci. 2017, 18, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Tzvetkov, N.T.; Stammler, H.-G.; Neumann, B.; Hristova, S.; Antonov, L.; Gastreich, M. Crystal Structures, Binding Interactions, and ADME Evaluation of Brain Penetrant N-Substituted Indazole-5-Carboxamides as Subnanomolar, Selective Monoamine Oxidase B and Dual MAO-A/B Inhibitors. Eur. J. Med. Chem. 2017, 127, 470–492. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Fitzgerald, J.C.; Stegen, K.; Westermeier, J.; Thost, A.-K.; Kato, H.; Mokranjac, D.; Sauerwald, J.; Martins, L.M.; Woitalla, D.; et al. Mitochondrial Proteolytic Stress Induced by Loss of Mortalin Function Is Rescued by Parkin and PINK1. Cell. Death Dis. 2014, 5, e1180. [Google Scholar] [CrossRef] [PubMed]
- Iarkov, A.; Barreto, G.E.; Grizzell, J.A.; Echeverria, V. Strategies for the Treatment of Parkinson’s Disease: Beyond Dopamine. Front. Aging Neurosci. 2020, 12, 4. [Google Scholar] [CrossRef]
- Tzvetkov, N.T.; Atanasov, A.G. Natural Product-Based Multitargeted Ligands for Alzheimer’s Disease Treatment? Future Med. Chem. 2018, 10, 1745–1748. [Google Scholar] [CrossRef] [PubMed]
- Tzvetkov, N.T.; Stammler, H.-G.; Hristova, S.; Atanasov, A.G.; Antonov, L. (Pyrrolo-Pyridin-5-Yl)Benzamides: BBB Permeable Monoamine Oxidase B Inhibitors with Neuroprotective Effect on Cortical Neurons. Eur. J. Med. Chem. 2019, 162, 793–809. [Google Scholar] [CrossRef] [PubMed]
- Van Bulck, M.; Sierra-Magro, A.; Alarcon-Gil, J.; Perez-Castillo, A.; Morales-Garcia, J.A. Novel Approaches for the Treatment of Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 719. [Google Scholar] [CrossRef]
- Ellis, J.M.; Fell, M.J. Current Approaches to the Treatment of Parkinson’s Disease. Bioorganic Med. Chem. Lett. 2017, 27, 4247–4255. [Google Scholar] [CrossRef]
- Abbott, A. Levodopa: The Story so Far. Nature 2010, 466, S6–S7. [Google Scholar] [CrossRef]
- Guan, J.-S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.-H.; Joseph, N.; Gao, J.; Nieland, T.J.F.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 Negatively Regulates Memory Formation and Synaptic Plasticity. Nature 2009, 459, 55–60. [Google Scholar] [CrossRef]
- Govindarajan, N.; Rao, P.; Burkhardt, S.; Sananbenesi, F.; Schlüter, O.M.; Bradke, F.; Lu, J.; Fischer, A. Reducing HDAC6 Ameliorates Cognitive Deficits in a Mouse Model for Alzheimer’s Disease. EMBO Mol. Med. 2013, 5, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Poplawski, S.G.; Garbett, K.A.; McMahan, R.L.; Kordasiewicz, H.B.; Zhao, H.; Kennedy, A.J.; Goleva, S.B.; Sanders, T.H.; Motley, S.T.; Swayze, E.E.; et al. An Antisense Oligonucleotide Leads to Suppressed Transcription of Hdac2 and Long-Term Memory Enhancement. Mol. Ther. Nucleic Acids 2020, 19, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Taliyan, R. Targeting Histone Deacetylases: A Novel Approach in Parkinson’s Disease. Park. Dis. 2015, 2015, e303294. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Tekwani, B.L. Histone Deacetylases Inhibitors in Neurodegenerative Diseases, Neuroprotection and Neuronal Differentiation. Front. Pharmacol. 2020, 11, 537. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, X.; Liu, F.; Li, S.; Shi, D. Rational Multitargeted Drug Design Strategy from the Perspective of a Medicinal Chemist. J. Med. Chem. 2021, 64, 10581–10605. [Google Scholar] [CrossRef]
- Bawa, P.; Pradeep, P.; Kumar, P.; Choonara, Y.E.; Modi, G.; Pillay, V. Multi-Target Therapeutics for Neuropsychiatric and Neurodegenerative Disorders. Drug Discov. Today 2016, 21, 1886–1914. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-Target-Directed Ligands To Combat Neurodegenerative Diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef]
- Jaiteh, M.; Zeifman, A.; Saarinen, M.; Svenningsson, P.; Bréa, J.; Loza, M.I.; Carlsson, J. Docking Screens for Dual Inhibitors of Disparate Drug Targets for Parkinson’s Disease. J. Med. Chem. 2018, 61, 5269–5278. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Gallo-Rodriguez, C.; Melman, N.; Fischer, B.; Maillard, M.; van Bergen, A.; van Galen, P.J.; Karton, Y. Structure-Activity Relationships of 8-Styrylxanthines as A2-Selective Adenosine Antagonists. J. Med. Chem. 1993, 36, 1333–1342. [Google Scholar] [CrossRef]
- Chen, J.-F.; Steyn, S.; Staal, R.; Petzer, J.P.; Xu, K.; der Schyf, C.J.V.; Castagnoli, K.; Sonsalla, P.K.; Castagnoli, N.; Schwarzschild, M.A. 8-(3-Chlorostyryl)Caffeine May Attenuate MPTP Neurotoxicity through Dual Actions of Monoamine Oxidase Inhibition and A2A Receptor Antagonism. J. Biol. Chem. 2002, 277, 36040–36044. [Google Scholar] [CrossRef]
- Perez-Castillo, Y.; Helguera, A.M.; Cordeiro, M.N.D.S.; Tejera, E.; Paz-y-Mino, C.; Sanchez-Rodriguez, A.; Borges, F.; Cruz-Monteagudo, M. Fusing Docking Scoring Functions Improves the Virtual Screening Performance for Discovering Parkinson’s Disease Dual Target Ligands. Curr. Neuropharmacol. 2017, 15, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Ganai, S.A.; Abdullah, E.; Rashid, R.; Altaf, M. Combinatorial In Silico Strategy towards Identifying Potential Hotspots during Inhibition of Structurally Identical HDAC1 and HDAC2 Enzymes for Effective Chemotherapy against Neurological Disorders. Front. Mol. Neurosci 2017, 10, 357. [Google Scholar] [CrossRef] [PubMed]
- Sadri-Vakili, G.; Cha, J.-H.J. Histone Deacetylase Inhibitors: A Novel Therapeutic Approach to Huntington’s Disease (Complex Mechanism of Neuronal Death). Curr. Alzheimer Res. 2006, 3, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Dai, X.-L.; Huang, H.-C.; Jiang, Z.-F. Targeting HDACs: A Promising Therapy for Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2011, 2011, e143269. [Google Scholar] [CrossRef] [PubMed]
- Khalid, S.; Zahid, M.A.; Ali, H.; Kim, Y.S.; Khan, S. Biaryl Scaffold-Focused Virtual Screening for Anti-Aggregatory and Neuroprotective Effects in Alzheimer’s Disease. BMC Neurosci. 2018, 19, 74. [Google Scholar] [CrossRef] [PubMed]
- Baig, M.H.; Ahmad, K.; Roy, S.; Ashraf, J.M.; Adil, M.; Siddiqui, M.H.; Khan, S.; Kamal, M.A.; Provazník, I.; Choi, I. Computer Aided Drug Design: Success and Limitations. Curr. Pharm. Des. 2016, 22, 572–581. [Google Scholar] [CrossRef]
- Song, C.M.; Lim, S.J.; Tong, J.C. Recent Advances in Computer-Aided Drug Design. Brief. Bioinform. 2009, 10, 579–591. [Google Scholar] [CrossRef]
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of Computer-Aided Drug Design in Modern Drug Discovery. Arch. Pharm. Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Aldrich, C.; Bertozzi, C.; Georg, G.I.; Kiessling, L.; Lindsley, C.; Liotta, D.; Merz, K.M., Jr.; Schepartz, A.; Wang, S. The Ecstasy and Agony of Assay Interference Compounds. J. Med. Chem. 2017, 60, 2165–2168. [Google Scholar] [CrossRef]
- Baell, J.; Walters, M.A. Chemistry: Chemical Con Artists Foil Drug Discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef]
- McGovern, S.L.; Caselli, E.; Grigorieff, N.; Shoichet, B.K. A Common Mechanism Underlying Promiscuous Inhibitors from Virtual and High-Throughput Screening. J. Med. Chem. 2002, 45, 1712–1722. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Saubern, S.; Guha, R.; Baell, J.B. KNIME Workflow to Assess PAINS Filters in SMARTS Format. Comparison of RDKit and Indigo Cheminformatics Libraries. Mol. Inform. 2011, 30, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Tzvetkov, N.T.; Stammler, H.-G.; Georgieva, M.G.; Russo, D.; Faraone, I.; Balacheva, A.A.; Hristova, S.; Atanasov, A.G.; Milella, L.; Antonov, L.; et al. Carboxamides vs. Methanimines: Crystal Structures, Binding Interactions, Photophysical Studies, and Biological Evaluation of (Indazole-5-Yl)Methanimines as Monoamine Oxidase B and Acetylcholinesterase Inhibitors. Eur. J. Med. Chem. 2019, 179, 404–422. [Google Scholar] [CrossRef] [PubMed]
- Bruns, R.F.; Watson, I.A. Rules for Identifying Potentially Reactive or Promiscuous Compounds. J. Med. Chem. 2012, 55, 9763–9772. [Google Scholar] [CrossRef]
- Hughes, J.; Rees, S.; Kalindjian, S.; Philpott, K. Principles of Early Drug Discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Blomme, E.A.G.; Will, Y. Toxicology Strategies for Drug Discovery: Present and Future. Chem. Res. Toxicol. 2016, 29, 473–504. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Cheung, J.; Gary, E.N.; Shiomi, K.; Rosenberry, T.L. Structures of Human Acetylcholinesterase Bound to Dihydrotanshinone I and Territrem B Show Peripheral Site Flexibility. ACS Med. Chem. Lett. 2013, 4, 1091–1096. [Google Scholar] [CrossRef]
- Lauffer, B.E.L.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone Deacetylase (HDAC) Inhibitor Kinetic Rate Constants Correlate with Cellular Histone Acetylation but Not Transcription and Cell Viability. J. Biol. Chem. 2013, 288, 26926–26943. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E.; Mattevi, A. Structures of Human Monoamine Oxidase B Complexes with Selective Noncovalent Inhibitors: Safinamide and Coumarin Analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef] [PubMed]
- Schneider, N.; Hindle, S.; Lange, G.; Klein, R.; Albrecht, J.; Briem, H.; Beyer, K.; Claußen, H.; Gastreich, M.; Lemmen, C.; et al. Substantial Improvements in Large-Scale Redocking and Screening Using the Novel HYDE Scoring Function. J. Comput. Aided Mol. Des. 2012, 26, 701–723. [Google Scholar] [CrossRef] [PubMed]
- Heitler, W.; London, F. Wechselwirkung neutraler Atome und homöopolare Bindung nach der Quantenmechanik. Z. Phys. 1927, 44, 455–472. [Google Scholar] [CrossRef]
- Labute, P. The Generalized Born/Volume Integral Implicit Solvent Model: Estimation of the Free Energy of Hydration Using London Dispersion Instead of Atomic Surface Area. J. Comput. Chem. 2008, 29, 1693–1698. [Google Scholar] [CrossRef]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A Fast Flexible Docking Method Using an Incremental Construction Algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef]
- Böhm, H.-J. The Development of a Simple Empirical Scoring Function to Estimate the Binding Constant for a Protein-Ligand Complex of Known Three-Dimensional Structure. J. Comput. Aided Mol. Des. 1994, 8, 243–256. [Google Scholar] [CrossRef]
- Schneider, N.; Lange, G.; Hindle, S.; Klein, R.; Rarey, M. A Consistent Description of HYdrogen Bond and DEhydration Energies in Protein–Ligand Complexes: Methods behind the HYDE Scoring Function. J. Comput. Aided Mol. Des. 2013, 27, 15–29. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
- Marchant, C.A.; Briggs, K.A.; Long, A. In Silico Tools for Sharing Data and Knowledge on Toxicity and Metabolism: Derek for Windows, Meteor, and Vitic. Toxicol. Mech. Methods 2008, 18, 177–187. [Google Scholar] [CrossRef]
- Judson, P.N.; Stalford, S.A.; Vessey, J. Assessing Confidence in Predictions Made by Knowledge-Based Systems. Toxicol. Res. 2013, 2, 70–79. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; Association for Computing Machinery: New York, NY, USA; p. 84. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme (PDB Code) | Docking Protocol | Docking Scores of the Reference Ligands, kcal/mol | ||
|---|---|---|---|---|
| Safinamide | Benzamide Derivative | Donepezil | ||
| AChE (4EY7) | MOE rigid | −13.05 ÷ −11.16 | −12.13 ÷ −11.43 | −14.71 ÷ −12.64 |
| MOE flexible | −7.35 ÷ −5.83 | −7.84 ÷ −6.62 | −8.52 ÷ −6.29 | |
| HDAC2 (4LYI) | MOE rigid | −11.72 ÷ −10.42 | −13.01 ÷ −11.30 | −13.24 ÷ −10.92 |
| MOE flexible | −7.17 ÷ −5.62 | −7.68 ÷ −6.74 | −8.51 ÷ −6.10 | |
| MAO-B (2V5Z) | MOE rigid | −13.56 ÷ −12.45 | −7.84 ÷ −4.66 | −9.12 ÷ −5.28 |
| MOE flexible | −8.34 ÷ −7.69 | −9.08 ÷ −8.17 | −9.96 ÷ −8.31 | |
| Virtual Screening Steps | Number of Docked Compounds | Number of Compounds Passing the Criteria for Inclusion in the Next Step |
|---|---|---|
| MOE rigid docking | 653,214 | 11,085 |
| MOE flexible docking | 11,085 | 1011 |
| SeeSAR flexible docking | 1011 | 445 |
| PAINS filtering | 445 | 377 |
| Affinity constraints | 377 | 16 |
| Pharmacophore filtering | 16 | 4 |
| No. | Name/Structure | Ki Predicted (nM) | Multi-Plication Product | ||
|---|---|---|---|---|---|
| AChE | HDAC2 | MAO-B | |||
| 1 | Specs AH-487/42478269 S(=O)(=O)(NCCc1ccccc1)c2ccc(cc2)CCC(=O)N3CC[NH+](CC3)CC | 131.0 | 5.971 | 0.6379 | 497.4 |
| 2 | Tripos 1503-03309 Clc1ncccc1OC[C@@H]2OCCN(C(=O)CSc3cc(OC)c(OC)cc3)C2 | 3.808 | 1615 | 1.784 | 10,970 |
| 3 | Chembridge 7905648 Clc1c(cc(OCC)c(OCC(=O)NCCc2ccccc2)c1)C[NH2+]CCO | 4346 | 6.160 | 0.5084 | 13,610 |
| 4 | Tripos 1526-25537 S=C(N1CCN(c2c(OCC)cccc2)CC1)Nc3cc4OCOc4cc3 | 17.04 | 868.2 | 0.9270 | 13,720 |
| 5 | EMC Microcollections 010F0838 O=C(N[C@H](CCCC(C)C)C)[C@@H]1[C@@H](C(=O)NCc2c3c(ccc2)cccc3)C[NH2+]C1 | 337.9 | 3339 | 0.1196 | 135,000 |
| 6 | Akos LT-1164 X 260 O=C(OC)c1cc(cc(c1)C=2OC(=CC2)C[NH2+]CCC=3c4c(NC3)cccc4)C(=O)OC | 59.21 | 937.0 | 3.000 | 166,400 |
| 7 | Chembridge 7928210 S(=O)(=O)(NCC(C)C)c1cc(c(OCC(=O)Nc2cc(ccc2)C(=O)C)cc1)C | 109.6 | 11.56 | 309.0 | 391,700 |
| 8 | Comgenex CGX-3274395 O=C(NCc1cc2OCOc2cc1)c3cc(ccc3)CN4c5c(OCC4=O)cccc5 | 14.15 | 13.76 | 3658 | 712,200 |
| 9 | Chem T&I SHCLME-048161 Clc1c(NC(=O)CSC2=NN=C(N2C)[C@@H](Oc3ccccc3)C)cc([N+]([O-])=O)cc1 | 34.19 | 72.17 | 439.6 | 1,085,000 |
| 10 | Chem T&I AMCLME-10390 O=C(Nc1c(OC)ccc(OC)c1)C[NH+]2CCN(c3c(OCC)cccc3)CC2 | 335.9 | 930.0 | 5.961 | 1,862,000 |
| 11 | Chemdiv 4378-0361 Clc1ccc(cc1)CS(=O)(=O)C[C@@H](O)CSc2nc3c(cc2C#N)CCCC3 | 88.81 | 517.3 | 93.30 | 4,287,000 |
| 12 | Tripos 1547-01361 O(Cc1ccccc1)C[C@@H](O)C[NH+]2CCN(c3c4c(nc(c3)C)cccc4)CC2 | 4785 | 201.3 | 9.489 | 9,142,000 |
| 13 | Chem T&I AMCLME-01759 S1C(NC(=O)C[NH+]2CC[NH+](Cc3cc(OC)ccc3)CC2)=C(C#N)C4=C1CCCCC4 | 727.6 | 2412 | 5.263 | 9,238,000 |
| 14 | Asinex ASN 04448308 CCN(CC)S(=O)(=O)c1ccc(cc1)S(=O)(=O)NCCc1ccc2OCOc2c1 | 2203 | 323.0 | 52.53 | 37,370,000 |
| 15 | Princeton Biomolecular OSSK_456453 [O-]C(=O)C(CCCC)NC(=O)C(C)Oc1ccc2c(c1)OC(=O)C=1CCCC2=1 | 65.99 | 1545 | 420.6 | 42,880,000 |
| 16 | Asinex BAS 07211091 O=S(=O)(NCc1ccccc1)c1ccc(cc1)OCC(=O)NCc1ccncc1 | 4794 | 6.599 | 2053 | 64,960,000 |
| Structure/Name | BBB Prediction | BBB Prediction (Consensus) | Derek Nexus Toxicity Prediction | |
|---|---|---|---|---|
| SwissADME | ACD/Percepta | |||
 1. Specs AH-487/42478269 | non-penetrant | penetrant | penetrant | No |
 2. Comgenex CGX-3274395 | penetrant | weak penetrant | penetrant | No |
 3. Chem T&I AMCLME-10390 | non-penetrant | penetrant | penetrant | hepatotoxicity cardiotoxicity a |
 4. Asinex BAS 07211091 | non-penetrant | weak penetrant | non-penetrant | No |
| Ligand | AChE | HDAC2 | MAO-B |
|---|---|---|---|
| Crystallographic ligand | −71.49 | −21.41 | −45.29 |
| Specs AH-487/42478269 | −76.01 | −30.16 | −77.37 |
| Comgenex CGX-3274395 | −72.83 | −45.41 | −83.35 |
| Chem T&I AMCLME-10390 | −82.14 | −48.94 | −74.57 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alov, P.; Stoimenov, H.; Lessigiarska, I.; Pencheva, T.; Tzvetkov, N.T.; Pajeva, I.; Tsakovska, I. In Silico Identification of Multi-Target Ligands as Promising Hit Compounds for Neurodegenerative Diseases Drug Development. Int. J. Mol. Sci. 2022, 23, 13650. https://doi.org/10.3390/ijms232113650
Alov P, Stoimenov H, Lessigiarska I, Pencheva T, Tzvetkov NT, Pajeva I, Tsakovska I. In Silico Identification of Multi-Target Ligands as Promising Hit Compounds for Neurodegenerative Diseases Drug Development. International Journal of Molecular Sciences. 2022; 23(21):13650. https://doi.org/10.3390/ijms232113650
Chicago/Turabian StyleAlov, Petko, Hristo Stoimenov, Iglika Lessigiarska, Tania Pencheva, Nikolay T. Tzvetkov, Ilza Pajeva, and Ivanka Tsakovska. 2022. "In Silico Identification of Multi-Target Ligands as Promising Hit Compounds for Neurodegenerative Diseases Drug Development" International Journal of Molecular Sciences 23, no. 21: 13650. https://doi.org/10.3390/ijms232113650
APA StyleAlov, P., Stoimenov, H., Lessigiarska, I., Pencheva, T., Tzvetkov, N. T., Pajeva, I., & Tsakovska, I. (2022). In Silico Identification of Multi-Target Ligands as Promising Hit Compounds for Neurodegenerative Diseases Drug Development. International Journal of Molecular Sciences, 23(21), 13650. https://doi.org/10.3390/ijms232113650