
Electrostatic Interactions Are the Primary Determinant of the Binding Affinity of SARS-CoV-2 Spike RBD to ACE2: A Computational Case Study of Omicron Variants
 ,
,
Abstract
1. Introduction
2. Results
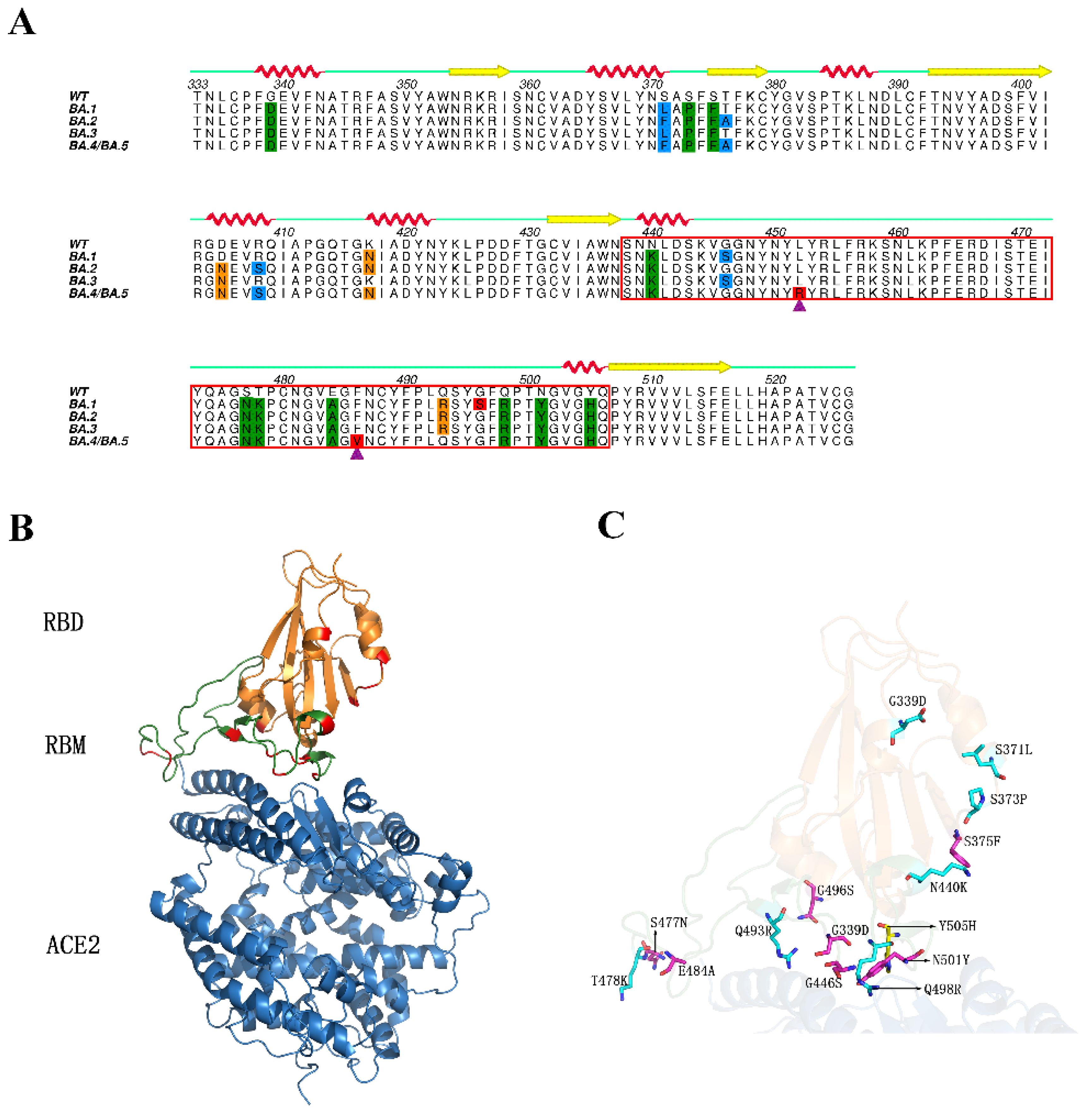
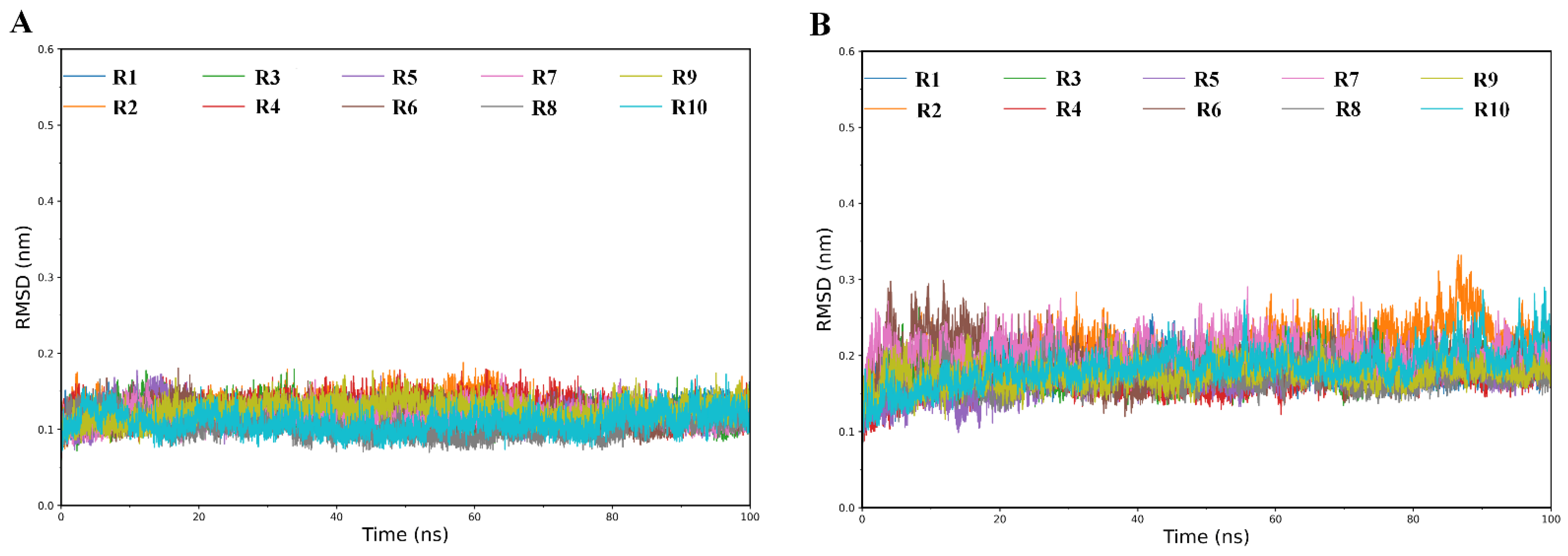
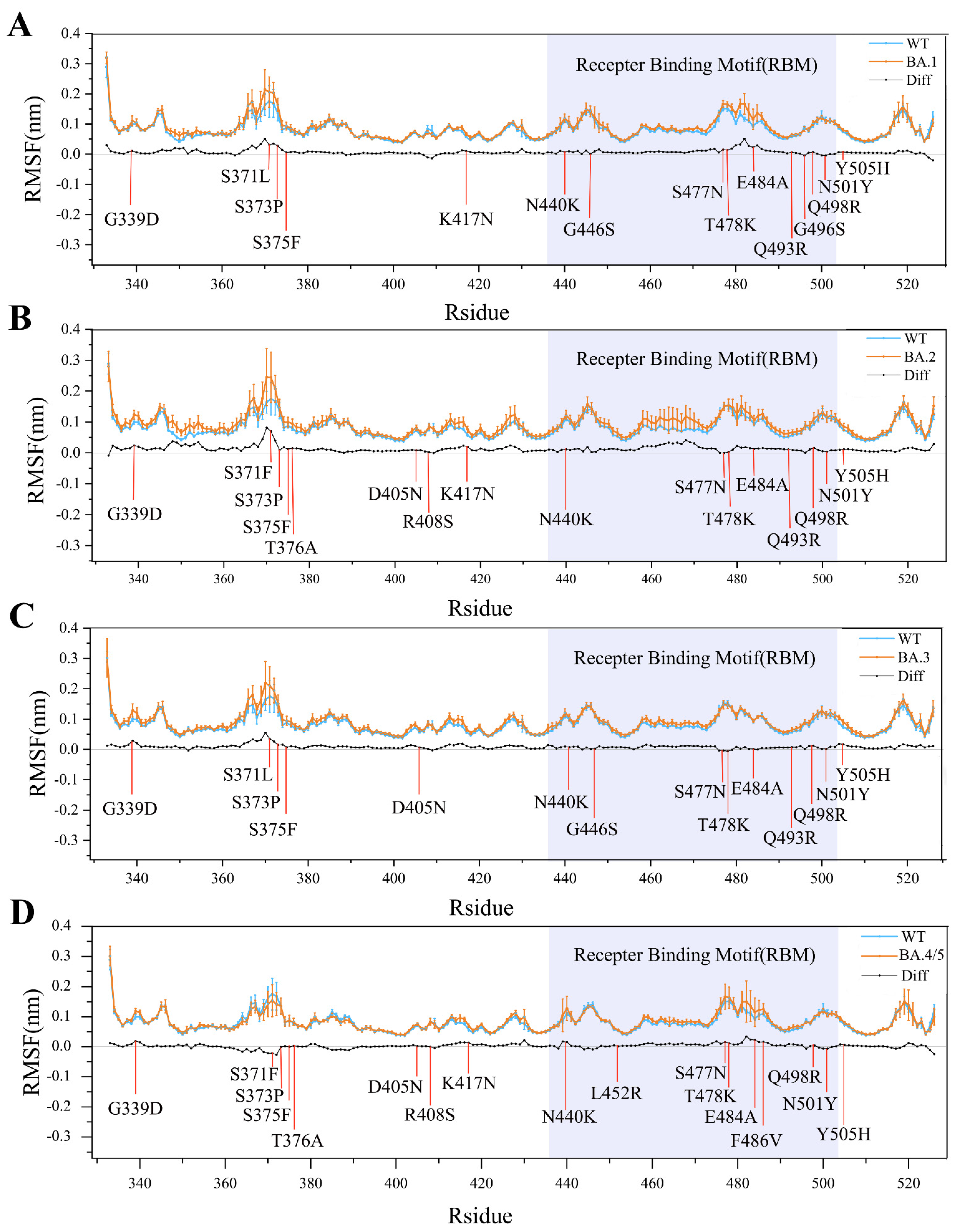
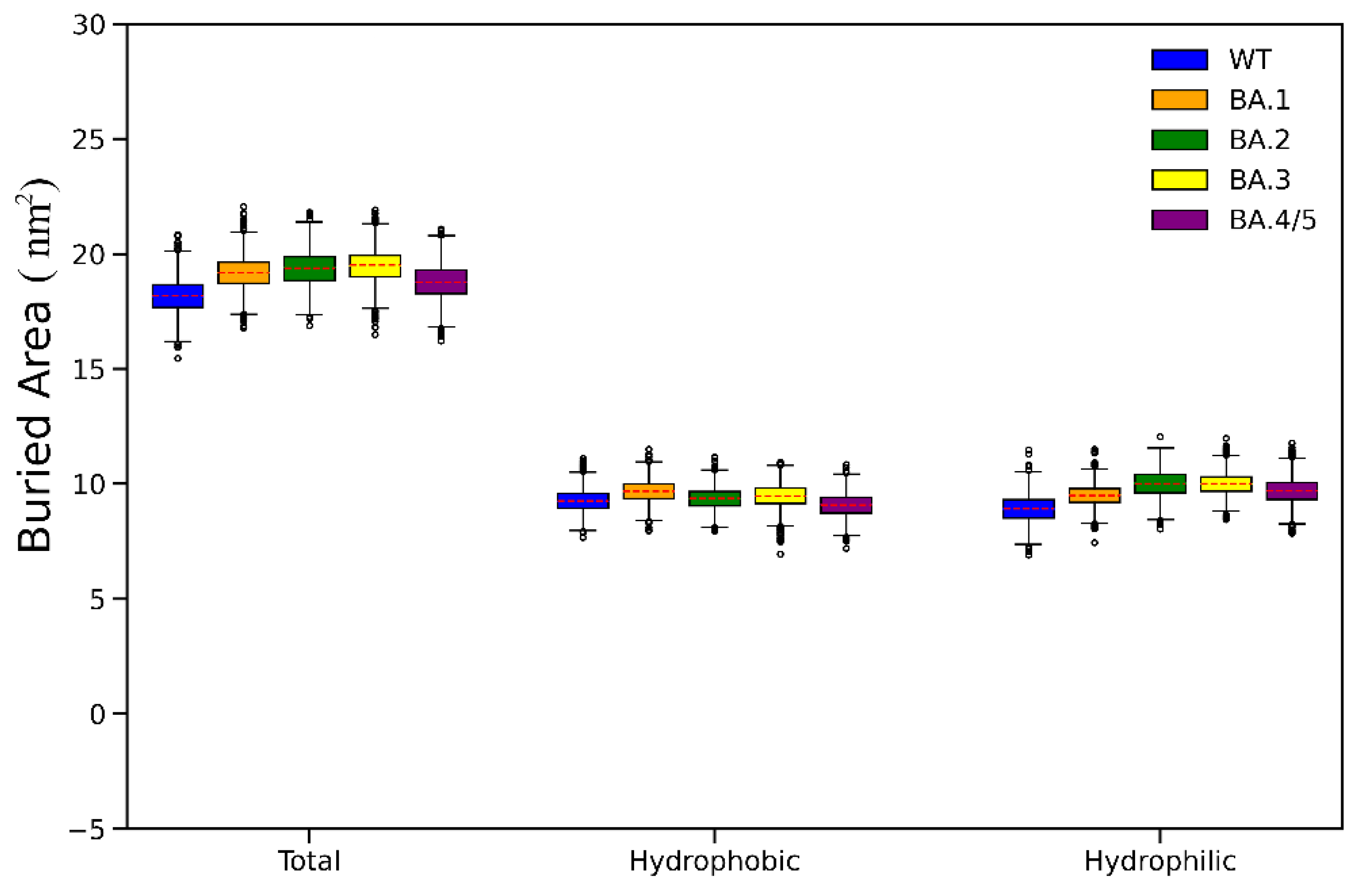
2.1. Structural Stability and Flexibility during Simulations
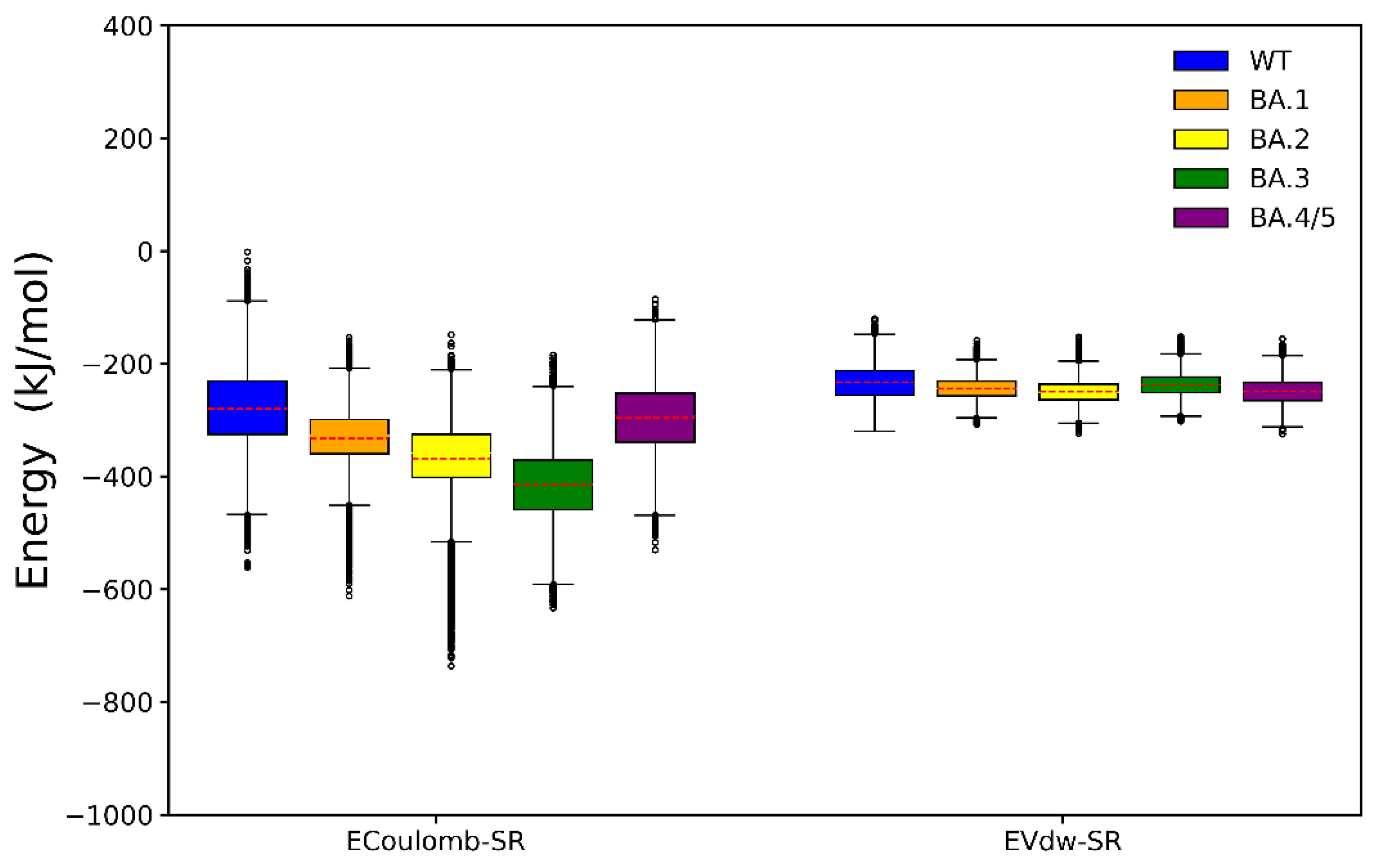
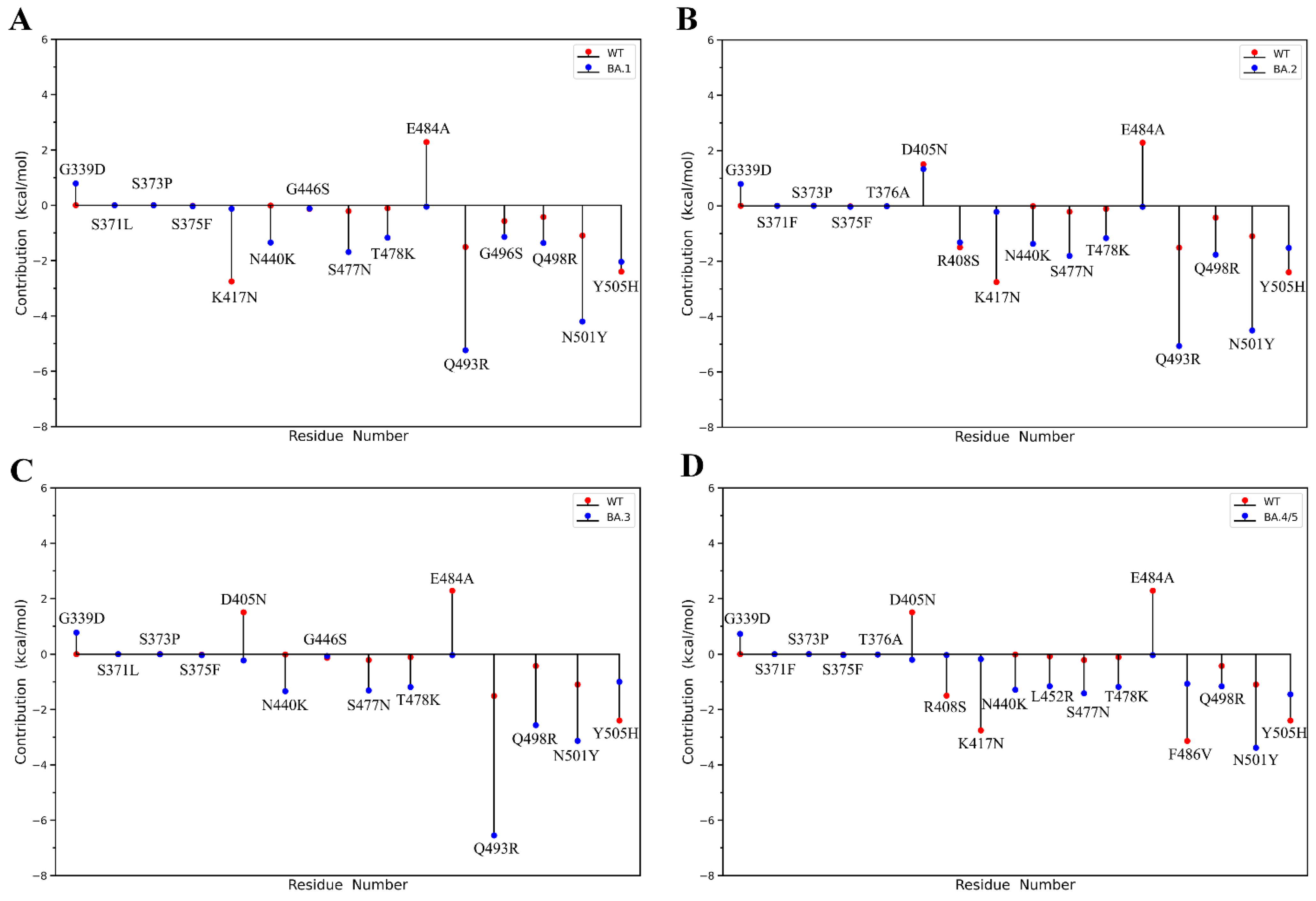
2.2. Binding Free Energy Calculation
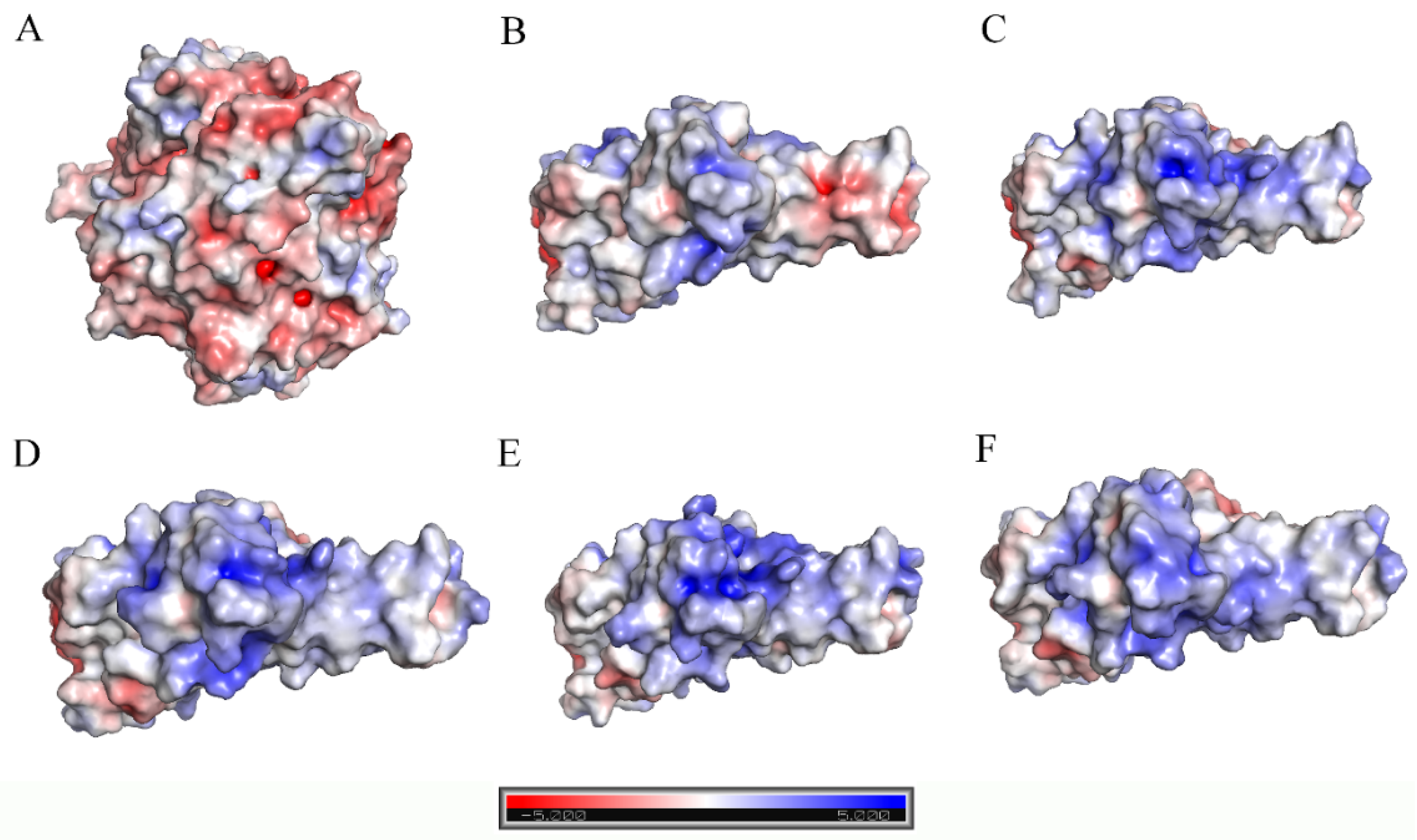
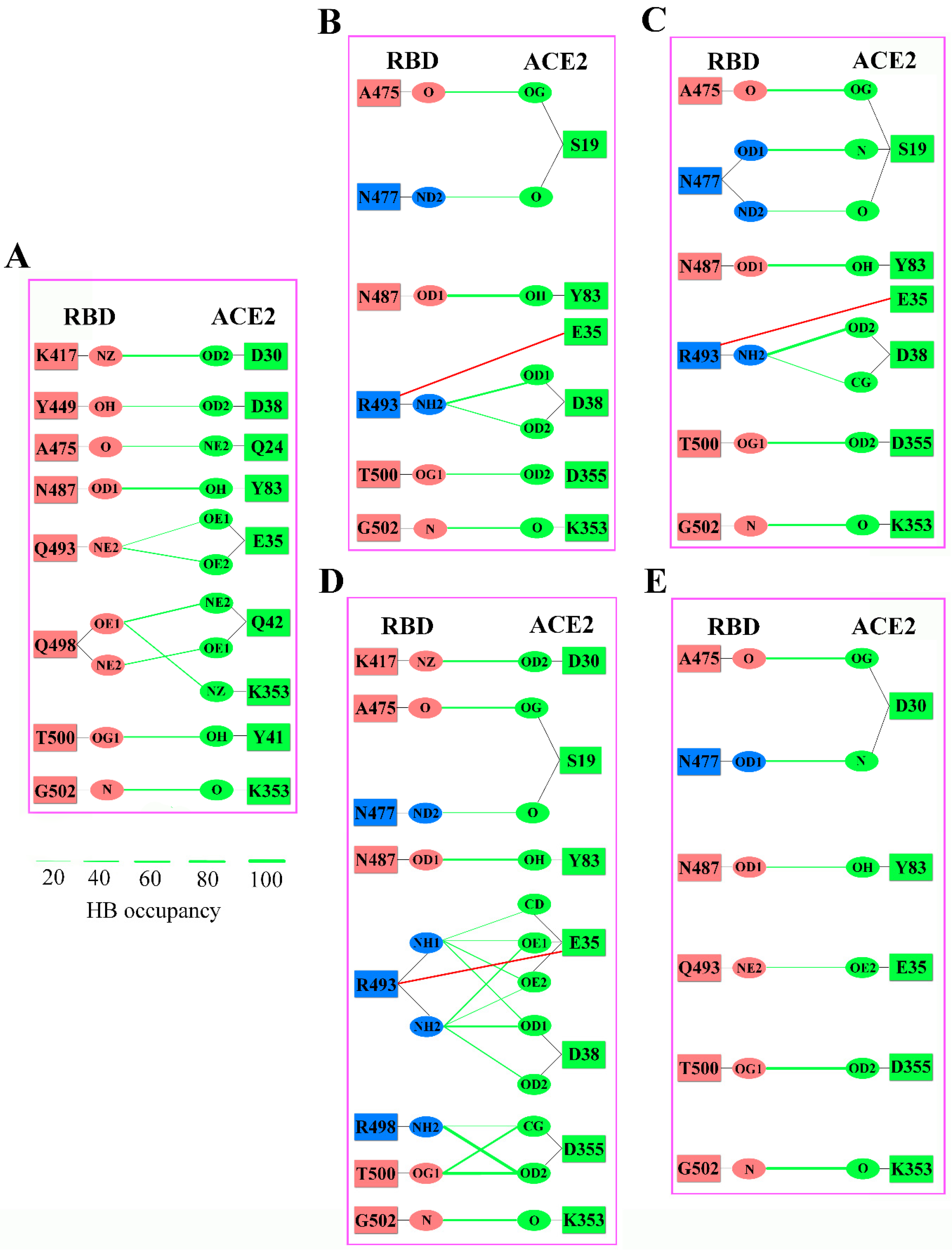
2.3. Analyses of Interfacial Interactions bwtween RBD and ACE2
3. Discussions
4. Materials and Methods
4.1. Structure Preparation
4.2. MD Simulation
4.3. Structural and Geometrical Properties
4.4. Binding Free Energy (BFE) Calculation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, M.; Gao, J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc. Natl. Acad. Sci. USA 2020, 117, 202008209. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Zhou, D.; Duyvesteyn, H.M.; Chen, C.-P.; Huang, C.-G.; Chen, T.-H.; Shih, S.-R.; Lin, Y.-C.; Cheng, C.-Y.; Cheng, S.-H.; Huang, Y.-C. Structural basis for the neutralization of SARS-CoV-2 by an antibody from a convalescent patient. Nat. Struct. Mol. Biol. 2020, 27, 950–958. [Google Scholar] [CrossRef]
- Zhang, Z.-B.; Xia, Y.-L.; Shen, J.-X.; Du, W.-W.; Fu, Y.-X.; Liu, S.-Q. Mechanistic origin of different binding affinities of SARS-CoV and SARS-CoV-2 spike RBDs to human ACE2. Cells 2022, 11, 1274. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Sorour, M.K.; Kasry, A. Comparing the binding interactions in the receptor binding domains of SARS-CoV-2 and SARS-CoV. J. Phys. Chem. Lett. 2020, 11, 4897–4900. [Google Scholar] [CrossRef]
- Delgado, J.M.; Duro, N.; Rogers, D.M.; Tkatchenko, A.; Pandit, S.A.; Varma, S. Molecular basis for higher affinity of SARS-CoV-2 spike RBD for human ACE2 receptor. Proteins Struct. Funct. Bioinform. 2021, 89, 1134–1144. [Google Scholar] [CrossRef]
- Bond, C.S.; Schüttelkopf, A.W. ALINE: A WYSIWYG protein-sequence alignment editor for publication-quality alignments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 510–512. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System. Available online: http://www.pymol.org (accessed on 16 February 2022).
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Thye, A.Y.-K.; Law, J.W.-F.; Pusparajah, P.; Letchumanan, V.; Chan, K.-G.; Lee, L.-H. Emerging SARS-CoV-2 variants of concern (VOCs): An impending global crisis. Biomedicines 2021, 9, 1303. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. How severe are Omicron infections. Nature 2021, 600, 577–578. [Google Scholar] [CrossRef]
- Fan, Y.; Li, X.; Zhang, L.; Wan, S.; Zhang, L.; Zhou, F. SARS-CoV-2 Omicron variant: Recent progress and future perspectives. Signal Transduct. Target. Ther. 2022, 7, 141. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 Omicron lineages BA. 4 and BA. 5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar] [CrossRef]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Y.; Iketani, S.; Nair, M.S.; Li, Z.; Mohri, H.; Wang, M.; Yu, J.; Bowen, A.D.; Chang, J.Y. Antibody evasion by SARS-CoV-2 Omicron subvariants BA. 2.12. 1, BA. 4, & BA. 5. Nature 2022, 608, 603–608. [Google Scholar] [PubMed]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef]
- Li, L.; Liao, H.; Meng, Y.; Li, W.; Han, P.; Liu, K.; Wang, Q.; Li, D.; Zhang, Y.; Wang, L.; et al. Structural basis of human ACE2 higher binding affinity to currently circulating Omicron SARS-CoV-2 sub-variants BA. 2 and BA. 1.1. Cell 2022, 185, 2952–2960. [Google Scholar] [CrossRef] [PubMed]
- Geng, Q.; Shi, K.; Ye, G.; Zhang, W.; Aihara, H.; Li, F. Structural Basis for Human Receptor Recognition by SARS-CoV-2 Omicron Variant BA. 1. J. Virol. 2022, 96, e00249-22. [Google Scholar] [CrossRef] [PubMed]
- Lupala, C.S.; Ye, Y.; Chen, H.; Su, X.-D.; Liu, H. Mutations on RBD of SARS-CoV-2 Omicron variant result in stronger binding to human ACE2 receptor. Biochem. Biophys. Res. Commun. 2022, 590, 34–41. [Google Scholar] [CrossRef]
- Shah, M.; Woo, H.G. Omicron: A heavily mutated SARS-CoV-2 variant exhibits stronger binding to ACE2 and potently escape approved COVID-19 therapeutic antibodies. Front. Immunol. 2022, 12, 6031. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhou, L.; Mo, M.; Liu, T.; Wu, C.; Gong, C.; Lu, K.; Gong, L.; Zhu, W.; Xu, Z. SARS-CoV-2 Omicron RBD shows weaker binding affinity than the currently dominant Delta variant to human ACE2. Signal Transduct. Target. Ther. 2022, 7, 8. [Google Scholar] [CrossRef]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.-Y. Binding Interactions between Receptor-Binding Domain of Spike Protein and Human Angiotensin Converting Enzyme-2 in Omicron Variant. J. Phys. Chem. Lett. 2022, 13, 3915–3921. [Google Scholar] [CrossRef]
- Nguyen, H.L.; Thai, N.Q.; Nguyen, P.H.; Li, M.S. SARS-CoV-2 omicron variant binds to human cells more strongly than the wild type: Evidence from molecular dynamics simulation. J. Phys. Chem. B 2022, 126, 4669–4678. [Google Scholar] [CrossRef]
- Dutta, S.; Panthi, B.; Chandra, A. All-atom simulations of human ACE2-spike protein RBD complexes for SARS-CoV-2 and some of its variants: Nature of interactions and free energy diagrams for dissociation of the protein complexes. J. Phys. Chem. B 2022, 126, 5375–5389. [Google Scholar] [CrossRef]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein–ACE2 complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640.e10. [Google Scholar] [CrossRef]
- Lan, J.; He, X.; Ren, Y.; Wang, Z.; Zhou, H.; Fan, S.; Zhu, C.; Liu, D.; Shao, B.; Liu, T.-Y.; et al. Structural insights into the SARS-CoV-2 Omicron RBD-ACE2 interaction. Cell Res. 2022, 32, 593–595. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Desingu, P.A.; Nagarajan, K. The emergence of Omicron lineages BA.4 and BA.5, and the global spreading trend. J. Med Virol. 2022, 94, 5077–5079. [Google Scholar] [CrossRef] [PubMed]
- Hirabara, S.M.; Serdan, T.D.A.; Gorjao, R.; Masi, L.N.; Pithon-Curi, T.C.; Covas, D.T.; Curi, R.; Durigon, E.L. SARS-COV-2 Variants: Differences and Potential of Immune Evasion. Front. Cell. Infect. Microbiol. 2022, 11, 1401. [Google Scholar] [CrossRef] [PubMed]
- Sztain, T.; Ahn, S.-H.; Bogetti, A.T.; Casalino, L.; Goldsmith, J.A.; Seitz, E.; McCool, R.S.; Kearns, F.L.; Acosta-Reyes, F.; Maji, S.; et al. A glycan gate controls opening of the SARS-CoV-2 spike protein. Nat. Chem. 2021, 13, 963–968. [Google Scholar] [CrossRef]
- Koehler, M.; Ray, A.; Moreira, R.A.; Juniku, B.; Poma, A.B.; Alsteens, D. Molecular insights into receptor binding energetics and neutralization of SARS-CoV-2 variants. Nat. Commun. 2021, 12, 6977. [Google Scholar] [CrossRef]
- Weikl, T.R.; Paul, F. Conformational selection in protein binding and function. Protein Sci. 2014, 23, 1508–1518. [Google Scholar] [CrossRef]
- Csermely, P.; Palotai, R.; Nussinov, R. Induced fit, conformational selection and independent dynamic segments: An extended view of binding events. Nat. Preced. 2010, 1–9. [Google Scholar] [CrossRef]
- Tian, F.; Tong, B.; Sun, L.; Shi, S.; Zheng, B.; Wang, Z.; Dong, X.; Zheng, P. N501Y mutation of spike protein in SARS-CoV-2 strengthens its binding to receptor ACE2. Elife 2021, 10, e69091. [Google Scholar] [CrossRef]
- Bhattarai, N.; Baral, P.; Gerstman, B.S.; Chapagain, P.P. Structural and dynamical differences in the spike protein RBD in the SARS-CoV-2 variants B. 1.1. 7 and B. 1.351. J. Phys. Chem. B 2021, 125, 7101–7107. [Google Scholar] [CrossRef]
- Socher, E.; Conrad, M.; Heger, L.; Paulsen, F.; Sticht, H.; Zunke, F.; Arnold, P. Mutations in the B. 1.1. 7 SARS-CoV-2 spike protein reduce receptor-binding affinity and induce a flexible link to the fusion peptide. Biomedicines 2021, 9, 525. [Google Scholar] [CrossRef] [PubMed]
- Roy, U. Comparative structural analyses of selected spike protein-RBD mutations in SARS-CoV-2 lineages. Immunol. Res. 2022, 70, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Tam, B.; Wang, S.M. RBD double mutations of SARS-CoV-2 strains increase transmissibility through enhanced interaction between RBD and ACE2 receptor. Viruses 2021, 14, 1. [Google Scholar] [CrossRef]
- Adhikari, P.; Jawad, B.; Podgornik, R.; Ching, W.-Y. Mutations of Omicron Variant at the Interface of the Receptor Domain Motif and Human Angiotensin-Converting Enzyme-2. Int. J. Mol. Sci. 2022, 23, 2870. [Google Scholar] [CrossRef] [PubMed]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Price, D.J.; Brooks, C.L., III. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef]
- Knapp, B.; Ospina-Forero, L.; Deane, C.M. Avoiding False Positive Conclusions in Molecular Simulation: The Importance of Replicas. J. Chem. Theory Comput. 2018, 14, 6127–6138. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Poma, A.B.; Cieplak, M.; Theodorakis, P.E. Combining the MARTINI and Structure-Based Coarse-Grained Approaches for the Molecular Dynamics Studies of Conformational Transitions in Proteins. J. Chem. Theory Comput. 2017, 13, 1366–1374. [Google Scholar] [CrossRef]
- Martínez, M.; Cooper, C.D.; Poma, A.B.; Guzman, H.V. Free Energies of the Disassembly of Viral Capsids from a Multiscale Molecular Simulation Approach. J. Chem. Inf. Model. 2019, 60, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T. What is the current value of MM/PBSA and MM/GBSA methods in drug discovery? Expert Opin. Drug Discov. 2021, 16, 1233–1237. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area Method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Wang, C.; Greene, D.; Xiao, L.; Qi, R.; Luo, R. Recent Developments and Applications of the MMPBSA Method. Front. Mol. Biosci. 2018, 4, 87. [Google Scholar] [CrossRef]
- Sang, P.; Tian, S.-H.; Meng, Z.-H.; Yang, L.-Q. Anti-HIV drug repurposing against SARS-CoV-2. RSC Adv. 2020, 10, 15775–15783. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy Components | WT (kcal/mol) | Omicron BA.1 (kcal/mol) | Omicron BA.2 (kcal/mol) | Omicron BA.3 (kcal/mol) | Omicron BA.4/5 (kcal/mol) |
|---|---|---|---|---|---|
| ΔEele | −615.20 (45.48) | −1368.55 (47.30) | −1381.35 (46.96) | −1948.76 (60.03) | −1285.69 (49.20) |
| ΔEvdw | −75.89 (6.53) | −81.71 (5.46) | −80.62 (5.50) | −78.74 (6.03) | −75.89 (5.04) |
| ΔGMM | −691.09 (46.95) | −1450.26 (47.39) | −1461.97 (46.85) | −2027.49 (59.95) | −1361.59 (50.12) |
| ΔGpolar | 655.95 (42.75) | 1393.51 (45.39) | 1402.02 (44.98) | 1958.08 (57.46) | 1319.13 (49.19) |
| ΔGnonpolar | −9.45 (0.58) | −9.80 (0.40) | −9.70 (0.38) | −10.37 (0.45) | −9.21 (0.45) |
| ΔGsol | 646.50 (42.47) | 1383.71 (45.26) | 1391.53 (44.88) | 1947.71 (57.69) | 1309.92 (49.01) |
| ΔGbind | −44.59 (10.69) | −66.55 (9.24) | −70.44 (9.33) | −79.78 (10.11) | −51.67 (9.38) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sang, P.; Chen, Y.-Q.; Liu, M.-T.; Wang, Y.-T.; Yue, T.; Li, Y.; Yin, Y.-R.; Yang, L.-Q. Electrostatic Interactions Are the Primary Determinant of the Binding Affinity of SARS-CoV-2 Spike RBD to ACE2: A Computational Case Study of Omicron Variants. Int. J. Mol. Sci. 2022, 23, 14796. https://doi.org/10.3390/ijms232314796
Sang P, Chen Y-Q, Liu M-T, Wang Y-T, Yue T, Li Y, Yin Y-R, Yang L-Q. Electrostatic Interactions Are the Primary Determinant of the Binding Affinity of SARS-CoV-2 Spike RBD to ACE2: A Computational Case Study of Omicron Variants. International Journal of Molecular Sciences. 2022; 23(23):14796. https://doi.org/10.3390/ijms232314796
Chicago/Turabian StyleSang, Peng, Yong-Qin Chen, Meng-Ting Liu, Yu-Ting Wang, Ting Yue, Yi Li, Yi-Rui Yin, and Li-Quan Yang. 2022. "Electrostatic Interactions Are the Primary Determinant of the Binding Affinity of SARS-CoV-2 Spike RBD to ACE2: A Computational Case Study of Omicron Variants" International Journal of Molecular Sciences 23, no. 23: 14796. https://doi.org/10.3390/ijms232314796
APA StyleSang, P., Chen, Y.-Q., Liu, M.-T., Wang, Y.-T., Yue, T., Li, Y., Yin, Y.-R., & Yang, L.-Q. (2022). Electrostatic Interactions Are the Primary Determinant of the Binding Affinity of SARS-CoV-2 Spike RBD to ACE2: A Computational Case Study of Omicron Variants. International Journal of Molecular Sciences, 23(23), 14796. https://doi.org/10.3390/ijms232314796