
Interplay between Autophagy and Herpes Simplex Virus Type 1: ICP34.5, One of the Main Actors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction

2. The HSV-1 Factor of Virulence ICP34.5
2.1. The HSV-1 γ34.5 Gene and the Infected Cell Protein 34.5
2.2. HSV-1 ICP34.5: A Catch-All
2.2.1. Functions of the Carboxyl Domain of HSV-1 ICP34.5
2.2.2. Functions of the Amino Domain of HSV-1 ICP34.5
2.2.3. Functions of the Amino and Carboxyl Domains of HSV-1 ICP34.5
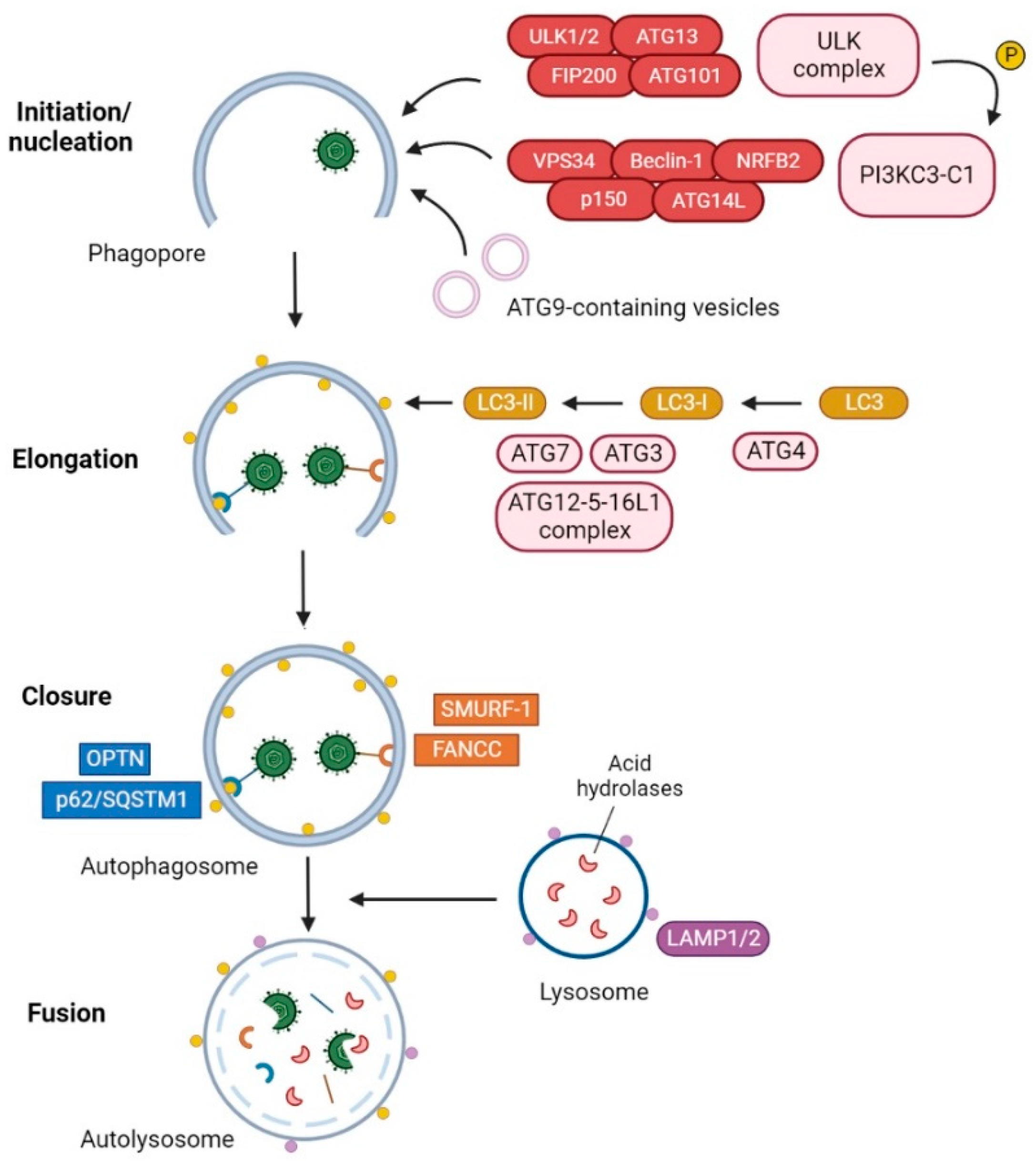
3. HSV-1 Modulation of Autophagy
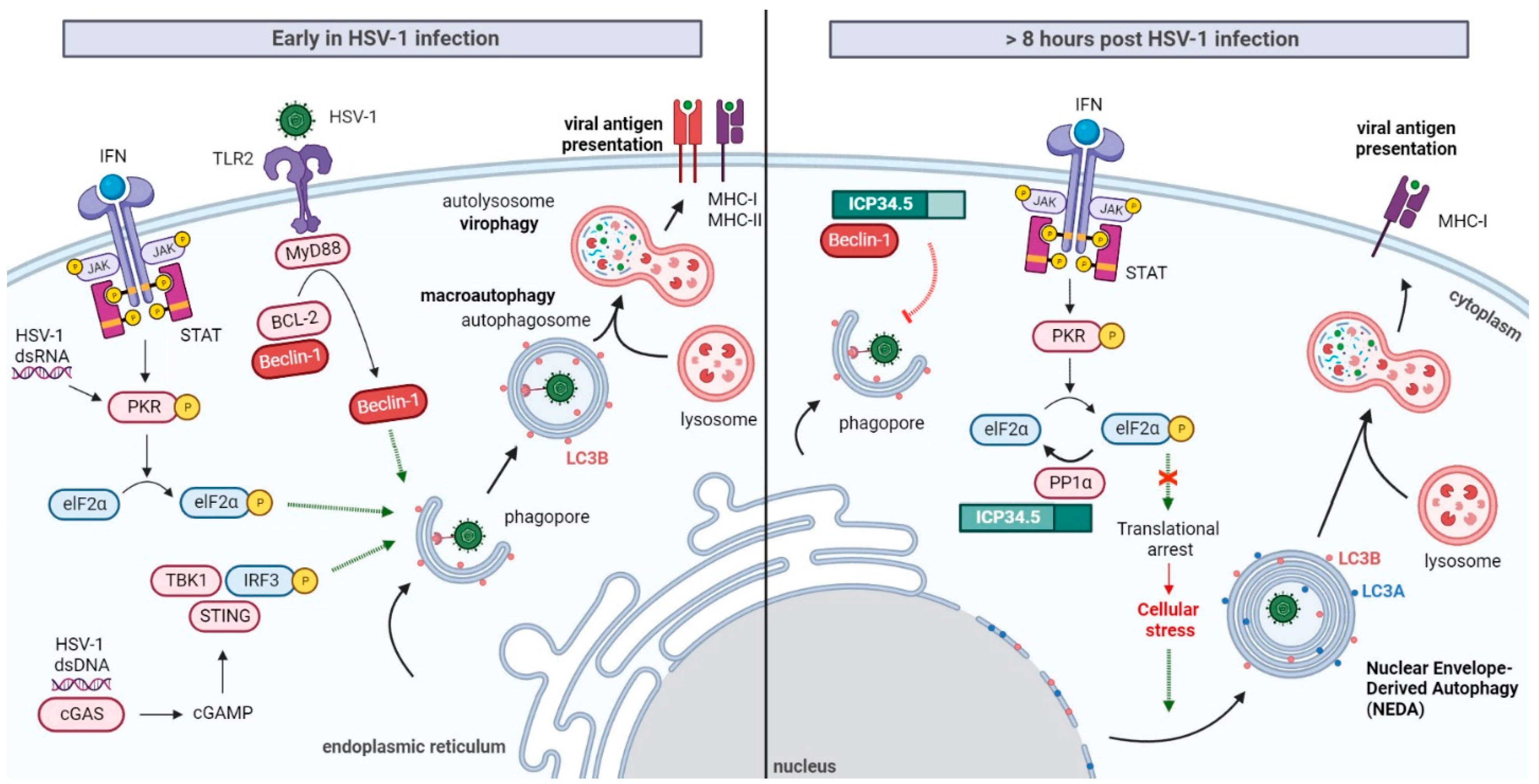
Induction of Autophagy at the Early Stages of HSV-1 Infection
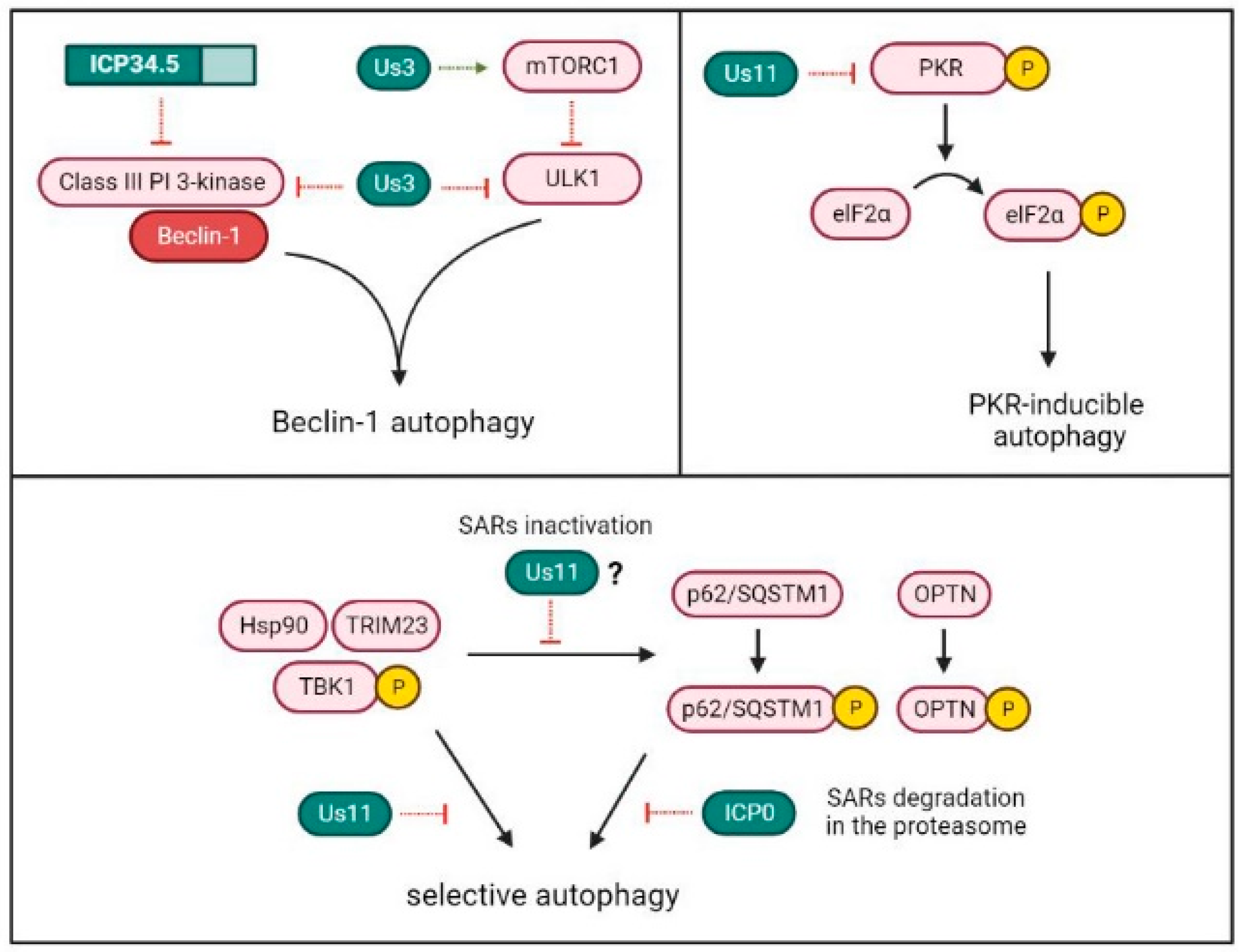
4. Selective Inhibition of Autophagy by HSV-1 ICP34.5
5. The Intricate Role of the Beclin-1-Binding Domain of ICP34.5 in HSV-1 Virulence
6. ICP34.5 Is Not Alone in HSV-1 Autophagy Inhibition
7. The Two Sides of Autophagy
8. Herpes Simplex Virus Type 2 and Autophagy
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ohsumi, Y. Historical Landmarks of Autophagy Research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Markaki, M.; Tavernarakis, N. Autophagy Mechanisms and Roles: Recent Advances and Implications. FEBS J. 2020, 287, 5024–5026. [Google Scholar] [CrossRef] [PubMed]
- Morishita, H.; Mizushima, N. Diverse Cellular Roles of Autophagy. Annu. Rev. Cell Dev. Biol. 2019, 35, 453–475. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B. Autophagy and Lysosomal Pathways in Nervous System Disorders. Mol. Cell Neurosci. 2018, 91, 167–208. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Tooze, S.A. Emerging Roles of ATG Proteins and Membrane Lipids in Autophagosome Formation. Cell Discov. 2020, 6, 32. [Google Scholar] [CrossRef]
- Nakatogawa, H. Mechanisms Governing Autophagosome Biogenesis. Nat. Rev. Mol. Cell Biol. 2020, 21, 439–458. [Google Scholar] [CrossRef] [PubMed]
- Melia, T.J.; Lystad, A.H.; Simonsen, A. Autophagosome Biogenesis: From Membrane Growth to Closure. J. Cell Biol. 2020, 219, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.A.; Ktistakis, N.T. Autophagosome Biogenesis Machinery. J. Mol. Biol. 2020, 432, 2449–2461. [Google Scholar] [CrossRef]
- Kawabata, T.; Yoshimori, T. Autophagosome Biogenesis and Human Health. Cell Discov. 2020, 6, 1–14. [Google Scholar] [CrossRef]
- Hu, Y.; Reggiori, F. Molecular Regulation of Autophagosome Formation. Biochem. Soc. Trans. 2022, 50, 55–69. [Google Scholar] [CrossRef]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A Subdomain of the Endoplasmic Reticulum Forms a Cradle for Autophagosome Formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria Supply Membranes for Autophagosome Biogenesis during Starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma Membrane Contributes to the Formation of Pre-Autophagosomal Structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Welter, E.; Borovsky, N.; Amar, N.; Mari, M.; Reggiori, F.; Elazar, Z. Lipid Droplets and Their Component Triglycerides and Steryl Esters Regulate Autophagosome Biogenesis. EMBO J. 2015, 34, 2117–2131. [Google Scholar] [CrossRef]
- Ge, L.; Melville, D.; Zhang, M.; Schekman, R. The ER-Golgi Intermediate Compartment Is a Key Membrane Source for the LC3 Lipidation Step of Autophagosome Biogenesis. eLife 2013, 2, e00947. [Google Scholar] [CrossRef]
- Puri, C.; Vicinanza, M.; Ashkenazi, A.; Gratian, M.J.; Zhang, Q.; Bento, C.F.; Renna, M.; Menzies, F.M.; Rubinsztein, D.C. The RAB11A-Positive Compartment Is a Primary Platform for Autophagosome Assembly Mediated by WIPI2 Recognition of PI3P-RAB11A. Dev. Cell 2018, 45, 114–131.e8. [Google Scholar] [CrossRef]
- Karanasios, E.; Walker, S.A.; Okkenhaug, H.; Manifava, M.; Hummel, E.; Zimmermann, H.; Ahmed, Q.; Domart, M.C.; Collinson, L.; Ktistakis, N.T. Autophagy Initiation by ULK Complex Assembly on ER Tubulovesicular Regions Marked by ATG9 Vesicles. Nat. Commun. 2016, 7, 12420. [Google Scholar] [CrossRef]
- Orsi, A.; Razi, M.; Dooley, H.C.; Robinson, D.; Weston, A.E.; Collinson, L.M.; Tooze, S.A. Dynamic and Transient Interactions of Atg9 with Autophagosomes, but Not Membrane Integration, Are Required for Autophagy. Mol. Biol. Cell 2012, 23, 1860–1873. [Google Scholar] [CrossRef]
- Geng, J.; Klionsky, D.J. The Atg8 and Atg12 Ubiquitin-like Conjugation Systems in Macroautophagy. “Protein Modifications: Beyond the Usual Suspects” review series. EMBO Rep. 2008, 9, 859–864. [Google Scholar] [CrossRef]
- Nakamura, S.; Yoshimori, T. New Insights into Autophagosome-Lysosome Fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef]
- Orvedahl, A.; Sumpter, R.; Xiao, G.; Ng, A.; Zou, Z.; Tang, Y.; Narimatsu, M.; Gilpin, C.; Sun, Q.; Roth, M.; et al. Image-Based Genome-Wide SiRNA Screen Identifies Selective Autophagy Factors. Nature 2011, 480, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Ames, J.; Yadavalli, T.; Suryawanshi, R.; Hopkins, J.; Agelidis, A.; Patil, C.; Fredericks, B.; Tseng, H.; Valyi-Nagy, T.; Shukla, D. OPTN Is a Host Intrinsic Restriction Factor against Neuroinvasive HSV-1 Infection. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sumpter, R.; Sirasanagandla, S.; Fernández, Á.F.; Wei, Y.; Dong, X.; Franco, L.; Zou, Z.; Marchal, C.; Lee, M.Y.; Clapp, D.W.; et al. Fanconi Anemia Proteins Function in Mitophagy and Immunity. Cell 2016, 165, 867–881. [Google Scholar] [CrossRef]
- Gubas, A.; Dikic, I. A Guide to the Regulation of Selective Autophagy Receptors. FEBS J. 2022, 289, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Johansen, T. Mechanisms of Selective Autophagy. Annu. Rev. Cell Dev. Biol. 2021, 37, 143–169. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo Recognition and Degradation by Selective Autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Reggio, A.; Buonomo, V.; Grumati, P. Eating the Unknown: Xenophagy and ER-Phagy Are Cytoprotective Defenses against Pathogens. Exp. Cell Res. 2020, 396, 1–8. [Google Scholar] [CrossRef]
- Mao, J.; Lin, E.; He, L.; Yu, J.; Tan, P.; Zhou, Y. Autophagy and Viral Infection. Adv. Exp. Med. Biol. 2019, 1209, 55–78. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Luo, X.; Ren, M. Clearance or Hijack: Universal Interplay Mechanisms Between Viruses and Host Autophagy From Plants to Animals. Front. Cell. Infect. Microbiol. 2022, 11, 1–15. [Google Scholar] [CrossRef]
- Singh, N.; Tscharke, D.C. Herpes Simplex Virus Latency Is Noisier the Closer We Look. J. Virol. 2020, 94, 1–7. [Google Scholar] [CrossRef]
- Gnann, J.W.; Whitley, R.J. Herpes Simplex Encephalitis: An Update. Curr. Infect Dis. Rep. 2017, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, R.; Sicurella, M.; Agostini, S.; Marconi, P.; Clerici, M. Herpes Simplex Virus Type 1 and Alzheimer’s Disease: Link and Potential Impact on Treatment. Expert Rev. Anti Infect. Ther. 2019, 17, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Buscarinu, M.C.; Fornasiero, A.; Romano, S.; Ferraldeschi, M.; Renié, R.; Trasimeni, G.; Salvetti, M.; Ristori, G. Coincident Onset of Multiple Sclerosis and Herpes Simplex Virus 1 Encephalitis: A Case Report. Mult. Scler. Demyelinating Disord. 2017, 2, 10–13. [Google Scholar] [CrossRef]
- Zhu, S.; Viejo-Borbolla, A. Pathogenesis and Virulence of Herpes Simplex Virus. Virulence 2021, 12, 2670–2702. [Google Scholar] [CrossRef]
- Wilcox, D.R.; Longnecker, R. The Herpes Simplex Virus Neurovirulence Factor Γ34.5: Revealing Virus–Host Interactions. PLoS Pathog. 2016, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Kern, E.R.; Whitley, R.J.; Roizman, B. Mapping of Herpes Simplex Virus-1 Neurovirulence to γ134.5, a Gene Nonessential for Growth in Culture. Science 1990, 250, 1262–1266. [Google Scholar] [CrossRef]
- Pasieka, T.J.; Baas, T.; Carter, V.S.; Proll, S.C.; Katze, M.G.; Leib, D.A. Functional Genomic Analysis of Herpes Simplex Virus Type 1 Counteraction of the Host Innate Response. J. Virol. 2006, 80, 7600–7612. [Google Scholar] [CrossRef]
- Chou, J.; Roizman, B.; Marjorie Kovler, T.B. The Terminal a Sequence of the Herpes Simplex Virus Genome Contains the Promoter of a Gene Located in the Repeat Sequences of the L Component. J. Virol. 1986, 57, 629–637. [Google Scholar] [CrossRef]
- Fornace, A.J.; Nebert, D.W.; Hollander, M.C.; Luethy, J.D.; Papathanasiou, M.; Fargnoli, J.; Holbrook3, N.J. Mammalian Genes Coordinately Regulated by Growth Arrest Signals and DNA-Damaging Agents. Mol. Cell Biol. 1989, 9, 4196–4203. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Barnett, B.C. Neurovirulence Factor. Nature 1991, 353, 609. [Google Scholar] [CrossRef]
- He, B.; Chou, J.; Liebermann, D.A.; Hoffman, B.; Roizman, B. The Carboxyl Terminus of the Murine MyD116 Gene Substitutes for the Corresponding Domain of the γ134.5Gene of Herpes Simplex Virus To Preclude the Premature Shutoff of Total Protein Synthesis in Infected Human Cells. J. Virol. 1996, 70, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Brett, M.-E.; He, B. Signals That Dictate Nuclear, Nucleolar, and Cytoplasmic Shuttling of the γ134.5 Protein of Herpes Simplex Virus Type 1. J. Virol. 2002, 76, 9434–9445. [Google Scholar] [CrossRef] [PubMed]
- Harland, J.; Dunn, P.; Cameron, E.; Conner, J.; Brown, S.M. The Herpes Simplex Virus (HSV) Protein ICP34.5 Is a Virion Component That Forms a DNA-Binding Complex with Proliferating Cell Nuclear Antigen and HSV Replication Proteins. J. Neurovirol. 2003, 9, 477–488. [Google Scholar] [CrossRef]
- Mao, H.; Rosenthal, K.S. An N-Terminal Arginine-Rich Cluster and a Proline-Alanine-Threonine Repeat Region Determine the Cellular Localization of the Herpes Simplex Virus Type 1 ICP34.5 Protein and Its Ligand, Protein Phosphatase 1. J. Biol. Chem. 2002, 277, 11423–11431. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Rosenthal, K.S. Strain-Dependent Structural Variants of Herpes Simplex Virus Type 1 ICP34.5 Determine Viral Plaque Size, Efficiency of Glycoprotein Processing, and Viral Release and Neuroinvasive Disease Potential. J. Virol. 2003, 77, 3409–3417. [Google Scholar] [CrossRef] [PubMed]
- Bolovan, C.A.; Sawtell, N.M.; Thompson’, R.L. ICP34.5 Mutants of Herpes Simplex Virus Type 1 Strain 17syn+ Are Attenuated for Neurovirulence in Mice and for Replication in Confluent Primary Mouse Embryo Cell Cultures. J. Virol. 1994, 68, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.M.; Harland, J.; Maclean, A.R.; Podlecht, J.; Clements, J.B. Cell Type and Cell State Determine Differential in Vitro Growth of Non-Neurovirulent ICP34.5-Negative Herpes Simplex Virus Types 1 and 2. J. Gen. Virol. 1994, 75, 2367–2377. [Google Scholar] [CrossRef] [PubMed]
- Kesari, S.; Lasner, T.M.; Balsara, K.R.; Randazzo, B.P.; Lee, M.V.; Trojanowski, J.Q.; Fraser, N.W. A Neuroattenuated ICP34.5-Deficient Herpes Simplex Virus Type 1 Replicates in Ependymal Cells of the Murine Central Nervous System. J. Gen. Virol. 1998, 79, 525–536. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern Recognition Receptors in Health and Diseases. Signal Transduct. Target Ther. 2021, 6, 1–24. [Google Scholar] [CrossRef]
- Danastas, K.; Miranda-Saksena, M.; Cunningham, A.L. Herpes Simplex Virus Type 1 Interactions with the Interferon System. Int. J. Mol. Sci. 2020, 21, 5150. [Google Scholar] [CrossRef]
- Verzosa, A.L.; McGeever, L.A.; Bhark, S.J.; Delgado, T.; Salazar, N.; Sanchez, E.L. Herpes Simplex Virus 1 Infection of Neuronal and Non-Neuronal Cells Elicits Specific Innate Immune Responses and Immune Evasion Mechanisms. Front Immunol. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Gal-Ben-Ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A Kinase to Remember. Front. Mol. Neurosci. 2019, 11, 1–20. [Google Scholar] [CrossRef]
- Leib, D.A.; Machalek, M.A.; Williams, B.R.G.; Silverman, R.H.; Virgin, H.W. Specific Phenotypic Restoration of an Attenuated Virus by Knockout of a Host Resistance Gene. Proc. Natl. Acad. Sci. USA 2000, 97, 6097–6101. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Gross, M.; Roizman, B. The Γ134.5 Protein of Herpes Simplex Virus 1 Complexes with Protein Phosphatase 1α to Dephosphorylate the α Subunit of the Eukaryotic Translation Initiation Factor 2 and Preclude the Shutoff of Protein Synthesis by Double-Stranded RNA-Activated Protein Ki. Proc. Natl. Acad. Sci. USA 1997, 94, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, C.; Chen, X.; Yu, J.; Wang, Y.; Yang, Y.; Du, M.; Jin, H.; Ma, Y.; He, B.; et al. ICP34.5 Protein of Herpes Simplex Virus Facilitates the Initiation of Protein Translation by Bridging Eukaryotic Initiation Factor 2α (EIF2α) and Protein Phosphatase 1. J. Biol. Chem. 2011, 286, 24785–24792. [Google Scholar] [CrossRef]
- Meng, W.; Han, S.C.; Li, C.C.; Dong, H.J.; Wang, X.J. Multifunctional Viral Protein Γ34.5 Manipulates Nucleolar Protein NOP53 for Optimal Viral Replication of HSV-1 Article. Cell Death Dis. 2018, 9, 103. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Alexander, D.; Tallóczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 Confers Neurovirulence by Targeting the Beclin 1 Autophagy Protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Manivanh, R.; Mehrbach, J.; Charron, A.J.; Grassetti, A.; Cerón, S.; Taylor, S.A.; Cabrera, J.R.; Gerber, S.; Leib, D.A. Herpes Simplex Virus 1 ICP34.5 Alters Mitochondrial Dynamics in Neurons. J. Virol. 2020, 94, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Jin, H.; Valyi-Nagy, T.; Cao, Y.; Yan, Z.; He, B. Inhibition of TANK Binding Kinase 1 by Herpes Simplex Virus 1 Facilitates Productive Infection. J. Virol. 2012, 86, 2188–2196. [Google Scholar] [CrossRef]
- Verpooten, D.; Yijie, M.; Hou, S.; Yan, Z.; He, B. Control of TANK-Binding Kinase 1-Mediated Signaling by the Γ1 34.5 Protein of Herpes Simplex Virus 1. J. Biol. Chem. 2009, 284, 1097–1105. [Google Scholar] [CrossRef]
- Manivanh, R.; Mehrbach, J.; Knipe, D.M.; Leib, D.A. Role of Herpes Simplex Virus γ134.5 in the Regulation of IRF3 Signaling. J. Virol. 2017, 91, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zheng, C. A Tug of War: DNA-Sensing Antiviral Innate Immunity and Herpes Simplex Virus Type I Infection. Front. Microbiol. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Liu, X.; Ma, Y.; Cao, Y.; He, B. Herpes Simplex Virus 1 γ134.5 Protein Inhibits STING Activation That Restricts Viral Replication. J. Virol. 2018, 26, e01015-18. [Google Scholar] [CrossRef]
- Brown, S.M.; Maelean, A.R.; Aitken, J.D.; Harland, J. ICP34.5 Influences Herpes Simplex Virus Type 1 Maturation and Egress from Infected Cells in Vitro. J. Gen. Virol. 1994, 75, 3679–3686. [Google Scholar] [CrossRef] [PubMed]
- Mou, F.; Wills, E.G.; Park, R.; Baines, J.D. Effects of Lamin A/C, Lamin B1, and Viral US3 Kinase Activity on Viral Infectivity, Virion Egress, and the Targeting of Herpes Simplex Virus U L 34-Encoded Protein to the Inner Nuclear Membrane. J. Virol. 2008, 82, 8094–8104. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Wu, S.; Pan, S.; Zhou, C.; Ma, Y.; Ru, Y.; Dong, S.; He, B.; Zhang, C.; et al. P32 Is a Novel Target for Viral Protein ICP34.5 of Herpes Simplex Virus Type 1 and Facilitates Viral Nuclear Egress. J. Biol. Chem. 2014, 289, 35795–35805. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Pan, S.; Zhang, L.; Baines, J.; Roller, R.; Ames, J.; Yang, M.; Wang, J.; Chen, D.; Liu, Y.; et al. Herpes Simplex Virus 1 Induces Phosphorylation and Reorganization of Lamin A/C through the γ134.5 Protein That Facilitates Nuclear Egress. J. Virol. 2016, 90, 10414–10422. [Google Scholar] [CrossRef]
- Jin, H.; Ma, Y.; Prabhakar, B.S.; Feng, Z.; Valyi-Nagy, T.; Yan, Z.; Verpooten, D.; Zhang, C.; Cao, Y.; He, B. The γ134.5 Protein of Herpes Simplex Virus 1 Is Required To Interfere with Dendritic Cell Maturation during Productive Infection. J. Virol. 2009, 83, 4984–4994. [Google Scholar] [CrossRef]
- Jin, H.; Yan, Z.; Ma, Y.; Cao, Y.; He, B. A Herpesvirus Virulence Factor Inhibits Dendritic Cell Maturation through Protein Phosphatase 1 and IκB Kinase. J. Virol. 2011, 85, 3397–3407. [Google Scholar] [CrossRef]
- Liu, X.; Ma, Y.; Voss, K.; van Gent, M.; Chan, Y.K.; Gack, M.U.; Gale, M.; He, B. The Herpesvirus Accessory Protein Γ134.5 Facilitates Viral Replication by Disabling Mitochondrial Translocation of RIG-I. PLoS Pathog. 2021, 17, 1–23. [Google Scholar] [CrossRef]
- Humeau, J.; Leduc, M.; Cerrato, G.; Loos, F.; Kepp, O.; Kroemer, G. Phosphorylation of Eukaryotic Initiation Factor-2α (EIF2α) in Autophagy. Cell Death Dis. 2020, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Tallóczy, Z.; Jiang, W.; Virgin IV, H.W.; Leib, D.A.; Scheuner, D.; Kaufman, R.J.; Eskelinen, E.-L.; Levine, B. Regulation of Starvation-and Virus-Induced Autophagy by the EIF2 Kinase Signaling Pathway. Proc. Nat. Acad. Sci. USA 2002, 99, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Tallóczy, Z.; Virgin, H.W.; Levine, B. PKR-Dependent Xenophagic Degradation of Herpes Simplex Virus Type 1. Autophagy 2006, 2, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Turan, A.; Grosche, L.; Krawczyk, A.; Mühl-Zürbes, P.; Drassner, C.; Düthorn, A.; Kummer, M.; Hasenberg, M.; Voortmann, S.; Jastrow, H.; et al. Autophagic Degradation of Lamins Facilitates the Nuclear Egress of Herpes Simplex Virus Type 1. J. Cell Biol. 2019, 218, 508–523. [Google Scholar] [CrossRef]
- Delgado, M.A.; Elmaoued, R.A.; Davis, A.S.; Kyei, G.; Deretic, V. Toll-like Receptors Control Autophagy. EMBO J. 2008, 27, 1110–1121. [Google Scholar] [CrossRef]
- Uyangaa, E.; Choi, J.Y.; Patil, A.M.; Hossain, F.M.A.; Park, S.O.K.; Kim, B.; Kim, K.; Eo, S.K. Dual TLR2/9 Recognition of Herpes Simplex Virus Infection Is Required for Recruitment and Activation of Monocytes and NK Cells and Restriction of Viral Dissemination to the Central Nervous System. Front. Immunol. 2018, 9, 1–21. [Google Scholar] [CrossRef]
- Lima, G.K.; Zolini, G.P.; Mansur, D.S.; Lima, B.H.F.; Wischhoff, U.; Astigarraga, R.G.; Dias, M.F.; Silva, M.D.G.; Béla, S.R.; Antonelli, L.R.D.V.; et al. Toll-Like Receptor (TLR) 2 and TLR9 Expressed in Trigeminal Ganglia Are Critical to Viral Control during Herpes Simplex Virus 1 Infection. Am. J. Pathol. 2010, 177, 2433–2445. [Google Scholar] [CrossRef]
- Brun, P.; Scarpa, M.; Marchiori, C.; Conti, J.; Kotsafti, A.; Porzionato, A.; de Caro, R.; Scarpa, M.; Calistri, A.; Castagliuolo, I. Herpes Simplex Virus Type 1 Engages Toll like Receptor 2 to Recruit Macrophages during Infection of Enteric Neurons. Front. Microbiol. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Oh, J.E.; Lee, H.K. Pattern Recognition Receptors and Autophagy. Front. Immunol. 2014, 5, 1–7. [Google Scholar] [CrossRef]
- Siracusano, G.; Venuti, A.; Lombardo, D.; Mastino, A.; Esclatine, A.; Sciortino, M.T. Early Activation of MyD88-Mediated Autophagy Sustains HSV-1 Replication in Human Monocytic THP-1 Cells. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Rasmussen, S.B.; Horan, K.A.; Holm, C.K.; Stranks, A.J.; Mettenleiter, T.C.; Simon, A.K.; Jensen, S.B.; Rixon, F.J.; He, B.; Paludan, S.R. Activation of Autophagy by α-Herpesviruses in Myeloid Cells Is Mediated by Cytoplasmic Viral DNA through a Mechanism Dependent on Stimulator of IFN Genes. J. Immunol. 2011, 187, 5268–5276. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, S.; Aitken, J.; Sutherland, J.S.; Nicholl, M.J.; Preston, V.G.; Preston, C.M. Early Induction of Autophagy in Human Fibroblasts after Infection with Human Cytomegalovirus or Herpes Simplex Virus 1. J. Virol. 2011, 85, 4212–4221. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, L.; Mashbat, B.; Leung, C.; Brookes, C.; Hamad, S.; Krokowski, S.; Shenoy, A.R.; Lorenzo, L.; Levin, M.; O’Hare, P.; et al. Human TANK-Binding Kinase 1 Is Required for Early Autophagy Induction upon Herpes Simplex Virus 1 Infection. J. Allergy Clin. Immunol. 2019, 143, 765–769. [Google Scholar] [CrossRef]
- Liyana, A.; Vanessa, S.S. The Emerging Role of Human TBK1 in Virus-Induced Autophagy. Autophagy 2019, 15, 917–918. [Google Scholar] [CrossRef]
- Menon, M.B.; Dhamija, S. Beclin 1 Phosphorylation—at the Center of Autophagy Regulation. Front. Cell Dev. Biol. 2018, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Leib, D.A.; Gobeil, P.A.M. Herpes Simplex Virus γ134.5 Interferes with Autophagosome Maturation and Antigen Presentation in Dendritic Cells. mBio 2012, 3, e00267-12. [Google Scholar] [CrossRef]
- Yakoub, A.M.; Shukla, D. Herpes Simplex Virus-1 Fine-Tunes Host’s Autophagic Response to Infection: A Comprehensive Analysis in Productive Infection Models. PLoS ONE 2015, 10, e0124646. [Google Scholar] [CrossRef]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplante, A.; Gingras, D.; Alexander, D.; Leib, D.; Norbury, C.; Lippé, R.; et al. Autophagy Enhances the Presentation of Endogenous Viral Antigens on MHC Class I Molecules during HSV-1 Infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef]
- Budida, R.; Stankov, M.V.; Döhner, K.; Buch, A.; Panayotova-Dimitrova, D.; Tappe, K.A.; Pohlmann, A.; Sodeik, B.; Behrens, G.M.N. Herpes Simplex Virus 1 Interferes with Autophagy of Murine Dendritic Cells and Impairs Their Ability to Stimulate CD8+ T Lymphocytes. Eur. J. Immunol. 2017, 47, 1819–1834. [Google Scholar] [CrossRef]
- Leib, D.A.; Alexander, D.E.; Cox, D.; Yin, J.; Ferguson, T.A. Interaction of ICP34.5 with Beclin 1 Modulates Herpes Simplex Virus Type 1 Pathogenesis through Control of CD4 + T-Cell Responses. J. Virol. 2009, 83, 12164–12171. [Google Scholar] [CrossRef]
- Radtke, K.; English, L.; Rondeau, C.; Leib, D.; Lippé, R.; Desjardins, M. Inhibition of the Host Translation Shutoff Response by Herpes Simplex Virus 1 Triggers Nuclear Envelope-Derived Autophagy. J. Virol. 2013, 87, 3990–3997. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.E.; Ward, S.L.; Mizushima, N.; Levine, B.; Leib, D.A. Analysis of the Role of Autophagy in Replication of Herpes Simplex Virus in Cell Culture. J. Virol. 2007, 81, 12128–12134. [Google Scholar] [CrossRef] [PubMed]
- Cassady, K.A.; Gross, M. The Herpes Simplex Virus Type 1 US11 Protein Interacts with Protein Kinase R in Infected Cells and Requires a 30-Amino-Acid Sequence Adjacent to a Kinase Substrate Domain. J. Virol. 2002, 76, 2029–2035. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.A.; Khoo, D.; Mohr, I.; Sen, G.C. Inhibition of PACT-Mediated Activation of PKR by the Herpes Simplex Virus Type 1 Us11 Protein. J. Virol. 2002, 76, 11054–11064. [Google Scholar] [CrossRef] [PubMed]
- Poppers, J.; Mulvey, M.; Khoo, D.; Mohr, I. Inhibition of PKR Activation by the Proline-Rich RNA Binding Domain of the Herpes Simplex Virus Type 1 Us11 Protein. J. Virol. 2000, 74, 11215–11221. [Google Scholar] [CrossRef] [PubMed]
- Lussignol, M.; Queval, C.; Bernet-Camard, M.-F.; Cotte-Laffitte, J.; Beau, I.; Codogno, P.; Esclatine, A. The Herpes Simplex Virus 1 Us11 Protein Inhibits Autophagy through Its Interaction with the Protein Kinase PKR. J. Virol. 2013, 87, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Matrenec, R.; Gack, M.U.; He, B. Disassembly of the TRIM23-TBK1 Complex by the Us11 Protein of Herpes Simplex Virus 1 Impairs Autophagy. J. Virol. 2019, 93, e00497-19. [Google Scholar] [CrossRef]
- Sparrer, K.M.J.; Gack, M.U. TRIM Proteins: New Players in Virus-Induced Autophagy. PLoS Pathog. 2018, 14, e1006787. [Google Scholar] [CrossRef]
- Sparrer, K.M.J.; Gableske, S.; Zurenski, M.A.; Parker, Z.M.; Full, F.; Baumgart, G.J.; Kato, J.; Pacheco-Rodriguez, G.; Liang, C.; Pornillos, O.; et al. TRIM23 Mediates Virus-Induced Autophagy via Activation of TBK1. Nat. Microbiol. 2017, 2, 1543–1557. [Google Scholar] [CrossRef]
- Ylä-Anttila, P. Autophagy Receptors as Viral Targets. Cell. Mol. Biol. Lett. 2021, 26, 1–11. [Google Scholar] [CrossRef]
- Waisner, H.; Kalamvoki, M. The ICP0 Protein of Herpes Simplex Virus 1 (HSV-1) Downregulates Major Autophagy Adaptor Proteins Sequestosome 1 and Optineurin during the Early Stages of HSV-1 Infection. J. Virol. 2019, 1, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 Enhances Its Binding to Ub Chains and Promotes Selective Autophagy of Damaged Mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.V.; Mills, J.; Lapierre, L.R. Selective Autophagy Receptor P62/SQSTM1, a Pivotal Player in Stress and Aging. Front. Cell Dev. Biol. 2022, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.A.; Tumbarello, D.A. Optineurin: A Coordinator of Membrane-Associated Cargo Trafficking and Autophagy. Front. Immunol. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Rubio, R.M.; Mohr, I. Inhibition of ULK1 and Beclin1 by an α-Herpesvirus Akt-like Ser/Thr Kinase Limits Autophagy to Stimulate Virus Replication. Proc. Natl. Acad. Sci. USA 2019, 116, 26941–26950. [Google Scholar] [CrossRef]
- Chuluunbaatar, U.; Roller, R.; Feldman, M.E.; Brown, S.; Shokat, K.M.; Mohr, I. Constitutive mTORC1 Activation by a Herpesvirus Akt Surrogate Stimulates mRNA Translation and Viral Replication. Genes Dev. 2010, 24, 2627–2639. [Google Scholar] [CrossRef]
- Holczer, M.; Hajdú, B.; Lőrincz, T.; Szarka, A.; Bánhegyi, G.; Kapuy, O. Fine-Tuning of AMPK–ULK1–mTORC1 Regulatory Triangle Is Crucial for Autophagy Oscillation. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Dupont, N.; Codogno, P. Non-Canonical Autophagy: Facts and Prospects. Curr. Pathobiol. Rep. 2013, 1, 263–271. [Google Scholar] [CrossRef][Green Version]
- Chawla, K.; Subramanian, G.; Rahman, T.; Fan, S.; Chakravarty, S.; Gujja, S.; Demchak, H.; Chakravarti, R.; Chattopadhyay, S. Autophagy in Virus Infection: A Race between Host Immune Response and Viral Antagonism. Immuno 2022, 2, 153–169. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, T.; Hu, J.; Jin, S.; Wu, J.; Guan, X.; Wu, Y.; Cui, J. Targeting Selective Autophagy as a Therapeutic Strategy for Viral Infectious Diseases. Front. Microbiol. 2022, 13, 1–15. [Google Scholar] [CrossRef]
- Yakoub, A.M.; Shukla, D. Autophagy Stimulation Abrogates Herpes Simplex Virus-1 Infection. Sci. Rep. 2015, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Gu, Y.; Ta, L.; Wang, K.; Xu, Z. Induction of Autophagy by the MG-132 Proteasome Inhibitor Is Associated with Endoplasmic Reticulum Stress in MCF-7 Cells. Mol. Med. Rep. 2016, 13, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Movaqar, A.; Abdoli, A.; Aryan, E.; Meshkat, Z. The Propagation of HSV-1 in High Autophagic Activity. Microb. Pathog. 2021, 152, 104599. [Google Scholar] [CrossRef]
- Yordy, B.; Iijima, N.; Huttner, A.; Leib, D.; Iwasaki, A. A Neuron-Specific Role for Autophagy in Antiviral Defense against Herpes Simplex Virus. Cell Host Microbe 2012, 12, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during Viral Infection—A Double-Edged Sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Dou, Z.; Xu, C.; Donahue, G.; Shimi, T.; Pan, J.A.; Zhu, J.; Ivanov, A.; Capell, B.C.; Drake, A.M.; Shah, P.P.; et al. Autophagy Mediates Degradation of Nuclear Lamina. Nature 2015, 527, 105–109. [Google Scholar] [CrossRef]
- Bello-Morales, R.; López-Guerrero, J.A. Extracellular Vesicles in Herpes Viral Spread and Immune Evasion. Front. Microbiol. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Bello-morales, R.; Ripa, I.; López-guerrero, J.A. Extracellular Vesicles in Viral Spread and Antiviral Response. Viruses 2020, 12, 623. [Google Scholar] [CrossRef]
- Yang, L.; Li, J.; Li, S.; Dang, W.; Xin, S.; Long, S.; Zhang, W.; Cao, P.; Lu, J. Extracellular Vesicles Regulated by Viruses and Antiviral Strategies. Front. Cell Dev. Biol. 2021, 9, 1–11. [Google Scholar] [CrossRef]
- Bello-Morales, R.; López-Guerrero, J.A. Isolation/Analysis of Extracellular Microvesicles from HSV-1-Infected Cells. Methods Mol. Biol. Biol. 2020, 2060, 305–317. [Google Scholar] [CrossRef]
- Bello-Morales, R.; Praena, B.; de La Nuez, C.; Rejas, M.T.; Guerra, M.; Galán-Ganga, M.; Izquierdo, M.; Calvo, V.; Krummenacher, C.; Antonio López-Guerrero, J.; et al. Role of Microvesicles in the Spread of Herpes Simplex Virus 1 in Oligodendrocytic Cells. J. Virol. 2018, 92, e00088-18. [Google Scholar] [CrossRef] [PubMed]
- le Sage, V.; Banfield, B.W. Dysregulation of Autophagy in Murine Fibroblasts Resistant to HSV-1 Infection. PLoS ONE 2012, 7, e042636. [Google Scholar] [CrossRef] [PubMed]
- Deepak, S.; Abraam, Y. Fine-Tuning of Autophagy Is Required for a Productive Herpes Simplex Virus Ocular Infection. Investig. Ophthalmol. Vis. Sci. 2015, 56, 940. [Google Scholar]
- Hofstetter, A.M.; Rosenthal, S.L.; Stanberry, L.R. Current Thinking on Genital Herpes. Curr. Opin. Infect. Dis. 2014, 27, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Liu, T.; Wang, W.; Duan, Q.; Liu, T.; Xu, L.; Huang, G.; Chen, Z. The Chinese Herbal Prescription JZ-1 Induces Autophagy to Protect against Herpes Simplex Virus-2 in Human Vaginal Epithelial Cells by Inhibiting the PI3K/Akt/mTOR Pathway. J. Ethnopharmacol. 2020, 254, 1–12. [Google Scholar] [CrossRef]
- Shao, Q.; Wu, F.; Liu, T.; Wang, W.; Liu, T.; Jin, X.; Xu, L.; Ma, Y.; Huang, G.; Chen, Z. JieZe-1 Alleviates HSV-2 Infection-Induced Genital Herpes in Balb/c Mice by Inhibiting Cell Apoptosis via Inducing Autophagy. Front. Pharmacol. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Yakoub, A.M.; Shukla, D. Basal Autophagy Is Required for Herpes Simplex Virus-2 Infection. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef]
- Taha, M.Y.; Clements, G.B.; Moira Brown, S. The Herpes Simplex Virus Type 2 (HG52) Variant JH2604 Has a 1488 Bp Deletion Which Eliminates Neurovirulence in Mice. J. Gen. Virol. 1989, 70, 3073–3078. [Google Scholar] [CrossRef]
- Tang, S.; Guo, N.; Patel, A.; Krause, P.R. Herpes Simplex Virus 2 Expresses a Novel Form of ICP34.5, a Major Viral Neurovirulence Factor, through Regulated Alternative Splicing. J. Virol. 2013, 87, 5820–5830. [Google Scholar] [CrossRef]
- Ravi, V.; Kennedy, P.G.; Maclean, A.R. Functional Analysis of the Herpes Simplex Virus Type 2 Strain HG52 RL1 Gene: The Intron Plays No Role in Virulence. J. Gen. Virol. 1998, 79, 1613–1617. [Google Scholar] [CrossRef]
- Korom, M.; Davis, K.L.; Morrison, L.A. Up to Four Distinct Polypeptides Are Produced from the γ134.5 Open Reading Frame of Herpes Simplex Virus 2. J. Virol. 2014, 88, 11284–11296. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Patel, A.; Krause, P.R. Novel Less-Abundant Viral MicroRNAs Encoded by Herpes Simplex Virus 2 Latency-Associated Transcript and Their Roles in Regulating ICP34.5 and ICP0 MRNAs. J. Virol. 2009, 83, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Bertke, A.S.; Patel, A.; Wang, K.; Cohen, J.I.; Krause, P.R.; Shenk, T.E. An Acutely and Latently Expressed Herpes Simplex Virus 2 Viral MicroRNA Inhibits Expression of ICP34.5, a Viral Neurovirulence Factor. Proc. Nat. Acad. Sci. USA 2008, 105, 10931–10936. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ripa, I.; Andreu, S.; López-Guerrero, J.A.; Bello-Morales, R. Interplay between Autophagy and Herpes Simplex Virus Type 1: ICP34.5, One of the Main Actors. Int. J. Mol. Sci. 2022, 23, 13643. https://doi.org/10.3390/ijms232113643
Ripa I, Andreu S, López-Guerrero JA, Bello-Morales R. Interplay between Autophagy and Herpes Simplex Virus Type 1: ICP34.5, One of the Main Actors. International Journal of Molecular Sciences. 2022; 23(21):13643. https://doi.org/10.3390/ijms232113643
Chicago/Turabian StyleRipa, Inés, Sabina Andreu, José Antonio López-Guerrero, and Raquel Bello-Morales. 2022. "Interplay between Autophagy and Herpes Simplex Virus Type 1: ICP34.5, One of the Main Actors" International Journal of Molecular Sciences 23, no. 21: 13643. https://doi.org/10.3390/ijms232113643
APA StyleRipa, I., Andreu, S., López-Guerrero, J. A., & Bello-Morales, R. (2022). Interplay between Autophagy and Herpes Simplex Virus Type 1: ICP34.5, One of the Main Actors. International Journal of Molecular Sciences, 23(21), 13643. https://doi.org/10.3390/ijms232113643