Abstract
Higher power conversion efficiencies for photovoltaic devices can be achieved through simple and low production cost processing of perovskites. Due to their limited long-term stability, however, there is an urgent need to find alternative structural combinations for this family of materials. In this study, we propose to investigate the prospects of cation-substitution within the A-site of the perovskite by selecting nine substituting organic and inorganic cations to enhance the stability of the material. The tolerance and the octahedral factors are calculated and reported as two of the most critical geometrical features, in order to assess which perovskite compounds can be experimentally designed. Our results showed an improvement in the thermal stability of the organic cation substitutions in contrast to the inorganic cations, with an increase in the power conversion efficiency of the Hydroxyl-ammonium (NH3OH) substitute to η = 25.84%.
1. Introduction
Perovskite materials with the ABX3 (A = cations, B = Pb, and X = I) formula have a variety of valuable properties, including both in application-oriented uses and fundamental physics [1,2,3]. One of the main reasons perovskites are so successful is their adaptability—they can be tailored to have a variety of different properties, which allows them to meet specific requirements such as an impressive series of improvements in light to electricity efficiency [4,5,6,7,8]. In just over five years of exhaustive research, the efficiency of (CH3NH3)PbI3 has been around 25% [9,10]. In this family, the perovskite architecture is largely retained, with one ion substituted by an organic ion (usually A or X). This makes the perovskite a hybrid with A and B as the cations, and X as a halogen or oxygen ion—two of the most common anions—while different metal cations can form halide and oxide perovskites [11,12] where the radii of A is larger than that of B. Both these perovskites have crystal structures that are very similar, with BX6 octahedra located in between the A atoms [13,14]. Studies from first-principles calculations showed that adding substituents such as Br, Cl and F to MAPbI3 can help facilitate the transfer of charge from the hybrid perovskite to the electrodes [2]. On the other hand, B-site substitutions change the bandgap. In our study, we investigated how the cation substitution at the A-site might affect the organic perovskite’s properties. Our analysis suggests that substituting the A cation may be a way to obtain an optimal band gap (1.1–1.4 eV) and high power conversion efficiency [9,15]. The Goldschmidt tolerance factor (t) has been a key element in the development of perovskites for many years, and it has been expanded to the growing field of organic-inorganic perovskites [4,16,17,18]. The t value should range between 0.8 and 1.11—otherwise, the crystal structure will be distorted, and might be destroyed. A smaller t value will result in a tetragonal or orthotopic shape with low symmetry. The t-value of an ideal cubic structure is from 0.89 to 1.0. The ideal value of the octahedral factor (μ), on the other hand (044 to 0.90), affects the stability of the octahedron, and further influences the stability of the perovskite structure [19,20].
As shown in Figure 1, recent machine learning studies generated about 2352 cations from different protonated amines. Only 742 of these cations showed a tolerance factor (t) of between 0.8 and 1.11, from which about 140 materials are better identified, such as well-characterized hybrid perovskites [13,21].
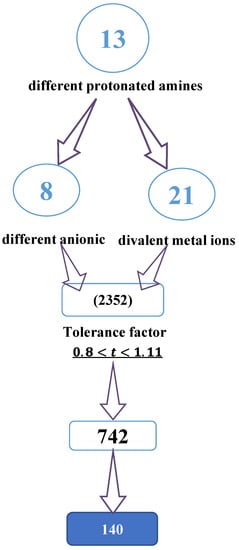
Figure 1.
Diagram showing the selection process for cations using the tolerance factor.
In our paper, we limited our study to nine cations (CH4, PH4, Li3O, CH3OH, Li3S, CH3NH3, CH(NH2)2, CH3C(NH2)2 and C(NH2)3), whose chemical formulas are detailed in Figure 2, and which have ionic radii of 146, 167, 170, 216, 220, 222, 253, 277 and 278 pm, respectively [22,23].
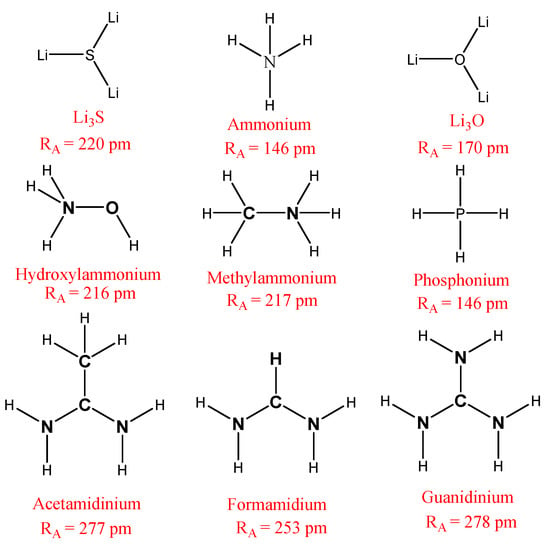
Figure 2.
The chemical formula for the cations studied in this paper.
For all substitutions (except for Li3O and Li3S), the short circuit current (JSC), the open-circuit voltage (VOC) and the power conversion efficiency (PCE) are also calculated. We hypothesized that every photon energy above E is absorbed, always promoting the perovskite to generate electron-hole pairs [20,24,25,26,27,28,29,30,31].
VOC was calculated, as well as the maximum theoretical limit of the PCE for all configurations, where the efficiency is regarded as a function of the band gap energy (Eg) and the total incident power density (PS), which can be calculated using solar spectrum data. PCE was obtained by using Jsc, Voc, and the fill factor (FF), as well as PS [32,33,34,35].
2. Results and Discussion
We used density functional theory (DFT) and ab-initio molecular dynamics (AIMD) to investigate the crystal structure, electronic structure, thermal and dynamic stability, and the power conversion efficiency of the substituted cubic MAPbI3 (MA = CH3NH3+).
2.1. Optimization
We optimized the structures after substituting the cations while observing the distortions in the nascent crystallites relative to the pristine crystal of MAPbI3. We noticed clear distortions in some structures, especially those with larger cations, or inorganic (Li3S, Li3O) size cations, while the other structures did not change significantly. Figure 3a,b shows the engineered structures before and after optimization for all the studied systems. Table 1 gives an exhaustive list of all calculated parameters [14,36]. This is attributed to many reasons, such as the difference in electrical polarization between the anion and the cation, as well as the size of the cation in relation to a unit cell.
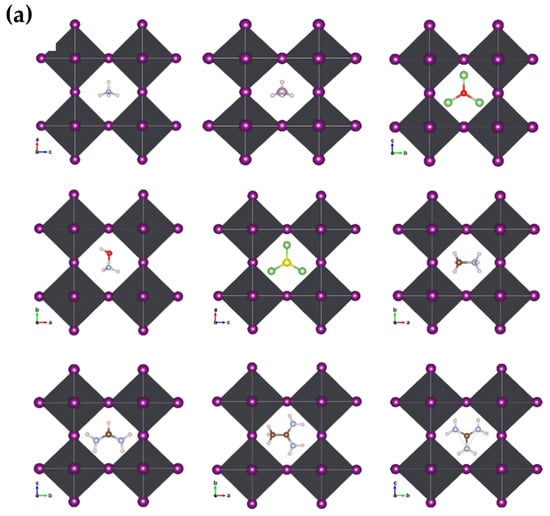
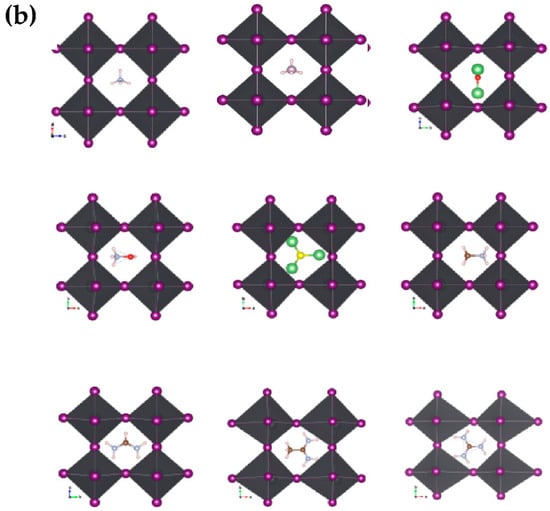
Figure 3.
Geometric structures of AiPbI3 with different cation: (a) before optimization; and (b) after optimization with Van der Waals correlation.

Table 1.
The theoretical lattice constants a, b, c, α, β, γ, and space groups of AiPbI3 with the different cations.
2.2. Tolerance Factor and Octahedral Factor and Parameters Optimization
In our study, we substituted the MA cation with a number of different-sized organic cations and their constituting atoms within the cubic phase, and for a first examination of the stability of the crystal lattice with each cation, we calculated the tolerance factor (t) and the octahedral stability factor (μ), and matched them with the permissible ranges, which were arranged in an ascending order of size, as shown in Table 2. The results indicated that every one of these falls inside the stable range [37].
Table 2.
Estimated radii (RA), band gap (Eg), formation energy (Ef), polyhedral factor (μ) and tolerance factor (t) of (Ai)PbI3 Perovskites with GGA-PBE (vdW) by theoretical calculations.
2.3. Stability
Formation Energy:
We can clearly see that this perovskite can be produced in a laboratory based on the calculated formation energy values. Additionally, we also noticed that the formation energy rises as the cation size increases in all structures (see Table 1). The reason may be attributed to the poor thermal stability. The formation energies were calculated by Equation (1)
where Ai is the different cation, and x and y are the numbers of Ai and BX3 atoms in a unit cell, respectively.
2.4. Dynamical Stability
The total phonon density of states (Ph-DOS) is calculated at the equilibrium volumes for different cations of Ai, and displayed in Figure 4. For all these cations, a small imaginary frequency was observed for the CH4, PH4, CH3OH, CH3NH3, CH(NH2)2, CH3C(NH2)2, and C(NH2)3, indicating that these structures are stable or at least relatively stable at ambient conditions. Other than this, a clear imaginary frequency was observed in the alkali (Li3O, and Li3S) structures, indicating that these structures are unstable at ambient conditions.
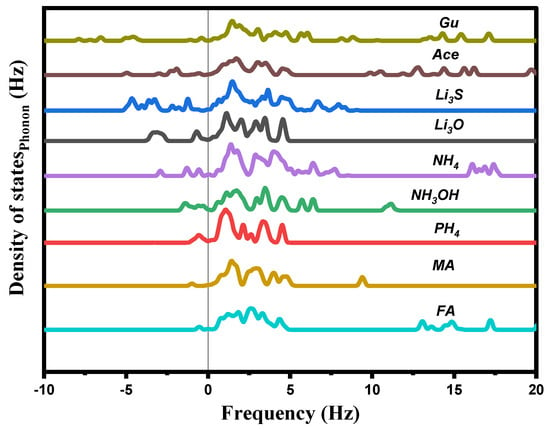
Figure 4.
Calculated total phonon density of states Ph−DOS for AiPbI3 with the different substituting cations.
2.5. Thermal Stability
Furthermore, we simulated the chemical reaction using an ab-initio molecular dynamics simulation that was based on the force calculations from density functional theory. The simulations were run on models with 3 × 3 × 3 supercells, for a total of 25 ps per case (see Figure 5). The smaller the organic component of a system, the more it is over-correlated with other parts of the system. This suggests that the smaller system is more easily moved around by electrostatic forces. A large-scale system mitigates this finite-size artifact, and produces a more reliable explanation of the anisotropic rotational behavior of cations. To compare the dynamics performance of the perovskites with the cations, the AIMD simulations are performed for the cubic NH4PbI3, PH4PbI3, Li3OPbI3, NH3OPbI3, Li3SPbI3, MAPbI3, AMPbI3, AcPbI3 and GuPbI3 perovskites with a 3 × 3 × 3 supercell. Figure 6 shows the AIMD simulation results for cubic Li3OPbI3 and Li3SPbI3 perovskites after 25 ps.

Figure 5.
The cubic APbI3 perovskites’ structure changed with a 3 × 3 × 3 supercell after ab initio molecular dynamics simulation of 50 ps at 300 K. Atomic colors are: Pb (grey); C (brown); H (white); F (blue); Li (cyan); Cl (green); N (silver); O (red); S (yellow); P (lavender); and I (purple).
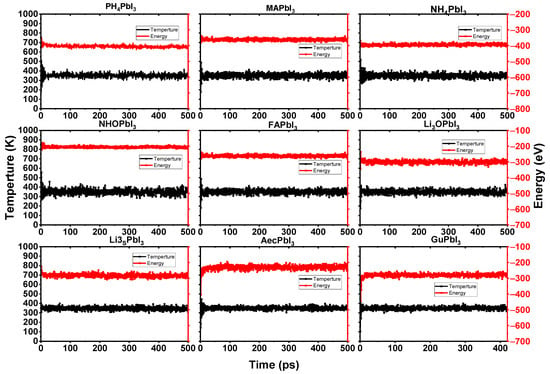
Figure 6.
Fluctuation of the temperature and total energy of the considered perovskite systems from 0 to 500 ps using AIMD simulations at 300 K.
According to the AIMD simulation of the AiPbI3 perovskite depicted in Figure 6, the AiPbI3 perovskites exhibit no evident structural change after a 500 ps AIMD simulation at 300 K.
The Ai+ ions in the AiPbI3 perovskites do not congregate, the PbI3 frame does not crumble (see Figure 5), and the temperature and energy curves indicate balance with a little shock (see Figure 6).
Furthermore, given the strong interaction between Li3O+ and Li3S+ ions in super-alkali perovskites, as well as the large-size cations, investigating the dynamics performance of perovskites based on various cations (Ai) is critical for identifying perovskites with stable dynamics performance [38]. This may be due to the hydrogen bonding between the cation and anions (PbI3 frame) and the difficulty of movement of the cation inside the cavity of the structure, due to its large size, and might also be attributed to the electrostatic forces resulting from the transfer of charges between positive cations A+ and negative anions PbI6- (see Figure S3).
2.6. Electronic
In photovoltaic applications, determining the absorber layer material with appropriate electronic features plays an important role in improving device performance. The size of a band gap is important in estimating the number of photons that are absorbed, and the rate of photogenerated charge carriers with minimal optical losses. To generate and transport free charge carriers efficiently, low exciton binding energy and low effective masses of holes and electrons are necessary. Moreover, DFT studies reveal that, apart from LiO3, LiS3 and Gu, the electrical structures of the examined substituted perovskites were like those of MAPbI3, although their bandgaps marginally decreased or increased compared to those of MAPbI3. Our findings imply that adjusting the band gap and enhancing thermal and dynamical stability may both be accomplished by substitution in halide perovskite. To help us better understand the electronic structure of the optimized AiPbI3 perovskite structures, we calculated their band structures using the GGA-PBE method for Ai = NH4, PH4, Li3O, NH3O, Li3S, CH3NH3, CH(NH2)2, Ace, and Gu along the high symmetry points of the Brillouin zone, which has taken three space groups—cubic, triclinic and orthorhombic—as shown in Figure 7a. Here, the Fermi level is set to zero. The maximum energy of valence band (VBM) and the minimum energy of conduction band (CBM) for all computed band structures are clearly shown in the figure, to be located along points R, Q and E of the Brillouin zone for (PH4, Li3O, CH(NH2)2), (Li3S, Ace, Gu-NH4) and (NH3O, MA), respectively. Since these band gaps represent the direct nature of the band gaps, these materials can be called as having good optical absorption properties (Figure 7b,c). The calculated band gaps for NH4PbI3, PH4PbI3, Li3OPbI3, NH3OPbI3, Li3SPbI3, CH3NH3PbI3, CH(NH2)2PbI3, AcPbI3 and GuPbI3 are 1.7611, 1.6101, 0.5932, 1.5348, 0.4146, 1.5971, 1.4815, 1.9645 and 2.1517 eV, as given in Table 3 respectively, which are in fair agreement with the experimental results and other theoretical calculations.
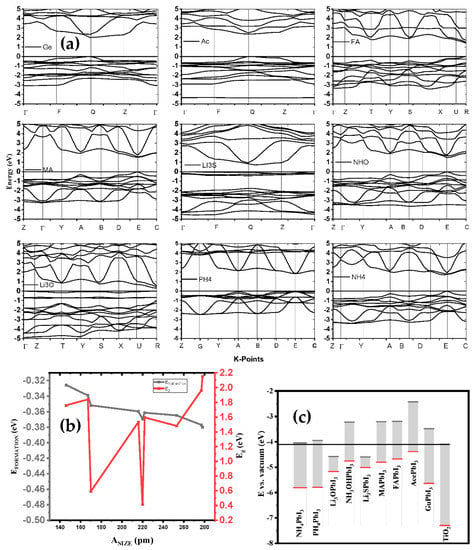
Figure 7.
(a) The variation of formation energy and bandgap function of the different cations; (b) the potential energy with AiPbI3; and (c) band structure of AiPbI3 in comparison with Van der Waals.
Table 3.
Calculated open-circuit voltages (VOC), short circuit currents (JSC) and power conversion efficiencies (η) for the hypothetical perovskites AiPbI3 and MAPbI3.
2.7. Optic
The optical properties of the solar cell, including the absorption edge, the intensity of the absorption at various wavelengths and the reflection coefficient, are crucial to determine its external and internal quantum efficiencies (see Equation (S5) in the Supplementary Materials). These optical properties are the result of light waves interacting with its valence electrons. The optical response of a material when it is interacted with an electromagnetic radiation, such as light waves, is given by the complex dielectric functions ε1(ω) and ε2(ω). For energies greater than Eg, our predicted absorption (α) compares favorably to a recent absorption measurement, providing important validation of our technique. According to the experiment (see Figure 8), the calculated α is 104 cm−1 at the start, and increases linearly (on a logarithmic scale) with energy. The visible slope variation around α = 2.5–3 eV replicates a comparable characteristic in the experimental spectrum [39]. The absorption curves of AiPbI3 and GaAs are identical throughout a large energy range, except for the energy onset corresponding to the different Eg of the studied materials.
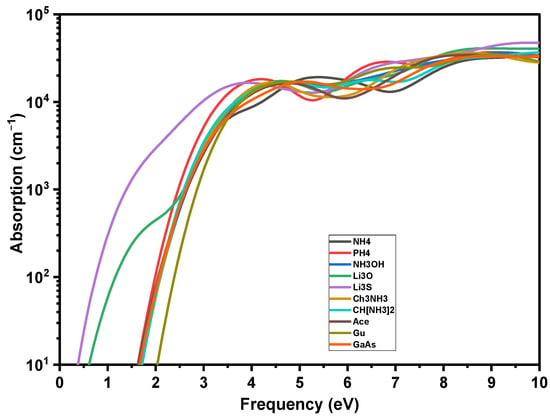
Figure 8.
Absorption coefficients of crystalline GaAs and AiPbI3 perovskite.
2.8. Power Conversion Efficiency
Despite the cation substitution in the identical structures, these power conversion efficiencies were observed to be substantially closer to those of single-junction solar cells (shown in Table 3), which were estimated using Equation (S5) [40]. For each simulation, we considered a power input of PS = 100 mW/cm2, which is the power under the maximum AM1.5 solar illumination spectrum with TCell = 300 k and TS = 6000 k.
The electron-hole pairs are easily separated and moved in the CBM and VBM states, which suggests that the perovskite-based solar cells studied so far have the potential to have high PCE, which may give them an edge over other cells in terms of power output. Figure 9a shows the PCE of solar cells based on these perovskites as a function of the band gaps. The cubic HAPbI3 perovskite-based solar cells with a PCE of 25.84% are the best option, followed by the MAPbI3 perovskites with a PCE of 24.63%. Experimental reports have shown that the efficiency of solar cells tends to be around 25.2% [10], while the maximum efficiency conversion is observed for a 1.4–1.6 eV bandgap. Moreover, the cubic GuPbI3 perovskite with a direct maximum band gap of 2.15 eV also shows a PCE of 16.52%. The PCE results of these perovskites indicate that they are a good choice for single-junction solar cells. We have calculated PCE for our intrinsic perovskite solar cell with Eg of perovskite structures shown in Table 3. The efficiency of these structures, according to the Shockley–Queisser model, is represented in Figure 9b. Based on these results, the considered perovskites can reach an efficiency limit of 25.84% in ideal conditions, with TC = 300 k and TS = 6000 k for the AM1.5 spectrum.
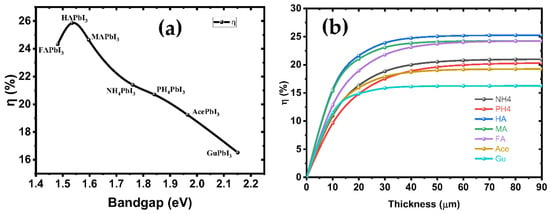
Figure 9.
(a) The variation theoretical efficiencies under AM1.5 illumination in dependence of the bandgap energy according to the substitution of cation in A-site; and (b) the variation in efficiencies function in thickness of all structures.
Figure 9b shows the variation of the PV parameters as a function of the absorber’s thickness. Our results depicted from the simulations show that at a thickness of 60 nm, the efficiency rose as high as 24.63%, while FF = 68.28%, JSC = 26.04 mA/cm2 and VOC = 1.09 V. The defects were considered in the simulated absorber layer. We found that the efficiency increases up to 25.84% as the thickness increases from 10 nm to 90 nm, thus representing the increased electron-hole pair generation rate in the absorber MAPbI3 [41], while at higher thickness, the recombination of charges starts to occur earlier in the metal, before they reach the contact points—this results in a saturation of charges at thicker metal layers [42]. It was found that the amount of VOC decreased as the thickness of the absorber material, MAPbI3, was increased. This was related to the short-circuit current (JSC) and dark saturation current (J0), and at higher thickness the dark saturation current rises, which causes the recombination probability of the charge carriers to increase. The Shockley-Queisser Equation (S10) clearly describes the relationship between VOC, JSC and J0 [43]. Figure 10 illustrates the J-V curves of all structures, and the structure parameters are shown in Table 3.
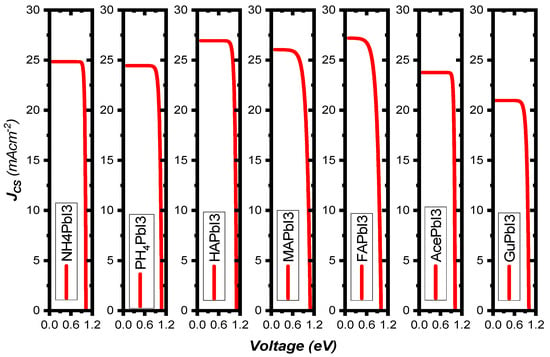
Figure 10.
The curves of short-circuit current function open-circuit voltage of AiPbI3 with different cations under AM 1.5 G illumination at 100 mW/cm2.
3. Conclusions
In conclusion, we found that there is a high probability of the formation of 3D perovskite structures by calculating tolerance factors and octahedral factors for 486 permutations of ABX3 organic-inorganic hybrid compounds reported in the literature. Nine different organic monoammonium cations were studied in combination with Pb metal ions and I halide. We used CH3NH3+ as a reference to estimate the steric bulk of molecular cations. We have found that the structures are dynamically stable and thermally efficient, and as a consequence, the adjustment in A+ revealed a significantly increased PCE of 25.84% in Hydroxylammonium (NH3OH), compared to 24.63% in the MA of the control device, as well as an obvious improvement in its operational and thermal stabilities. There are many previously unknown compounds that could form stable perovskite phases at ambient conditions. This study is expected to lead to increased research efforts in designing perovskite structures with specific optoelectronic properties, as well as their great structural flexibility. This, combined with their promising physical properties, makes perovskites a powerful chemical structure for ferroelectrics and future photovoltaics technologies.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms232113556/s1, Figure S1: The process of merging a cation A+ into the PbI6- framework to produce perovskite structures; Figure S2: Ionic packing in an ideal cubic perovskite structure, with (a) a loosely packed crystal structure with a small A radius with t < 1, (b) an ideal cubic perovskite structure with 0.8 < t < 1.11 and (c) a tightly packed crystal structure with a large A radius with t > 1; Figure S3 Difference in charge between the A+ cation and the PbI6- anion, where the yellow area represents charge accumulation, whereas the blue area represents charge depletion. References [44,45,46,47,48,49,50,51,52] are cited in Supplementary Materials.
Author Contributions
A.E.M. and O.M. conceived/planned the simulations and supervised the findings of this work. A.A-S. and A.S. contributed to the configuration of the models and experiments. A.E.K. and A.B. verified the results and contributed to the interpretation of results. A.A.-S., A.E.M. and O.M. wrote the manuscript with insights from all the authors. A.E.M. secured the funds for this research. All authors discussed the results and contributed to the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partly funded by United Arab Emirates University UPAR project, grant number 31N393.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Used data and equations are available in the Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Glazer, A.M. The Classification of Tilted Octahedra in Perovskites. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1972, 28, 3384–3392. [Google Scholar] [CrossRef]
- Peña, M.A.; Fierro, J.L.G. Chemical Structures and Performance of Perovskite Oxides. Chem. Rev. 2001, 101, 1981–2017. [Google Scholar] [CrossRef] [PubMed]
- Jonathan, L.; Diguna, L.J.; Samy, O.; Muqoyyanah, M.; Abu Bakar, S.; Birowosuto, M.D.; El Moutaouakil, A. Hybrid Organic–Inorganic Perovskite Halide Materials for Photovoltaics towards Their Commercialization. Polymers 2022, 14, 1059. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Li, C.; Li, B.; Zhang, J.; Sun, Y.; Guo, W.; Zhou, Z.; Pang, S.; Yan, Y. Interaction Engineering in Organic-Inorganic Hybrid Perovskite Solar Cells. Mater. Horiz. 2020, 7, 2208–2236. [Google Scholar] [CrossRef]
- Kumari, N.; Patel, S.R.; Gohel, J. V Superior Efficiency Achievement for FAPbI3-Perovskite Thin Film Solar Cell by Optimization with Response Surface Methodology Technique and Partial Replacement of Pb by Sn. Optik 2019, 176, 262–277. [Google Scholar] [CrossRef]
- Ye, F.; Lin, H.; Wu, H.; Zhu, L.; Huang, Z.; Ouyang, D.; Niu, G.; Choy, W.C.H. High-Quality Cuboid CH3NH3PbI3 Single Crystals for High Performance X-Ray and Photon Detectors. Adv. Funct. Mater. 2018, 29, 1806984. [Google Scholar] [CrossRef]
- Pbi, N.H.; Weller, M.T.; Weber, O.J.; Frost, J.M.; Walsh, A. Cubic Perovskite Structure of Black Formamidinium Lead Iodide, α-[HC(NH2)2]PbI3, at 298 K. J. Phys. Chem. Lett. 2015, 6, 3209–3212. [Google Scholar] [CrossRef]
- Chen, W.; Zhu, Y.; Xiu, J.; Chen, G.; Liang, H.; Liu, S.; Xue, H.; Birgersson, E.; Ho, J.W.; Qin, X.; et al. Monolithic Perovskite/Organic Tandem Solar Cells with 23.6% Efficiency Enabled by Reduced Voltage Losses and Optimized Interconnecting Layer. Nat. Energy 2022, 7, 229–237. [Google Scholar] [CrossRef]
- Jacak, J.E.; Jacak, W.A. Routes for Metallization of Perovskite Solar Cells. Materials 2022, 15, 2254. [Google Scholar] [CrossRef]
- Zeng, Z.; Xu, Y.; Zhang, Z.; Gao, Z.; Luo, M.; Yin, Z.; Zhang, C.; Xu, J.; Huang, B.; Luo, F.; et al. Rare-Earth-Containing Perovskite Nanomaterials: Design, Synthesis, Properties and Applications. Chem. Soc. Rev. 2020, 49, 1109–1143. [Google Scholar] [CrossRef]
- Takaba, H.; Kimura, S.; Alam, M.K. Crystal and Electronic Structures of Substituted Halide Perovskites Based on Density Functional Calculation and Molecular Dynamics. Chem. Phys. 2017, 485–486, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Luo, X. Tuning Bandgaps of Mixed Halide and Oxide Perovskites CsSnX3 (X=Cl, I), and SrBO3 (B=Rh, Ti). Appl. Sci. 2021, 11, 6862. [Google Scholar] [CrossRef]
- Kieslich, G.; Sun, S.; Cheetham, A.K. An Extended Tolerance Factor Approach for Organic-Inorganic Perovskites. Chem. Sci. 2015, 6, 3430–3433. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Yan, C.; Fong, P.W.K.; Yu, J.; Liu, H.; Yin, J.; Huang, J.; Lu, X.; Yan, H.; Li, G. In Situ and Ex Situ Investigations on Ternary Strategy and Co-Solvent Effects towards High-Efficiency Organic Solar Cells. Energy Environ. Sci. 2022, 15, 2479–2488. [Google Scholar] [CrossRef]
- Ali, R.; Hou, G.J.; Zhu, Z.G.; Yan, Q.B.; Zheng, Q.R.; Su, G. Predicted Lead-Free Perovskites for Solar Cells. Chem. Mater. 2018, 30, 718–728. [Google Scholar] [CrossRef]
- Fouladi, F.; Seyed, Y.; Kanjouri, F. Results in Physics MAPbI3 and FAPbI3 Perovskites as Solar Cells: Case Study on Structural, Electrical and Optical Properties. Results Phys. 2018, 10, 616–627. [Google Scholar] [CrossRef]
- Grote, C.; Berger, R.F. Strain Tuning of Tin-Halide and Lead-Halide Perovskites: A First-Principles Atomic and Electronic Structure Study. J. Phys. Chem. C 2015, 119, 22832–22837. [Google Scholar] [CrossRef]
- Li, D.; Li, D.; Zhang, H.; Yang, A.; Liang, C. High-Performance Photovoltaic Materials Based on the Superlattice Structures of Organic-Inorganic Halide Perovskite and Superhalogen Hybrid Perovskite. J. Phys. Chem. Lett. 2020, 11, 5282–5294. [Google Scholar] [CrossRef]
- Travis, W.; Glover, E.N.K.; Bronstein, H.; Scanlon, D.O.; Palgrave, R.G. On the Application of the Tolerance Factor to Inorganic and Hybrid Halide Perovskites: A Revised System. Chem. Sci. 2016, 7, 4548–4556. [Google Scholar] [CrossRef]
- Boubekraoui, A.; Moatassim, H.; Al-Shami, A.; Ez-Zahraouy, H. DFT Study of Structural, Electronic, and Thermoelectric Properties of Cs2PdX(X=Br2Be2Te2) Compound. Comput. Condens. Matter 2021, 29, e00600. [Google Scholar] [CrossRef]
- Stanić, D.; Kojić, V.; Čižmar, T.; Juraić, K.; Bagladi, L.; Mangalam, J.; Rath, T.; Gajović, A. Simulating the Performance of a Formamidinium Based Mixed Cation Lead Halide Perovskite Solar Cell. Materials 2021, 14, 6341. [Google Scholar] [CrossRef]
- Zhou, T.; Zhang, Y.; Wang, M.; Zang, Z.; Tang, X. Tunable Electronic Structures and High Efficiency Obtained by Introducing Superalkali and Superhalogen into AMX3-Type Perovskites. J. Power Sources 2019, 429, 120–126. [Google Scholar] [CrossRef]
- Adjogri, S.J.; Meyer, E.L. A Review on Lead-Free Hybrid Halide Perovskites as Light Absorbers for Photovoltaic Applications Based on Their Structural, Optical, and Morphological Properties. Molecules 2020, 25, 5039. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, Z.; Sibari, A.; Al-Shami, A.; Lahbabi, S.; El Kenz, A.; Benyoussef, A.; El Fatimy, A.; Mounkachi, O. Graphene/Phosphorene Nano-Heterostructure as a Potential Anode Material for (K/Na)-Ion Batteries: Insights from DFT and AIMD. Comput. Mater. Sci. 2022, 202, 110936. [Google Scholar] [CrossRef]
- EL Kassaoui, M.; Mansouri, Z.; Al-Shami, A.; Sibari, A.; Benyoussef, A.; El Kenz, A.; Mounkachi, O.; Loulidi, M. Design of Metal-Decorated Beryllium Carbide (Be2C) as a High-Capacity Hydrogen Storage Material with Strong Adsorption Characteristics. Appl. Surf. Sci. 2022, 589, 152960. [Google Scholar] [CrossRef]
- Al-Shami, A.; Lakhal, M.; Hamedoun, M.; El Kenz, A.; Benyoussef, A.; Loulidi, M.; Ennaoui, A.; Mounkachi, O. Tuning the Optical and Electrical Properties of Orthorhombic Hybrid Perovskite CH3NH3PbI3 by First-Principles Simulations: Strain-Engineering. Sol. Energy Mater. Sol. Cells 2018, 180, 266–270. [Google Scholar] [CrossRef]
- Hijazi, A.; Moutaouakil, A.E. Graphene and MoS2 Structures for THz Applications. In Proceedings of the 2019 44th International Conference on Infrared, Millimeter, IEEE, Paris, France, 1–6 September 2019, and Terahertz Waves (IRMMW-THz); pp. 1–2.
- Moutaouakil, A.E.; Kang, H.-C.; Handa, H.; Fukidome, H.; Suemitsu, T.; Sano, E.; Suemitsu, M.; Otsuji, T. Room Temperature Logic Inverter on Epitaxial Graphene-on-Silicon Device. Jpn. J. Appl. Phys. 2011, 50, 070113. [Google Scholar] [CrossRef]
- Moutaouakil, A.E.; Fukidome, H.; Otsuji, T. Investigation of Terahertz Properties in Graphene Ribbons. In Proceedings of the 2020 45th International Conference on Infrared, Millimeter, and Terahertz Waves (IRMMW-THz), Buffalo, NY, USA, 8–13 November 2020; pp. 1–2. [Google Scholar]
- Samy, O.; El Moutaouakil, A. A Review on MoS2 Energy Applications: Recent Developments and Challenges. Energies 2021, 14, 4586. [Google Scholar] [CrossRef]
- Tiouitchi, G.; Ali, M.A.; Benyoussef, A.; Hamedoun, M.; Lachgar, A.; Kara, A.; Ennaoui, A.; Mahmoud, A.; Boschini, F.; Oughaddou, H.; et al. Efficient Production of Few-Layer Black Phosphorus by Liquid-Phase Exfoliation. R. Soc. Open Sci. 2020, 7, 201210. [Google Scholar] [CrossRef]
- Nielsen, P. Coastal and Estuarine Processes. In Coastal And Estuarine Processes; World Scientific Publishing Company: Singapore, 2009; pp. 1–360. [Google Scholar]
- Muñoz-García, A.B.; Caputo, L.; Schiavo, E.; Baiano, C.; Maddalena, P.; Pavone, M. Ab Initio Study of Anchoring Groups for CuGaO2 Delafossite-Based p-Type Dye Sensitized Solar Cells. Front. Chem. 2019, 7, 158. [Google Scholar] [CrossRef]
- She, L.; Liu, M.; Zhong, D. Atomic Structures of CH3NH3PbI3 (001) Surfaces. ACS Nano 2016, 10, 1126–1131. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Vasudevan, V.; Lin, S.; Jasieniak, J.; Russo, S.P.; Birbilis, N.; Medhekar, N. V. Molecular Mechanisms of Thermal Instability in Hybrid Perovskite Light Absorbers for Photovoltaic Solar Cells. J. Mater. Chem. A 2020, 8, 17765–17779. [Google Scholar] [CrossRef]
- Jiang, X.; Chotard, P.; Luo, K.; Eckmann, F.; Tu, S.; Reus, M.A.; Yin, S.; Reitenbach, J.; Weindl, C.L.; Schwartzkopf, M.; et al. Revealing Donor–Acceptor Interaction on the Printed Active Layer Morphology and the Formation Kinetics for Nonfullerene Organic Solar Cells at Ambient Conditions. Adv. Energy Mater. 2022, 12, 2103977. [Google Scholar] [CrossRef]
- Wikipedia. Ionic Radius. Available online: https://en.wikipedia.org/wiki/Ionic_radius (accessed on 6 October 2022).
- Fang, H.; Jena, P. Li-Rich Antiperovskite Superionic Conductors Based on Cluster Ions. Proc. Natl. Acad. Sci. USA 2017, 114, 11046–11051. [Google Scholar] [CrossRef]
- Sell, D.D.; Casey, H.C. Optical Absorption and Photoluminescence Studies of Thin GaAs Layers in GaAsSingle Bond SignAlxGa1-XAs Double Heterostructures. J. Appl. Phys. 1974, 45, 800–807. [Google Scholar] [CrossRef]
- Hossain, M.I.; Qarony, W.; Ma, S.; Zeng, L.; Knipp, D.; Tsang, Y.H. Perovskite/Silicon Tandem Solar Cells: From Detailed Balance Limit Calculations to Photon Management. Nano-Micro Lett. 2019, 11, 58. [Google Scholar] [CrossRef]
- Tao, J.; Ali, N.; Chen, K.; Huai, Z.; Sun, Y.; Fu, G.; Kong, W.; Yang, S. Enhanced Efficiency in Perovskite Solar Cells by Eliminating the Electron Contact Barrier between the Metal Electrode and Electron Transport Layer. J. Mater. Chem. A 2019, 7, 1349–1355. [Google Scholar] [CrossRef]
- Christians, J.A.; Schulz, P.; Tinkham, J.S.; Schloemer, T.H.; Harvey, S.P.; Tremolet De Villers, B.J.; Sellinger, A.; Berry, J.J.; Luther, J.M. Tailored Interfaces of Unencapsulated Perovskite Solar Cells for >1000 Hour Operational Stability. Nat. Energy 2018, 3, 68–74. [Google Scholar] [CrossRef]
- Yan, L.; Wang, M.; Zhai, C.; Zhao, L.; Lin, S. Symmetry Breaking Induced Anisotropic Carrier Transport and Remarkable Thermoelectric Performance in Mixed Halide Perovskites CsPb(I1-xBrx)3. ACS Appl. Mater. Interfaces 2020, 12, 40453–40464. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A Modular and Open-Source Software Project for Quantum Simulations of Materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Dion, M.; Rydberg, H.; Schröder, E.; Langreth, D.C.; Lundqvist, B.I. Van Der Waals Density Functional for General Geometries. Phys. Rev. Lett. 2004, 92, 246401. [Google Scholar] [CrossRef]
- Singh, D.J.; Park, C.H. Polar Behavior in a Magnetic Perovskite from A-Site Size Disorder: A Density Functional Study. Phys. Rev. Lett. 2008, 100, 087601. [Google Scholar] [CrossRef]
- Sani, F.; Shafie, S.; Lim, H.N.; Musa, A.O. Advancement on Lead-Free Organic-Inorganic Halide Perovskite Solar Cells: A Review. Materials 2018, 11, 1008. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, J.; Bakr, O.M.; Sun, H.-T. Metal-Doped Lead Halide Perovskites: Synthesis, Properties, and Optoelectronic Applications. Chem. Mater. 2018, 30, 6589–6613. [Google Scholar] [CrossRef]
- Peng, C.; Chen, J.; Wang, H.; Hu, P. First-Principles Insight into the Degradation Mechanism of CH3NH3PbI3 Perovskite: Light-Induced Defect Formation and Water Dissociation. J. Phys. Chem. C 2018, 122, 27340–27349. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).