
The Accuracy of Semi-Empirical Quantum Chemistry Methods on Soot Formation Simulation †




Abstract
1. Introduction
2. Results and Discussion
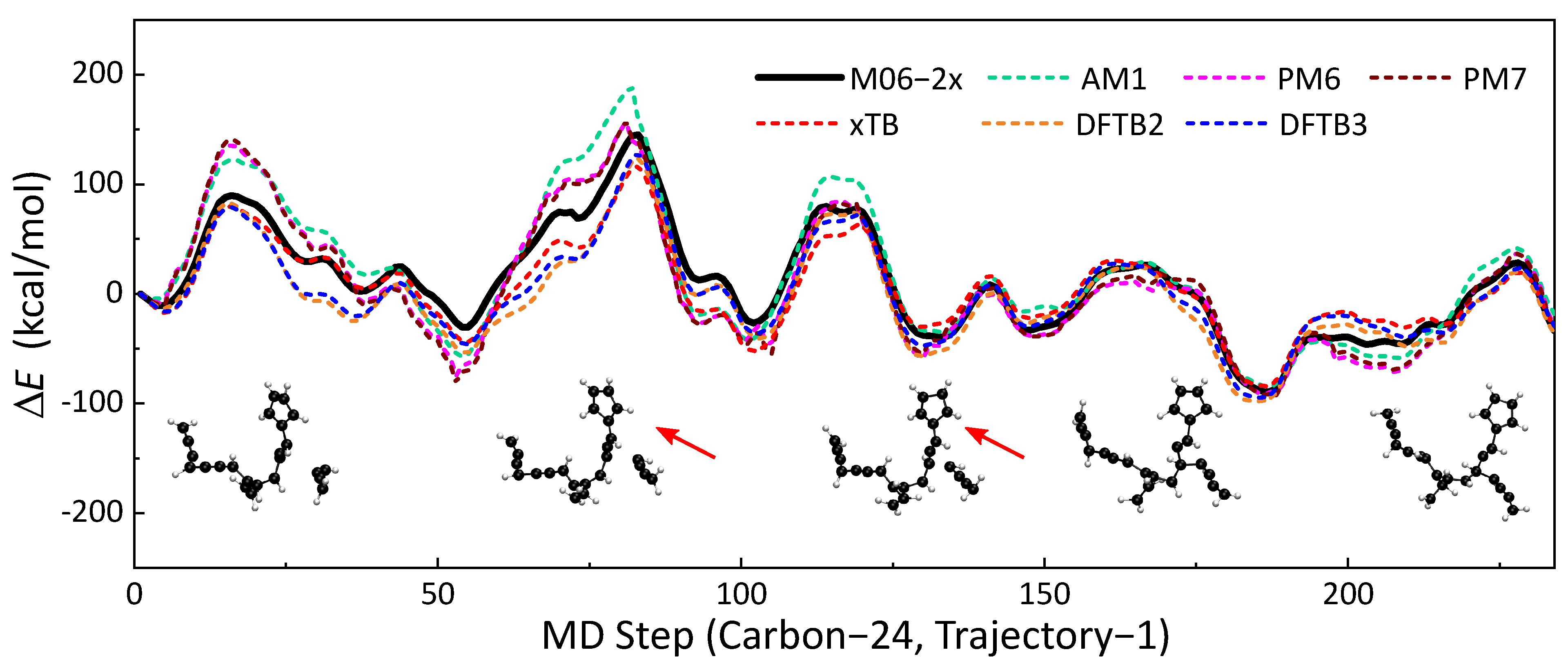
2.1. Similarity of Energy Profiles
2.2. Similarity of Optimized Structures
2.3. Intrinsic Reaction Coordinate Paths
2.4. Spin Density Distribution
3. Materials and Methods
3.1. SE Calculations
3.2. MD Simulations
3.3. Selection of the Benchmark DFT Method
4. Conclusions
- (1)
- The energy profiles calculated by SE methods are generally similar to those of reference DFT calculations. The GFN2-xTB method has significant advantages over other SE methods in describing the energy profile of MD trajectories. Compared with HF-based methods (AM1, PM6, and PM7), DFTB series methods have better performances. We also find that the consideration of spin polarization does not significantly improve the performance of DFBT2.
- (2)
- SE methods can optimize the structures of the soot precursor well. DFTB methods generally perform better than HF-based SE methods for both small soot-relevant test sets and sets with larger structures. The structures of cyclic or polycyclic rigid molecules are described well using SE methods. Yet, HF-based methods have larger offsets relative to the reference for molecules with flexible side chains.
- (3)
- In the validation of IRC energy profiles, SE methods can give accurate relative energies, which can be used for the qualitative analysis of reaction paths in most cases, but are still not suitable for quantitative calculations. In addition, in the case of calculating H-transfer in gaseous PAH precursors, both GFN2-xTB and DFTB methods were observed to underestimate the transition state energies because of the fractional charge error.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SE | semi-empirical |
| DFT | density functional theory |
| DFTB | density functional tight binding |
| MD | molecular dynamic |
| RMSE | root-mean-square error |
| AIMD | ab initio molecular dynamic |
| IRC | intrinsic reaction coordinate |
References
- Bond, T.C.; Doherty, S.J.; Fahey, D.W.; Forster, P.M.; Berntsen, T.; DeAngelo, B.J.; Flanner, M.G.; Ghan, S.; Kärcher, B.; Koch, D.; et al. Bounding the role of black carbon in the climate system: A scientific assessment. J. Geophys. Res. Atmos. 2013, 118, 5380–5552. [Google Scholar] [CrossRef]
- Straif, K.; Keil, U.; Taeger, D.; Holthenrich, D.; Sun, Y.; Bungers, M.; Weiland, S.K. Exposure to Nitrosamines, Carbon Black, Asbestos, and Talc and Mortality from Stomach, Lung, and Laryngeal Cancer in a Cohort of Rubber Workers. Am. J. Epidemiol. 2000, 152, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.R.; Jerrett, M.; Anderson, H.R.; Burnett, R.T.; Stone, V.; Derwent, R.; Atkinson, R.W.; Cohen, A.; Shonkoff, S.B.; Krewski, D.; et al. Public health benefits of strategies to reduce greenhouse-gas emissions: Health implications of short-lived greenhouse pollutants. Lancet 2009, 374, 2091–2103. [Google Scholar] [CrossRef]
- Haynes, B.; Wagner, H. Soot formation. Prog. Energy Combust. Sci. 1981, 7, 229–273. [Google Scholar] [CrossRef]
- Richter, H.; Howard, J. Formation of polycyclic aromatic hydrocarbons and their growth to soot—A review of chemical reaction pathways. Prog. Energy Combust. Sci. 2000, 26, 565–608. [Google Scholar] [CrossRef]
- McEnally, C.S.; Pfefferle, L.D.; Atakan, B.; Kohse-Höinghaus, K. Studies of aromatic hydrocarbon formation mechanisms in flames: Progress towards closing the fuel gap. Prog. Energy Combust. Sci. 2006, 32, 247–294. [Google Scholar] [CrossRef]
- Knyazev, V.D.; Slagle, I.R. Kinetics of the Reaction between Propargyl Radical and Acetylene. J. Phys. Chem. A 2002, 106, 5613–5617. [Google Scholar] [CrossRef]
- Johansson, K.O.; Head-Gordon, M.P.; Schrader, P.E.; Wilson, K.R.; Michelsen, H.A. Resonance-stabilized hydrocarbon-radical chain reactions may explain soot inception and growth. Science 2018, 361, 997–1000. [Google Scholar] [CrossRef]
- Jin, H.; Xing, L.; Yang, J.; Zhou, Z.; Qi, F.; Farooq, A. Continuous Butadiyne Addition to Propargyl: A Radical-Efficient Pathway for Polycyclic Aromatic Hydrocarbons. J. Phys. Chem. Lett. 2021, 12, 8109–8114. [Google Scholar] [CrossRef] [PubMed]
- Frenklach, M.; Mebel, A.M. On the mechanism of soot nucleation. Phys. Chem. Chem. Phys. 2020, 22, 5314–5331. [Google Scholar] [CrossRef]
- Zhao, L.; Prendergast, M.B.; Kaiser, R.I.; Xu, B.; Ablikim, U.; Ahmed, M.; Sun, B.J.; Chen, Y.L.; Chang, A.H.H.; Mohamed, R.K.; et al. Synthesis of Polycyclic Aromatic Hydrocarbons by Phenyl Addition?Dehydrocyclization: The Third Way. Angew. Chem. Int. Ed. 2019, 58, 17442–17450. [Google Scholar] [CrossRef]
- Jin, H.; Xing, L.; Liu, D.; Hao, J.; Yang, J.; Farooq, A. First aromatic ring formation by the radical-chain reaction of vinylacetylene and propargyl. Combust. Flame 2021, 225, 524–534. [Google Scholar] [CrossRef]
- Luehr, N.; Jin, A.G.B.; Martínez, T.J. Ab Initio Interactive Molecular Dynamics on Graphical Processing Units (GPUs). J. Chem. Theory Comput. 2015, 11, 4536–4544. [Google Scholar] [CrossRef]
- Chen, X.; Liu, M.; Gao, J. CARNOT: A Fragment-Based Direct Molecular Dynamics and Virtual-Reality Simulation Package for Reactive Systems. J. Chem. Theory Comput. 2022, 18, 1297–1313. [Google Scholar] [CrossRef]
- Pilsun, Y.; Sakano, M.; Saaketh, D.; Islam, M.M.; Peilin, L.; Strachan, A. Neural network reactive force field for C, H, N, and O systems. NPJ Comput. Mater. 2021, 7, 9. [Google Scholar]
- Nishimoto, Y.; Fedorov, D.G. The fragment molecular orbital method combined with density-functional tight-binding and periodic boundary conditions. J. Chem. Phys. 2021, 154, 111102. [Google Scholar] [CrossRef]
- Nishimoto, Y.; Fedorov, D.G.; Irle, S. Density-Functional Tight-Binding Combined with the Fragment Molecular Orbital Method. J. Chem. Theory Comput. 2014, 10, 4801–4812. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, Y.; Irle, S. The FMO-DFTB Method. In Recent Advances of the Fragment Molecular Orbital Method: Enhanced Performance and Applicability; Mochizuki, Y., Tanaka, S., Fukuzawa, K., Eds.; Springer: Singapore, 2021; pp. 459–485. [Google Scholar] [CrossRef]
- Zhu, J.; Vuong, V.Q.; Sumpter, B.G.; Irle, S. Artificial neural network correction for density-functional tight-binding molecular dynamics simulations. MRS Commun. 2019, 9, 867–873. [Google Scholar] [CrossRef]
- Gómez-Flores, C.L.; Maag, D.; Kansari, M.; Vuong, V.Q.; Irle, S.; Gräter, F.; Kubař, T.; Elstner, M. Accurate Free Energies for Complex Condensed-Phase Reactions Using an Artificial Neural Network Corrected DFTB/MM Methodology. J. Chem. Theory Comput. 2022, 18, 1213–1226. [Google Scholar] [CrossRef]
- Dral, P.O.; von Lilienfeld, O.A.; Thiel, W. Machine Learning of Parameters for Accurate Semiempirical Quantum Chemical Calculations. J. Chem. Theory Comput. 2015, 11, 2120–2125. [Google Scholar] [CrossRef]
- Zubatiuk, T.; Nebgen, B.; Lubbers, N.; Smith, J.S.; Zubatyuk, R.; Zhou, G.; Koh, C.; Barros, K.; Isayev, O.; Tretiak, S. Machine learned Hückel theory: Interfacing physics and deep neural networks. J. Chem. Phys. 2021, 154, 244108. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Liu, X.; Lei, T.; Teng, B.; Wen, X. A combined DFTB nanoreactor and reaction network generator approach for the mechanism of hydrocarbon combustion. Chem. Commun. 2021, 57, 11633–11636. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Shindo, S.; Irle, S.; Morokuma, K. Quantum Chemical Molecular Dynamics Simulations of Dynamic Fullerene Self-Assembly in Benzene Combustion. ACS Nano 2009, 3, 2241–2257. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, X.; Qian, H.J.; Ohta, Y.; Wu, X.; Eres, G.; Morokuma, K.; Irle, S. Quantum chemical simulations reveal acetylene-based growth mechanisms in the chemical vapor deposition synthesis of carbon nanotubes. Carbon 2014, 72, 22–37. [Google Scholar] [CrossRef]
- Ohta, Y.; Okamoto, Y.; Irle, S.; Morokuma, K. Rapid Growth of a Single-Walled Carbon Nanotube on an Iron Cluster: Density-Functional Tight-Binding Molecular Dynamics Simulations. ACS Nano 2008, 2, 1437–1444. [Google Scholar] [CrossRef]
- Qian, H.J.; van Duin, A.C.T.; Morokuma, K.; Irle, S. Reactive Molecular Dynamics Simulation of Fullerene Combustion Synthesis: ReaxFF vs. DFTB Potentials. J. Chem. Theory Comput. 2011, 7, 2040–2048. [Google Scholar] [CrossRef]
- Pople, J.A.; Santry, D.P.; Segal, G.A. Approximate Self-Consistent Molecular Orbital Theory. I. Invariant Procedures. J. Chem. Phys. 1965, 43, S129–S135. [Google Scholar] [CrossRef]
- Pople, J.A.; Segal, G.A. Approximate Self-Consistent Molecular Orbital Theory. II. Calculations with Complete Neglect of Differential Overlap. J. Chem. Phys. 1965, 43, S136–S151. [Google Scholar] [CrossRef]
- Pople, J.A.; Beveridge, D.L.; Dobosh, P.A. Approximate Self-Consistent Molecular Orbital Theory. V. Intermediate Neglect of Differential Overlap. J. Chem. Phys. 1967, 47, 2026–2033. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. Development and use of quantum mechanical molecular models. 76. AM1: A new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Stewart, J.J. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Thiel, W. Semiempirical quantum-chemical methods. WIREs Comput. Mol. Sci. 2014, 4, 145–157. [Google Scholar] [CrossRef]
- Christensen, A.S.; Kuba?, T.; Cui, Q.; Elstner, M. Semiempirical Quantum Mechanical Methods for Noncovalent Interactions for Chemical and Biochemical Applications. Chem. Rev. 2016, 116, 5301–5337. [Google Scholar] [CrossRef] [PubMed]
- Dewar, M.J.S.; Thiel, W. Ground states of molecules. 38. The MNDO method. Approximations and parameters. J. Am. Chem. Soc. 1977, 99, 4899–4907. [Google Scholar] [CrossRef]
- Porezag, D.; Frauenheim, T.; Köhler, T.; Seifert, G.; Kaschner, R. Construction of tight-binding-like potentials on the basis of density-functional theory: Application to carbon. Phys. Rev. B 1995, 51, 12947–12957. [Google Scholar] [CrossRef]
- Seifert, G.; Porezag, D.; Frauenheim, T. Calculations of molecules, clusters, and solids with a simplified LCAO-DFT-LDA scheme. Int. J. Quantum Chem. 1996, 58, 185–192. [Google Scholar] [CrossRef]
- Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.; Frauenheim, T.; Suhai, S.; Seifert, G. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B 1998, 58, 7260–7268. [Google Scholar] [CrossRef]
- Köhler, C.; Seifert, G.; Frauenheim, T. Density functional based calculations for Fen (n≤32). Chem. Phys. 2005, 309, 23–31. [Google Scholar] [CrossRef]
- Melix, P.; Oliveira, A.F.; Rüger, R.; Heine, T. Spin polarization in SCC-DFTB. Theor. Chem. Accounts 2016, 135, 232. [Google Scholar] [CrossRef]
- Grimme, S.; Bannwarth, C.; Shushkov, P. A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All spd-Block Elements (Z = 1–86). J. Chem. Theory Comput. 2017, 13, 1989–2009. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB - An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef]
- Spearman Rank Correlation Coefficient. In The Concise Encyclopedia of Statistics; Springer: New York, NY, USA, 2008; pp. 502–505. [CrossRef]
- Gao, C.W.; Allen, J.W.; Green, W.H.; West, R.H. Reaction Mechanism Generator: Automatic construction of chemical kinetic mechanisms. Comput. Phys. Commun. 2016, 203, 212–225. [Google Scholar] [CrossRef]
- Johnson, M.S.; Dong, X.; Dana, A.G.; Chung, Y.; Farina, D.; Gillis, R.J.; Liu, M.; Yee, N.W.; Blondal, K.; Mazeau, E.; et al. The RMG Database for Chemical Property Prediction. ChemRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Fujioka, K.; Nikolayev, A.A.; Zhao, L.; Doddipatla, S.; Azyazov, V.N.; Mebel, A.M.; Sun, R.; Kaiser, R.I. A chemical dynamics study of the reaction of the methylidyne radical (CH, X2Π) with dimethylacetylene (CH3CCCH3, X1A1g). Phys. Chem. Chem. Phys. 2022, 24, 578–593. [Google Scholar] [CrossRef]
- Da Silva, G.; Bozzelli, J.W. Indene Formation from Alkylated Aromatics: Kinetics and Products of the Fulvenallene + Acetylene Reaction. J. Phys. Chem. A 2009, 113, 8971–8978. [Google Scholar] [CrossRef] [PubMed]
- Frenklach, M.; Semenikhin, A.S.; Mebel, A.M. On the Mechanism of Soot Nucleation. III. The Fate and Facility of the E-Bridge. J. Phys. Chem. A 2021, 125, 6789–6795. [Google Scholar] [CrossRef]
- Semenikhin, A.S.; Savchenkova, A.S.; Chechet, I.V.; Matveev, S.G.; Frenklach, M.; Mebel, A.M. On the mechanism of soot nucleation. II. E-bridge formation at the PAH bay. Phys. Chem. Chem. Phys. 2020, 22, 17196–17204. [Google Scholar] [CrossRef]
- Simon, A.; Rapacioli, M.; Rouaut, G.; Trinquier, G.; Gadéa, F.X. Dissociation of polycyclic aromatic hydrocarbons: Molecular dynamics studies. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2017, 375, 20160195. [Google Scholar] [CrossRef]
- Lundberg, M.; Nishimoto, Y.; Irle, S. Delocalization errors in a hubbard-like model: Consequences for density-functional tight-binding calculations of molecular systems. Int. J. Quantum Chem. 2012, 112, 1701–1711. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Stewart, J.J.P. MOPAC2016. 2016. Available online: http://OpenMOPAC.net (accessed on 15 September 2022).
- Throssell, K.T. Evaluating and Improving Approximate LCAO-MO Theory with Restored Overlap and Bond Order Bond Energy Corrections. Ph.D. Thesis, Wesleyan University, Middletown, CT, USA, 2018. [Google Scholar]
- Hourahine, B.; Aradi, B.; Blum, V.; Bonafé, F.; Buccheri, A.; Camacho, C.; Cevallos, C.; Deshaye, M.Y.; Dumitrică, T.; Dominguez, A.; et al. DFTB+, a software package for efficient approximate density functional theory based atomistic simulations. J. Chem. Phys. 2020, 152, 124101. [Google Scholar] [CrossRef]
- Elstner, M.; Frauenheim, T.; Kaxiras, E.; Seifert, G.; Suhai, S. A Self-Consistent Charge Density-Functional Based Tight-Binding Scheme for Large Biomolecules. Phys. Status Solidi (B) 2000, 217, 357–376. [Google Scholar] [CrossRef]
- Gaus, M.; Goez, A.; Elstner, M. Parametrization and Benchmark of DFTB3 for Organic Molecules. J. Chem. Theory Comput. 2013, 9, 338–354. [Google Scholar] [CrossRef]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; in ’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Becke, A.D. Density?functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Xu, X.; Goddard, W.A. The X3LYP extended density functional for accurate descriptions of nonbond interactions, spin states, and thermochemical properties. Proc. Natl. Acad. Sci. USA 2004, 101, 2673–2677. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865, Erratum in Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Goerigk, L.; Grimme, S. Efficient and Accurate Double-Hybrid-Meta-GGA Density Functionals—Evaluation with the Extended GMTKN30 Database for General Main Group Thermochemistry, Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2011, 7, 291–309. [Google Scholar] [CrossRef]
- Watts, J.D.; Gauss, J.; Bartlett, R.J. Coupled-cluster methods with noniterative triple excitations for restricted open?shell Hartree?Fock and other general single determinant reference functions. Energies and analytical gradients. J. Chem. Phys. 1993, 98, 8718–8733. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AM1 | PM6 | PM7 | GFN2-xTB | DFTB2-SP | DFTB2 | DFTB3 | |
|---|---|---|---|---|---|---|---|
| C15H11 | 0.0456 | 0.0915 | 0.3517 | 0.0563 | 0.0602 | 0.0602 | 0.0536 |
| C18H14 | 0.0824 | 0.1040 | 0.0943 | 0.0674 | - | 0.0887 | 0.0761 |
| C21H17 | 0.5073 | 0.5068 | 0.5143 | 0.1463 | 0.1689 | 0.2087 | 0.2087 |
| C23H15O1 | 0.1819 | 0.1710 | 0.5080 | 0.0497 | 0.0642 | 0.0609 | 0.0656 |
| C26H19 | 0.3568 | 0.6439 | 0.6862 | 0.1690 | 0.1931 | 0.1929 | 0.2592 |
| C26H22 | 1.2711 | 2.4898 | 2.5142 | 0.9765 | - | 0.7924 | 0.7377 |
| C28H18 | 0.2743 | 0.2878 | 0.2850 | 0.3956 | 0.3964 | 0.3883 | 0.3957 |
| C34H23 | 0.7308 | 0.8423 | 0.8707 | 0.2865 | 0.2803 | 0.2870 | 0.2595 |
| C47H32 | 0.3241 | 0.5465 | 0.6677 | 0.4234 | - | 0.0998 | 0.1037 |
| C50H35 | 0.3511 | 0.4144 | 0.5725 | 0.2471 | 0.1177 | 0.1183 | 0.0671 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cong, Y.; Zhai, Y.; Chen, X.; Li, H. The Accuracy of Semi-Empirical Quantum Chemistry Methods on Soot Formation Simulation. Int. J. Mol. Sci. 2022, 23, 13371. https://doi.org/10.3390/ijms232113371
Cong Y, Zhai Y, Chen X, Li H. The Accuracy of Semi-Empirical Quantum Chemistry Methods on Soot Formation Simulation. International Journal of Molecular Sciences. 2022; 23(21):13371. https://doi.org/10.3390/ijms232113371
Chicago/Turabian StyleCong, Yang, Yu Zhai, Xin Chen, and Hui Li. 2022. "The Accuracy of Semi-Empirical Quantum Chemistry Methods on Soot Formation Simulation" International Journal of Molecular Sciences 23, no. 21: 13371. https://doi.org/10.3390/ijms232113371
APA StyleCong, Y., Zhai, Y., Chen, X., & Li, H. (2022). The Accuracy of Semi-Empirical Quantum Chemistry Methods on Soot Formation Simulation. International Journal of Molecular Sciences, 23(21), 13371. https://doi.org/10.3390/ijms232113371