
Design, Synthesis and Biological Evaluation of Novel 1,3,5-Triazines: Effect of Aromatic Ring Decoration on Affinity to 5-HT7 Receptor

 ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Results
2.1. Chemistry
2.2. Radioligand Binding and SAR
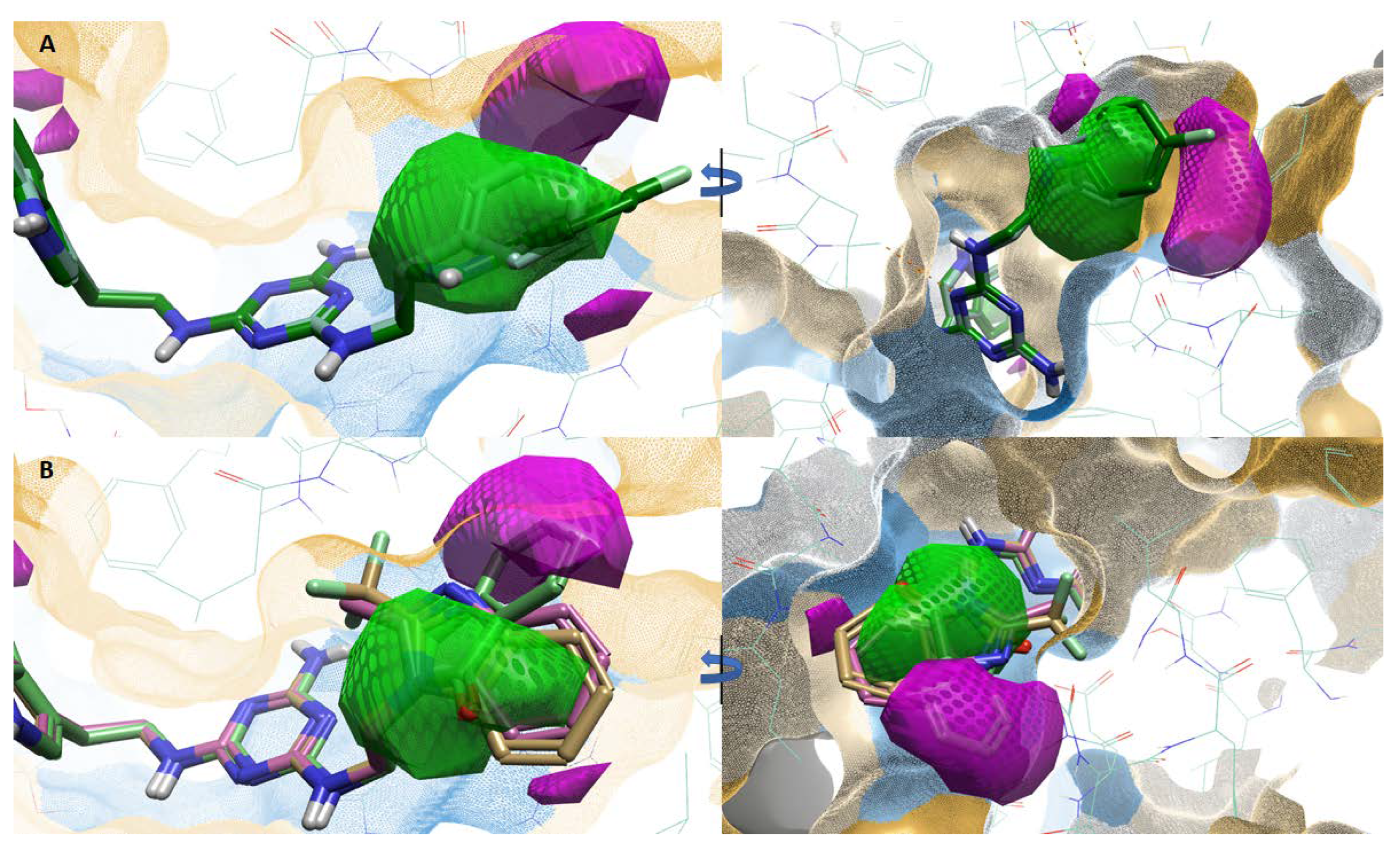
2.3. Atlas Activity Analysis: 3D-SAR
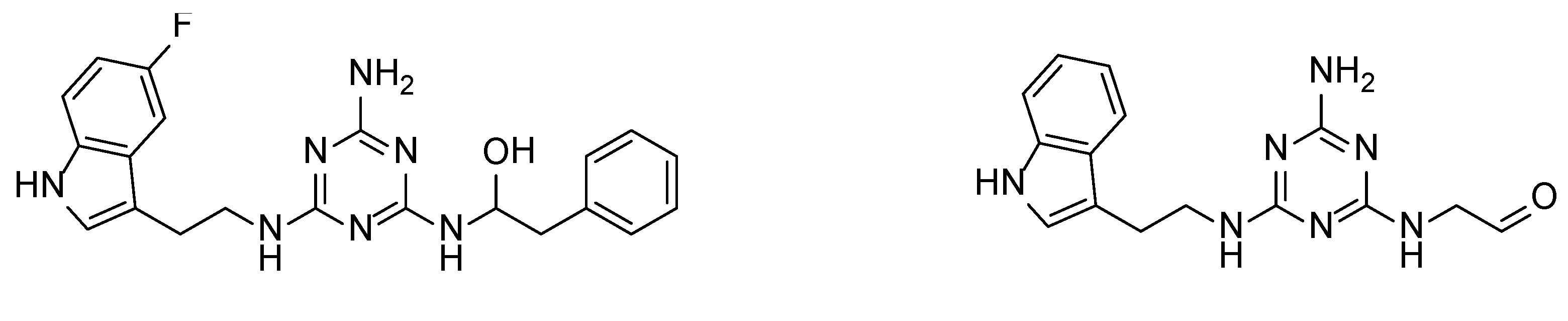
2.4. Molecular Modelling
2.5. Metabolic Stability
2.6. CYP3A4 Interaction
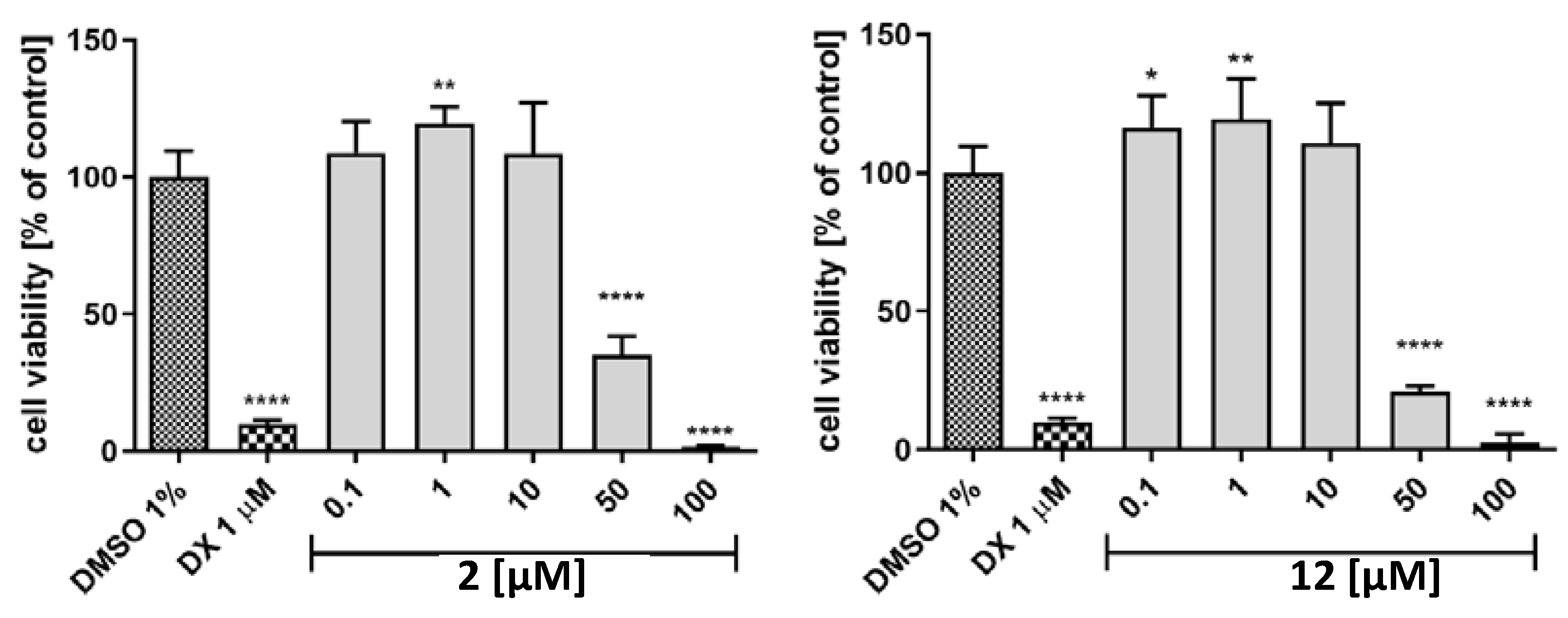
2.7. Hepatotoxicity
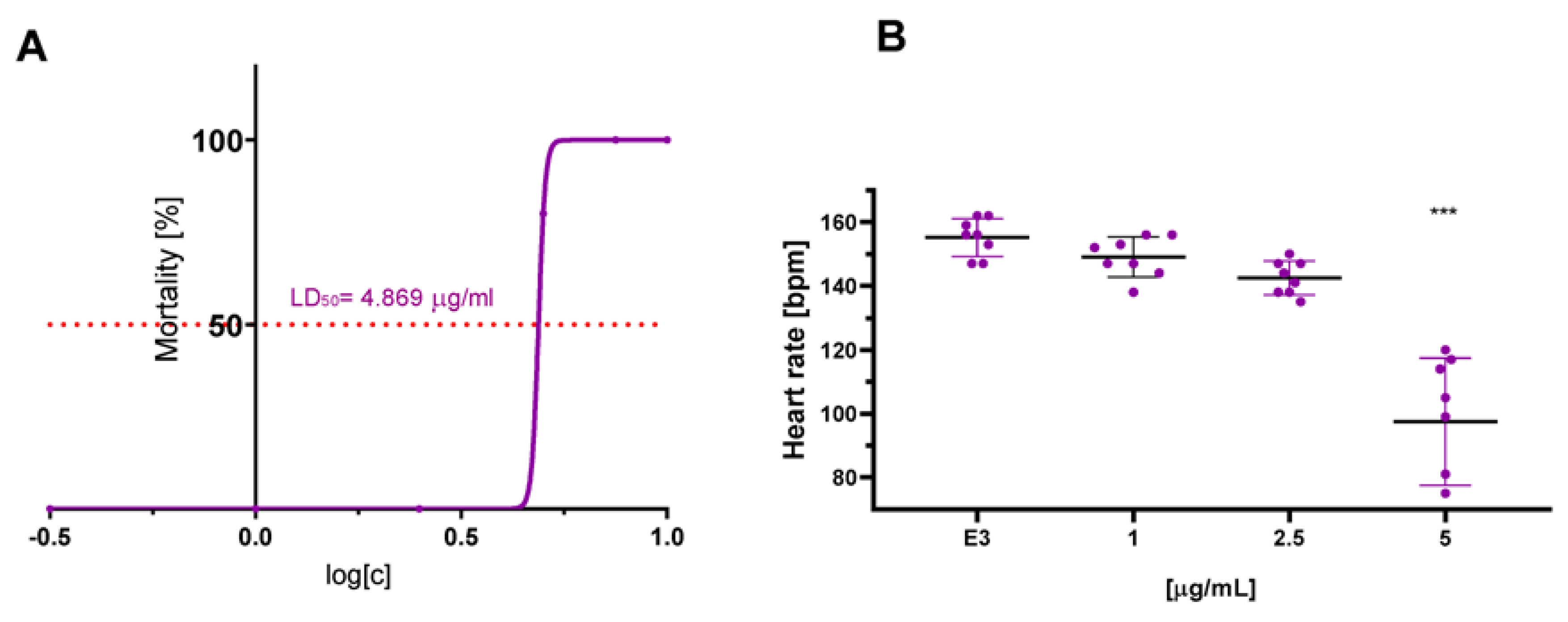
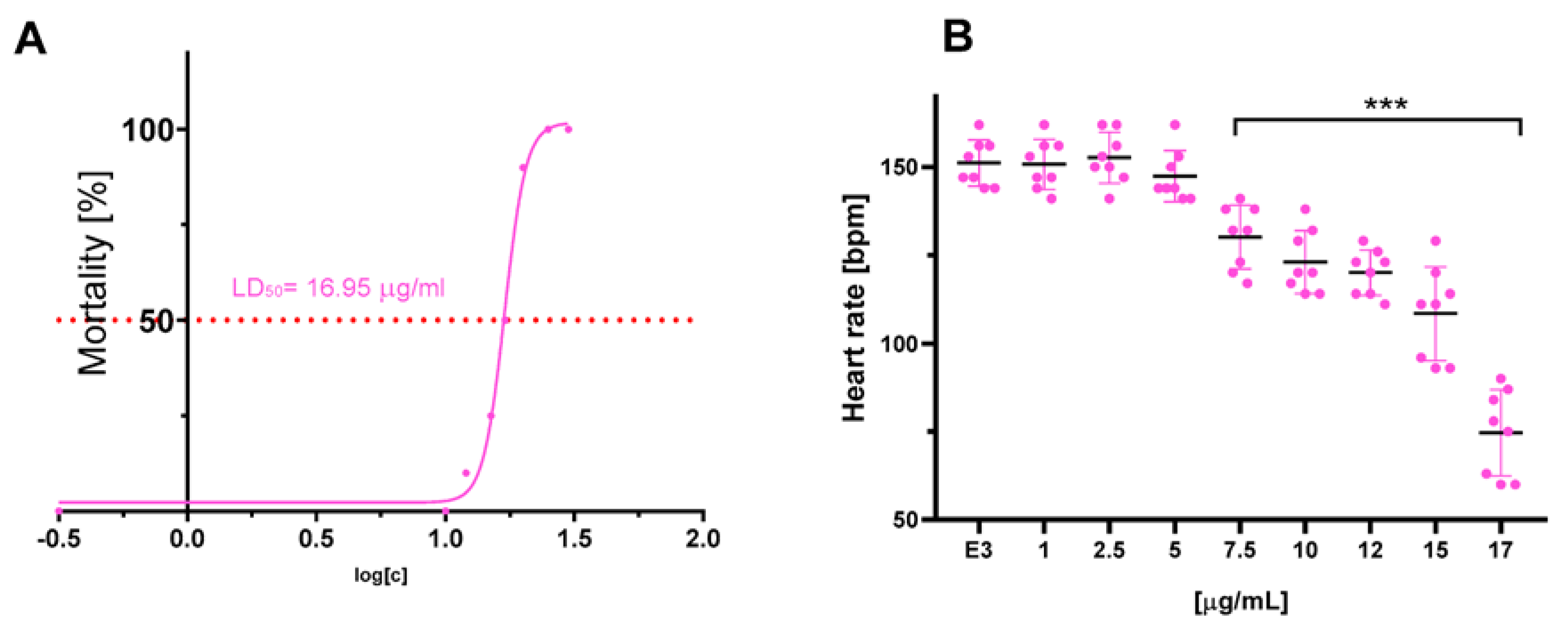
2.8. In Vivo Cardiotoxicity
3. Discussion and Conclusions
4. Materials and Methods
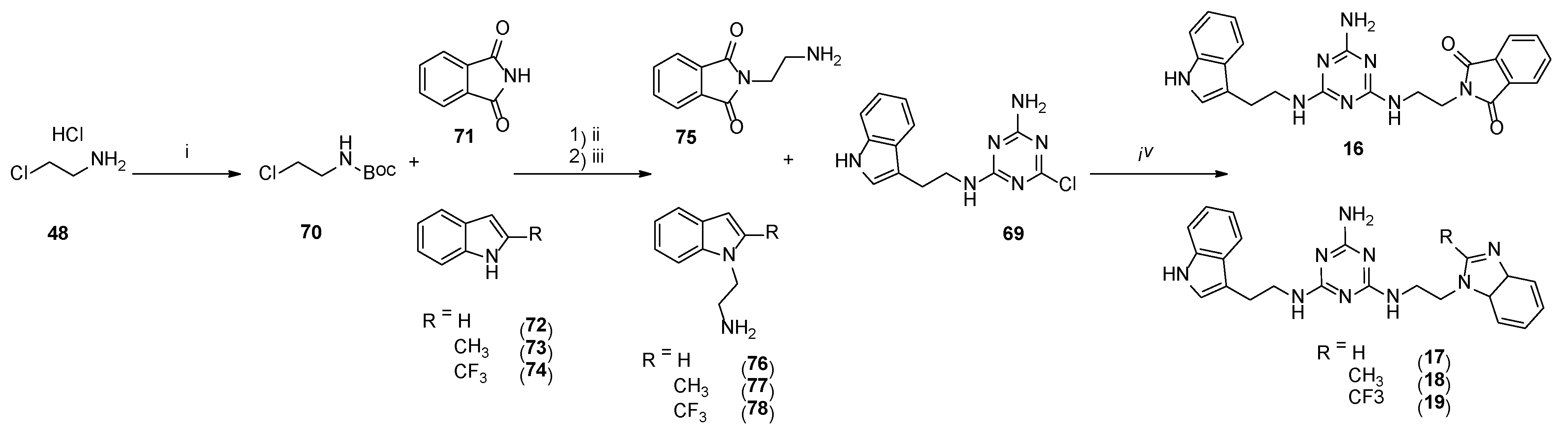
4.1. Chemistry
4.1.1. General
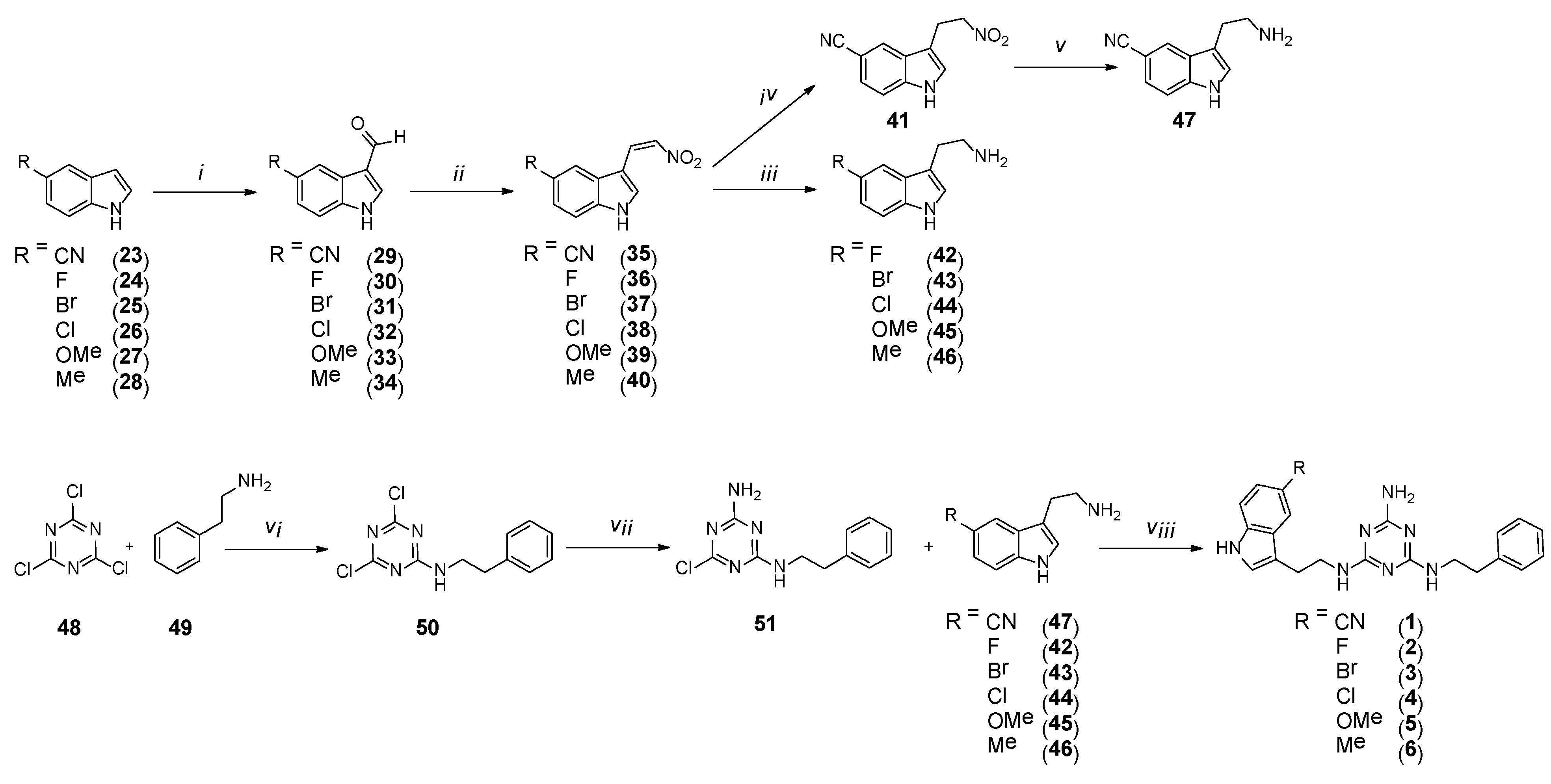
4.1.2. General Procedure for the Synthesis of Compounds 29–34
4.1.3. General Procedure for the Synthesis of Compounds 35–40
4.1.4. Synthesis of 3-(2-Nitroethyl)-1H-Indole-5-Carbonitrile (41)
4.1.5. General Procedure for the Synthesis of Compounds 42–46
4.1.6. Synthesis of 3-(2-Aminoethyl)-1H-Indole-5-Carbonitrile (47)
4.1.7. Synthesis of 4,6-Dichloro-N-Phenethyl-1,3,5-Triazin-2-Amine (50)
4.1.8. Synthesis of 6-Chloro-N2-Phenethyl-1,3,5-Triazine-2,4-Diamine (51)
4.1.9. General Procedure for the Synthesis of Final Compounds 1–6 (Microwave-Assisted)
4.1.10. 3-(2-((4-Amino-6-(Phenethylamino)-1,3,5-Triazin-2-yl)Amino)Ethyl)-1H-Indole-5-Carbonitrile Hydrochloride (1)
4.1.11. N2-(2-(5-Fluoro-1H-Indol-3-yl)ethyl)-N4-Phenethyl-1,3,5-Triazine-2,4,6-Triamine Hydrochloride (2)
4.1.12. N2-(2-(5-Bromo-1H-Indol-3-yl)ethyl)-N4-Phenethyl-1,3,5-Triazine-2,4,6-Triamine Hydrochloride (3)
4.1.13. N2-(2-(5-Chloro-1H-Indol-3-yl)ethyl)-N4-Phenethyl-1,3,5-Triazine-2,4,6-Triamine Hydrochloride (4)
4.1.14. N2-(2-(5-Methoxy-1H-Indol-3-yl)ethyl)-N4-Phenethyl-1,3,5-Triazine-2,4,6-Triamine Hydrochloride (5)
4.1.15. N2-(2-(5-Methyl-1H-Indol-3-yl)ethyl)-N4-Phenethyl-1,3,5-Triazine-2,4,6-Triamine Hydrochloride (6)
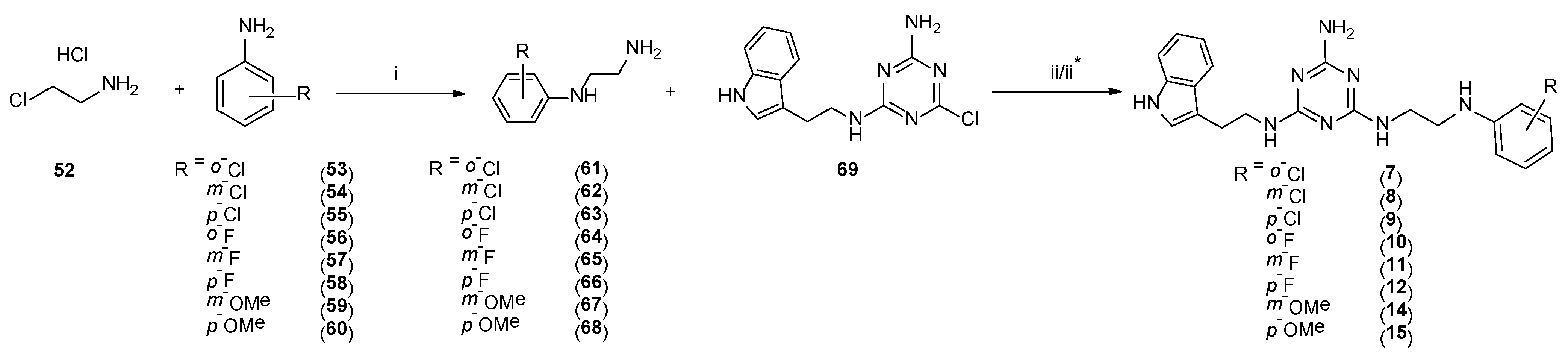
4.1.16. General Procedure for the Synthesis of Compounds 61–68
4.1.17. General Procedure for the Synthesis of Final Compounds 7, 9–11, and 14 (microwave-assisted)
4.1.18. N2-(2-(1H-Indol-3-yl)ethyl)-N4-(2-((2-Chlorophenyl)amino)ethyl)-1,3,5-Triazine-2,4,6-Triamine Hydrochloride (7)
4.1.19. N2-(2-(1H-Indol-3-yl)ethyl)-N4-(2-((4-Chlorophenyl)amino)ethyl)-1,3,5-Triazine-2,4,6-Triamine Hydrochloride (9)
4.1.20. N2-(2-(1H-Indol-3-yl)ethyl)-N4-(2-((2-Fluorophenyl)amino)ethyl)-1,3,5-Triazine-2,4,6-Triamine Hydrochloride (10)
4.1.21. N2-(2-(1H-Indol-3-yl)ethyl)-N4-(2-((3-Fluorophenyl)amino)ethyl)-1,3,5-Triazine-2,4,6-Triamine Hydrochloride (11)
4.1.22. N2-(2-(1H-Indol-3-yl)ethyl)-N4-(2-((3-Methoxyphenyl)amino)ethyl)-1,3,5-Triazine-2,4,6-Triamine Hydrochloride (14)
4.1.23. General Procedure for the Synthesis of Final Compounds 8, 12, and 15 (Microwave-Assisted)
4.1.24. N2-(2-(1H-Indol-3-yl)ethyl)-N4-(2-((3-Chlorophenyl)amino)ethyl)-1,3,5-Triazine-2,4,6-Triamine Hydrochloride (8)
4.1.25. N2-(2-(1H-Indol-3-yl)ethyl)-N4-(2-((4-Fluorophenyl)amino)ethyl)-1,3,5-Triazine-2,4,6-Triamine Hydrochloride (12)
4.1.26. N2-(2-(1H-Indol-3-yl)ethyl)-N4-(2-((4-Methoxyphenyl)amino)ethyl)-1,3,5-Triazine-2,4,6-Triamine Hydrochloride (15)
4.1.27. Synthesis of Tert-Butyl-(2-Chloroethyl)Carbamate (70)
4.1.28. General Procedure for the Synthesis of 75–78 (Microwave-Assisted)
4.1.29. General Procedure for the Synthesis of Final Compounds 16–19 (Microwave-Assisted)
4.1.30. 2-(2-((4-((2-(1H-Indol-3-yl)ethyl)amino)-6-Amino-1,3,5-Triazin-2-yl)amino)ethyl)isoindoline-1,3-Dione Hydrochloride (16)
4.1.31. N2-(2-(1H-Benzo[d]imidazol-1-yl)ethyl)-N4-(2-(1H-Indol-3-yl)ethyl)-1,3,5-Triazine-2,4,6-Triamine Hydrochloride (17)
4.1.32. N2-(2-(1H-indol-3-yl)ethyl)-N4-(2-(2-methyl-1H-benzo[d]imidazol-1-yl)ethyl)-1,3,5-triazine-2,4,6-triamine hydrochloride (18)
4.1.33. N2-(2-(1H-Indol-3-yl)ethyl)-N4-(2-(2-(Trifluoromethyl)-1H-Benzo[d]imidazol-1-yl)ethyl)-1,3,5-Triazine-2,4,6-Triamine Hydrochloride (19)
4.1.34. General Procedure for the Synthesis of Final Compounds 20–22 (Microwave-Assisted)
4.1.35. N2-(2-(1H-Indol-3-yl)ethyl)-N4-(3-Chlorophenyl)-1,3,5-Triazine-2,4,6-Triamine Hydrochloride (20)
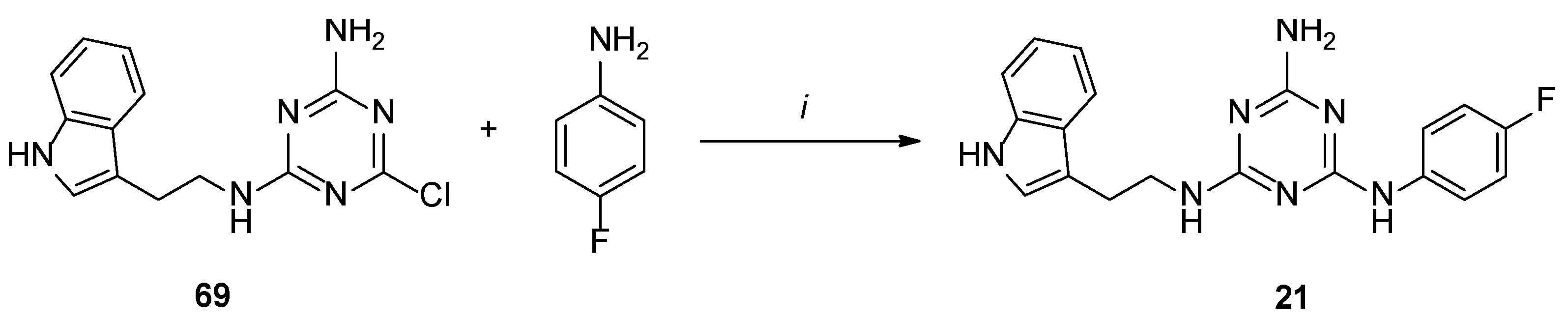
4.1.36. N2-(2-(1H-Indol-3-yl)ethyl)-N4-(4-Fluorophenyl)-1,3,5-Triazine-2,4,6-Triamine Hydrochloride (21)
4.1.37. N2-(2-(1H-Indol-3-yl)ethyl)-N4-(4-Methoxyphenyl)-1,3,5-Triazine-2,4,6-Triamine Hydrochloride (22)
4.2. Radioligand Assay
4.3. Atlas Activity
4.4. Molecular Modelling
4.5. Metabolic Stability
4.6. CYP3A4 Activity
4.7. Hepatotoxicity
4.8. In Vivo Cardiotoxicity
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vanhoenacker, P.; Haegeman, G.; Leysen, J.E. 5-HT7 receptors: Current knowledge and future prospects. Trends Pharmacol. Sci. 2000, 21, 70–77. [Google Scholar] [CrossRef]
- Naumenko, V.S.; Popova, N.K.; Lacivita, E.; Leopoldo, M.; Ponimaskin, E.G. Interplay between serotonin 5-HT1A and 5-HT7 receptors in depressive disorders. CNS Neurosci. Ther. 2014, 20, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.R.; Hedlund, P.B.; Roberts, A.J.; Sari, Y.; Bell, R.L.; Engleman, E.A. The 5-HT7 receptor as a potential target for treating drug and alcohol abuse. Front. Neurosci. 2015, 8, 448. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Khan, W.I. 5-HT7 receptor signaling: Improved therapeutic strategy in gut disorders. Front. Behav. Neurosci. 2014, 8, 396. [Google Scholar] [CrossRef]
- Stull, M.A.; Pai, V.; Vomachka, A.J.; Marshall, A.M.; Jacob, G.A.; Horseman, N.D. Mammary gland homeostasis employs serotonergic regulation of epithelial tight junctions. Proc. Natl. Acad. Sci. USA 2007, 104, 16708–16713. [Google Scholar] [CrossRef]
- Cadirci, E.; Halici, Z.; Bayir, Y.; Albayrak, A.; Karakus, E.; Polat, B.; Unal, D.; Atamanalp, S.S.; Aksak, S.; Gundogdu, C. Peripheral 5-HT7 receptors as a new target for prevention of lung injury and mortality in septic rats. Immunobiology 2013, 218, 1271–1283. [Google Scholar] [CrossRef]
- Cinar, I.; Sirin, B.; Halici, Z.; Palabiyik-Yucelik, S.S.; Akpinar, E.; Cadirci, E. 5-HT7 receptors as a new target for prostate cancer physiopathology and treatment: An experimental study on PC-3 cells and FFPE tissues. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Modica, M.N.; Lacivita, E.; Intagliata, S.; Salerno, L.; Romeo, G.; Pittalà, V.; Leopoldo, M. Structure-Activity Relationships and Therapeutic Potentials of 5-HT7 Receptor Ligands: An Update. J. Med. Chem. 2018, 61, 8475–8503. [Google Scholar] [CrossRef]
- Guseva, D.; Wirth, A.; Ponimaskin, E. Cellular mechanisms of the 5-HT7 receptor-mediated signaling. Front. Behav. Neurosci. 2014, 8, 306. [Google Scholar] [CrossRef]
- Ye, D.; Xu, H.; Tang, Q.; Xia, H.; Zhang, C.; Bi, F. The role of 5-HT metabolism in cancer. Biochim. Biophys. Acta. Rev. Cancer 2021, 1876, 188618. [Google Scholar] [CrossRef]
- Gautam, J.; Banskota, S.; Regmi, S.C.; Ahn, S.; Jeon, Y.H.; Jeong, H.; Kim, S.J.; Nam, T.G.; Jeong, B.S.; Kim, A.J. Tryptophan hydroxylase 1 and 5-HT7 receptor preferentially expressed in triple-negative breast cancer promote cancer progression through autocrine serotonin signaling. Mol. Cancer 2016, 15, 75. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wang, T.; Wang, Z.; Wu, X.; Gu, Y.; Huang, Q.; Wang, J.; Xie, J. 5-HT7 Receptor Contributes to Proliferation, Migration and Invasion in NSCLC Cells. Onco Targets Ther. 2020, 13, 2139–2151. [Google Scholar] [CrossRef] [PubMed]
- Quintero-Villegas, A.; Valdés-Ferrer, S.I. Role of 5-HT7 receptors in the immune system in health and disease. Mol. Med. 2020, 26, 2. [Google Scholar] [CrossRef] [PubMed]
- Albayrak, A.; Halici, Z.; Cadirci, E.; Polat, B.; Karakus, E.; Bayir, Y.; Unal, D.; Atasoy, M.; Dogrul, A. Inflammation and peripheral 5-HT7 receptors: The role of 5-HT7 receptors in carrageenan induced inflammation in rats. Eur. J. Pharmacol. 2013, 715, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Kułaga, D.; Drabczyk, A.K.; Satała, G.; Latacz, G.; Rózga, K.; Plażuk, D.; Jaśkowska, J. Design and synthesis of new potent 5-HT7 receptor ligands as a candidate for the treatment of central nervous system diseases. Eur. J. Med. Chem. 2022, 227, 113931. [Google Scholar] [CrossRef]
- Vermeulen, E.S.; Schmidt, A.W.; Sprouse, J.S.; Wikström, H.V.; Grol, C.J. Characterization of the 5-HT(7) receptor. Determination of the pharmacophore for 5-HT(7) receptor agonism and CoMFA-based modeling of the agonist binding site. J. Med. Chem. 2003, 46, 5365–5374. [Google Scholar] [CrossRef]
- Hogendorf, A.S.; Hogendorf, A.; Kurczab, R.; Satała, G.; Lenda, T.; Walczak, M.; Latacz, G.; Handzlik, J.; Kieć-Kononowicz, K.; Wierońska, J.M.; et al. Low-basicity 5-HT7 Receptor Agonists Synthesized Using the van Leusen Multicomponent Protocol. Sci. Rep. 2017, 7, 1444. [Google Scholar] [CrossRef]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef]
- Smith, D.A. Metabolism, Pharmacokinetics and Toxicity of Functional Groups: Impact of Chemical Building Blocks on ADMET; Drug Discovery; The Royal Society of Chemistry: London, UK, 2010; Chapter 2; pp. 61–98. [Google Scholar]
- Kułaga, D.; Jaśkowska, J.; Satała, G.; Latacz, G.; Śliwa, P. Aminotriazines with indole motif as novel, 5-HT7 receptor ligands with atypical binding mode. Bioorg. Chem. 2020, 104, 104254. [Google Scholar] [CrossRef]
- Mattson, R.J.; Denhart, D.J.; Catt, J.D.; Dee, M.F.; Deskus, J.A.; Ditta, J.L.; Epperson, J.; Dalton King, H.; Gao, A.; Poss, M.A.; et al. Aminotriazine 5-HT7 antagonists. Bioorg. Med. Chem. Lett. 2004, 14, 4245–4248. [Google Scholar] [CrossRef]
- Chen, Z.; Cohen, M.P.; Fisher, M.J.; Gillig, J.R.; McCowan, J.R.; Miller, S.C.; Schaus, J.M.; Giethlen, B. N-(2-arylethyl) Benzylamines as Antagonists of the 5-HT6 Receptor. EP1859798A1, 28 November 2007. [Google Scholar]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [PubMed]
- Flare, version 5, Cresset®, Litlington, Cambridgeshire, UK. Available online: http://www.cresset-group.com/flare (accessed on 2 May 2022).
- Schrödinger Release 2016-4: Maestro; Schrödinger; LLC: New York, NY, USA, 2016.
- Impellizzeri, A.A.R.; Pappalardo Basile, M.L.; Manfra, O.; Andressen, K.W.; Krobert, K.A.; Messina, A.; Levy, F.O.; Guccione, S. Identification of essential residues for binding and activation in the human 5-HT7(a) serotonin receptor by molecular modeling and site-directed mutagenesis. Front. Behav. Neurosci. 2015, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Wichur, T.; Godyń, J.; Góral, I.; Latacz, G.; Bucki, A.; Siwek, A.; Głuch-Lutwin, M.; Mordyl, B.; Śniecikowska, J.; Walczak, M.; et al. Development and crystallography-aided SAR studies of multifunctional BuChE inhibitors and 5-HT6 R antagonists with β-amyloid anti-aggregation properties. Eur. J. Med. Chem. 2021, 225, 113792. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Guenthner, T.; Gan, L.; Humphreys, W.G. CYP3A4 Induction by Xenobiotics: Biochemistry, Experimental Methods and Impact on Drug Discovery and Development. Curr. Drug Metab. 2004, 5, 483–505. [Google Scholar] [CrossRef]
- OECD. Test No. 236: Fish Embryo Acute Toxicity (FET) Test, OECD Guidelines for the Testing of Chemicals; Section 2; OECD Publishing: Paris, France, 2013. [Google Scholar] [CrossRef]
- Hedlund, P.B. The 5-HT7 receptor and disorders of the nervous system: An overview. Psychopharmacol. 2009, 206, 345–354. [Google Scholar] [CrossRef]
- Gregory, A.W.; Jakubec, P.; Turner, P.; Dixon, D.J. Gold and BINOL-Phosphoric Acid Catalyzed Enantioselective Hydroamination/N-Sulfonyliminium Cyclization Cascade. Org. Lett. 2013, 15, 4330–4333. [Google Scholar] [CrossRef]
- Kułaga, D.; Jaśkowska, J.; Satała, G. Design, synthesis and biological evaluation of novel serotonin and dopamine receptor ligands being 6-bromohexyl saccharine derivatives, Bioorg. Med. Chem. Lett. 2019, 29, 126667. [Google Scholar] [CrossRef]
- Latacz, G.; Hogendorf, G.A.S.; Hogendorf, A.; Lubelska, A.; Wieronska, J.M.; Wozniak, M.; Cieslik, P.; Kiec-Kononowicz, K.; Handzlik, J.; Bojarski, A.J. Search for a 5-CT alternative. In vitro and in vivo evaluation of novel pharmacological tools: 3-(1-alkyl-1H-imidazol-5-yl)-1H-indole-5-carboxamides, low-basicity 5-HT7 receptor agonists. Med. Chem. Comm. 2018, 9, 1882–1890. [Google Scholar] [CrossRef]
- Latacz, G.; Lubelska, A.; Jastrzebska-Wiesek, M.; Partyka, A.; Kucwaj-Brysz, K.; Wesołowska, A.; Kiec Kononowicz, K.; Handzlik, J. MF-8, a novel promising arylpiperazine-hydantoin based 5-HT7 receptor antagonist: In vitro drug-likeness studies and in vivo pharmacological evaluation. Bioorg. Med. Chem. Lett. 2018, 28, 878–883. [Google Scholar] [CrossRef]
- Latacz, G.; Lubelska, A.; Jastrzębska-Więsek, M.; Partyka, A.; Sobiło, A.; Olejarz, A.; Kucwaj-Brysz, K.; Satała, G.; Bojarski, A.J.; Wesołowska, A.; et al. In the search for a lead structure among series of potent and selective hydantoin 5-HT7R agents: The drug-likeness in vitro study. Chem. Biol. Drug Des. 2017, 90, 1295–1306. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type No. | Ligand No. | R1 | R2 | R3 | 5-HT1AR | 5-HT2AR | 5-HT6R | 5-HT7R | D2R |
|---|---|---|---|---|---|---|---|---|---|
 | 1 | CN | - | - | 12,980 ± 2354 | 927 ± 927 | 1563 ± 1563 | 2883 ± 351 | - |
| 2 | F | - | - | 13,900 ± 1759 | 413 ± 67 | 505 ± 61 | 8 ± 2 | - | |
| 3 | Br | - | - | 10,950 ± 1598 | 942 ± 112 | 234 ± 27 | 126 ± 17 | - | |
| 4 | Cl | - | - | 3913 ± 581 | 559 ± 41 | 702 ± 48 | 481 ± 23 | 14,530 ± 2697 | |
| 5 | OMe | - | - | 3128 ± 758 | 883 ± 108 | 509 ± 33 | 629 ± 148 | - | |
| 6 | Me | - | - | 16,560 ± 1943 | 835 ± 168 | 714 ± 112 | 100 ± 15 | - | |
 | 7 | - | o-Cl | - | 30,830 ± 4238 | 2125 ± 352 | 892 ± 144 | 423 ± 37 | 4294 ± 1028 |
| 8 | - | m-Cl | - | 15,630 ± 1927 | 1800 ± 264 | 4022 ± 719 | 131 ± 16 | 1238 ± 257 | |
| 9 | - | p-Cl | - | 16,160 ± 2566 | 2698 ± 581 | 641 ± 84 | 19 ± 4 | - | |
| 10 | - | o-F | - | 36,510 ± 8652 | 1514 ± 362 | 982 ± 115 | 132 ± 9 | 20,410 ± 3695 | |
| 11 | - | m-F | - | 34,580 ± 5581 | 1277 ± 168 | 979 ± 237 | 24 ± 5 | 13,760 ± 2491 | |
| 12 | - | p-F | - | 43,250 ± 9541 | 3570 ± 491 | 1630 ± 301 | 18 ± 3 | 2999 ± 679 | |
| 13 * | - | o-OMe | - | 37,040 | 14,390 | 2798 | 5823 | 3907 | |
| 14 | - | m-OMe | - | 108,900 ± 18,654 | 2449 ± 273 | 2790 ± 342 | 1036 ± 81 | 35,960 ± 8432 | |
| 15 | - | p-OMe | - | - | - | - | 756 ± 107 | - | |
 | 16 | - | - |  | 138,800 ± 31,821 | 22,350 ± 3546 | 8760 ± 1762 | 21,560 ± 5127 | 13,290 ± 1684 |
| 17 | - | - |  | 63,010 ± 13,591 | 11,150 ± 2571 | 2629 ± 427 | 227 ± 37 | - | |
| 18 | - | - |  | 180,200 ± 41,983 | 9355 ± 831 | 1958 ± 269 | 4128 ± 725 | - | |
| 19 | - | - |  | 76,200 ± 18,615 | 4035 ± 537 | 825 ± 72 | 3464 ± 284 | - | |
 | 20 | - | m-Cl | - | 14,100 ± 2594 | 2690 ± 549 | 2226 ± 528 | 1952 ± 261 | 2267 ± 153 |
| 21 | - | p-F | - | 249,500 ± 54,268 | 884 ± 73 | 1765 ± 243 | 2052 ± 419 | 7033 ± 439 | |
| 22 | - | p-OMe | - | 79,830 ± 16,252 | 3110 ± 438 | 3742 ± 539 | 727 ± 107 | - |
| Substrate | Molecular Mass (m/z) | % Remaining | Molecular Mass of the Metabolite (m/z) | Metabolic Pathway |
|---|---|---|---|---|
| 2 | 392.33 | 19.79 | 408.28 (M1) 406.28 (M2) 424.29 (M3) 390.27(M4) 390.27 (M5) 424.29 (M6) 408.34 (M7) 422.30 (M8) 424.29 (M9) | hydroxylation ketone formation double hydroxylation dehydrogenation dehydrogenation double hydroxylation hydroxylation ketone formation and hydroxylation hydroxylation |
| 12 | 407.35 | 29.12 | 312.25 (M1) 423.30 (M2) 405.29 (M3) 405.29 (M4) 439.31 (M5) 423.30 (M6) 405.02 (M7) | decomposition hydroxylation dehydrogenation dehydrogenation double hydroxylation hydroxylation dehydrogenation |
| Verapamil * | 455.54 | 23.93 | 441.42 (M1) | demethylation |
| 441.42 (M2) | demethylation | |||
| 291.35 (M3) | defragmentation | |||
| 293.34 (M4) | defragmentation/hydroxylation | |||
| 277.33 (M5) | defragmentation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kułaga, D.; Drabczyk, A.K.; Satała, G.; Latacz, G.; Boguszewska-Czubara, A.; Plażuk, D.; Jaśkowska, J. Design, Synthesis and Biological Evaluation of Novel 1,3,5-Triazines: Effect of Aromatic Ring Decoration on Affinity to 5-HT7 Receptor. Int. J. Mol. Sci. 2022, 23, 13308. https://doi.org/10.3390/ijms232113308
Kułaga D, Drabczyk AK, Satała G, Latacz G, Boguszewska-Czubara A, Plażuk D, Jaśkowska J. Design, Synthesis and Biological Evaluation of Novel 1,3,5-Triazines: Effect of Aromatic Ring Decoration on Affinity to 5-HT7 Receptor. International Journal of Molecular Sciences. 2022; 23(21):13308. https://doi.org/10.3390/ijms232113308
Chicago/Turabian StyleKułaga, Damian, Anna Karolina Drabczyk, Grzegorz Satała, Gniewomir Latacz, Anna Boguszewska-Czubara, Damian Plażuk, and Jolanta Jaśkowska. 2022. "Design, Synthesis and Biological Evaluation of Novel 1,3,5-Triazines: Effect of Aromatic Ring Decoration on Affinity to 5-HT7 Receptor" International Journal of Molecular Sciences 23, no. 21: 13308. https://doi.org/10.3390/ijms232113308
APA StyleKułaga, D., Drabczyk, A. K., Satała, G., Latacz, G., Boguszewska-Czubara, A., Plażuk, D., & Jaśkowska, J. (2022). Design, Synthesis and Biological Evaluation of Novel 1,3,5-Triazines: Effect of Aromatic Ring Decoration on Affinity to 5-HT7 Receptor. International Journal of Molecular Sciences, 23(21), 13308. https://doi.org/10.3390/ijms232113308