
Translational Implications for Radiosensitizing Strategies in Rhabdomyosarcoma

 , ,
, ,  , , , , ,
, , , , ,  ,
,  ,
,  and
and 
Abstract
1. Introduction to Rhabdomyosarcoma
1.1. Histological, Molecular Classification and Related Clinical and Prognostic Features
1.2. The Management of RMS Patients
2. Radiosensitizing Targets in RMS
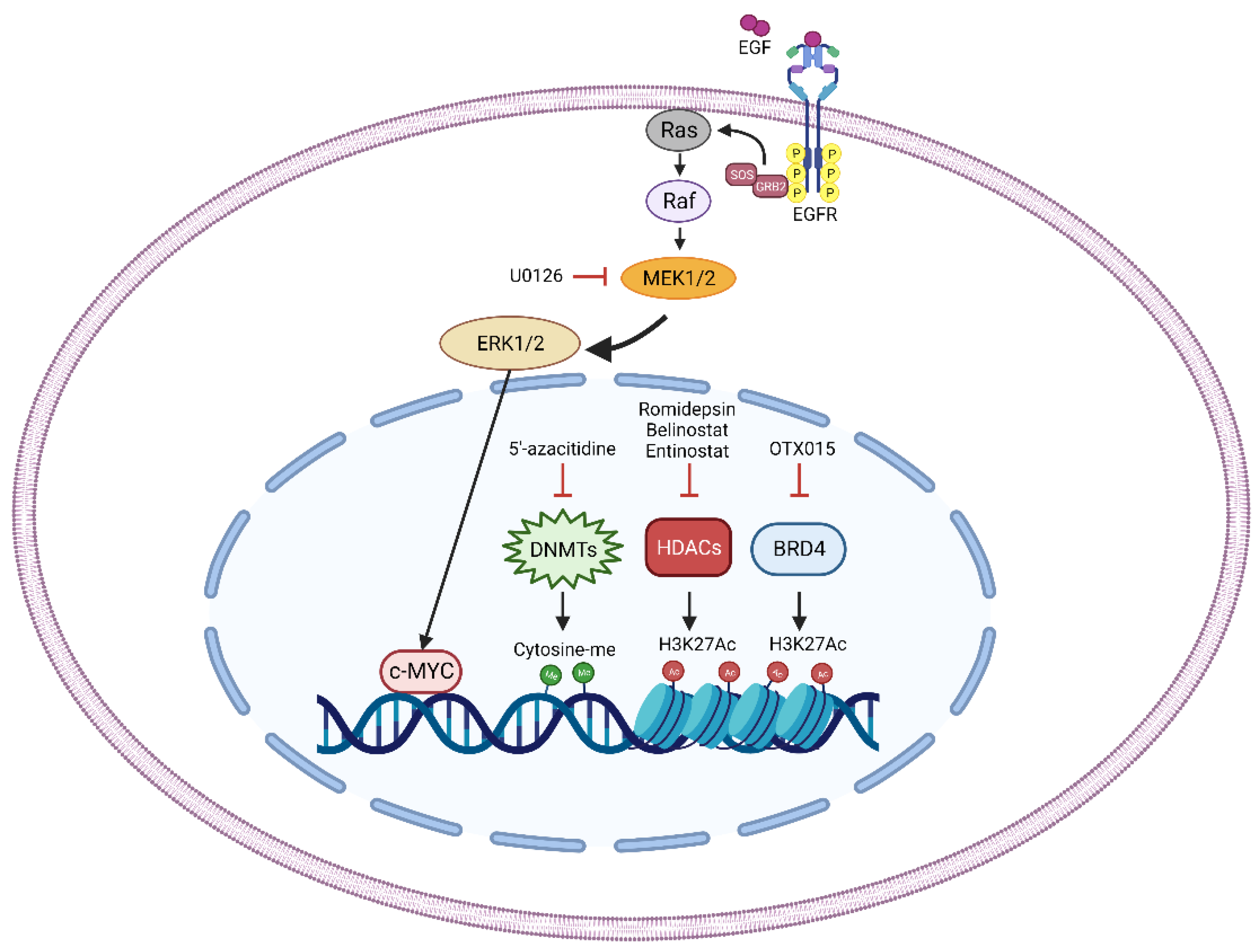
2.1. Epigenetic Targets
2.1.1. DNA Methyltransferases
2.1.2. Histone Deacetylases
2.1.3. Bromo- and Extra-Terminal Domain Proteins
2.2. Transcription Factors
2.2.1. SNAI2
2.2.2. C-MYC
2.2.3. NRF2
2.3. DNA Damage Effectors, Cell Cycle Regulators, and Cell Signaling Effectors
2.3.1. PARP
2.3.2. Caveolin
2.3.3. P53-MDM2 Pathway
2.3.4. MEK/ERK Pathway
2.3.5. PI3K/Akt Pathway
2.4. Genome Stability
2.4.1. KIF18B
2.4.2. FANCD2
2.5. Cytokines and Receptors
2.5.1. HGF
2.5.2. IFN-γ
2.5.3. Ephrin Receptor
2.6. Other Sensitizing Agents
2.6.1. Selenium
2.6.2. Resveratrol
2.6.3. Fenretinide
3. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rudzinski, E.R.; Anderson, J.R.; Chi, Y.-Y.; Gastier-Foster, J.M.; Astbury, C.; Barr, F.G.; Skapek, S.X.; Hawkins, D.S.; Weigel, B.J.; Pappo, A.; et al. Histology, Fusion Status, and Outcome in Metastatic Rhabdomyosarcoma: A Report from the Children’s Oncology Group. Pediatr. Blood Cancer 2017, 64, e26645. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.D.; Grosveld, G.C. Alveolar Rhabdomyosarcoma—The Molecular Drivers of PAX3/7-FOXO1-Induced Tumorigenesis. Skelet. Muscle 2012, 2, 25. [Google Scholar] [CrossRef] [PubMed]
- Newton, W.A.; Soule, E.H.; Hamoudi, A.B.; Reiman, H.M.; Shimada, H.; Beltangady, M.; Maurer, H. Histopathology of Childhood Sarcomas, Intergroup Rhabdomyosarcoma Studies I and II: Clinicopathologic Correlation. J. Clin. Oncol. 1988, 6, 67–75. [Google Scholar] [CrossRef]
- Davicioni, E.; Finckenstein, F.G.; Shahbazian, V.; Buckley, J.D.; Triche, T.J.; Anderson, M.J. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res. 2006, 66, 6936–6946. [Google Scholar] [CrossRef]
- Yechieli, R.L.; Mandeville, H.C.; Hiniker, S.M.; Bernier-Chastagner, V.; McGovern, S.; Scarzello, G.; Wolden, S.; Cameron, A.; Breneman, J.; Fajardo, R.D.; et al. Rhabdomyosarcoma. Pediatr. Blood Cancer 2021, 68, e28254. [Google Scholar] [CrossRef]
- Kelly, K.M.; Womer, R.B.; Sorensen, P.H.; Xiong, Q.B.; Barr, F.G. Common and Variant Gene Fusions Predict Distinct Clinical Phenotypes in Rhabdomyosarcoma. J. Clin. Oncol. 1997, 15, 1831–1836. [Google Scholar] [CrossRef]
- Williamson, D.; Missiaglia, E.; de Reyniès, A.; Pierron, G.; Thuille, B.; Palenzuela, G.; Thway, K.; Orbach, D.; Laé, M.; Fréneaux, P.; et al. Fusion Gene–Negative Alveolar Rhabdomyosarcoma Is Clinically and Molecularly Indistinguishable from Embryonal Rhabdomyosarcoma. J. Clin. Oncol. 2010, 28, 2151–2158. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.; McEvoy, J.; Wang, H.; Chen, X.; Honnell, V.; Ocarz, M.; Gordon, B.; Dapper, J.; Blankenship, K.; Yang, Y.; et al. Identification of Therapeutic Targets in Rhabdomyosarcoma through Integrated Genomic, Epigenomic, and Proteomic Analyses. Cancer Cell 2018, 34, 411–426.e9. [Google Scholar] [CrossRef]
- Seki, M.; Nishimura, R.; Yoshida, K.; Shimamura, T.; Shiraishi, Y.; Sato, Y.; Kato, M.; Chiba, K.; Tanaka, H.; Hoshino, N.; et al. Integrated Genetic and Epigenetic Analysis Defines Novel Molecular Subgroups in Rhabdomyosarcoma. Nat. Commun. 2015, 6, 7557. [Google Scholar] [CrossRef]
- Shern, J.F.; Chen, L.; Chmielecki, J.; Wei, J.S.; Patidar, R.; Rosenberg, M.; Ambrogio, L.; Auclair, D.; Wang, J.; Song, Y.K.; et al. Comprehensive Genomic Analysis of Rhabdomyosarcoma Reveals a Landscape of Alterations Affecting a Common Genetic Axis in Fusion-Positive and Fusion-Negative Tumors. Cancer Discov. 2014, 4, 216–231. [Google Scholar] [CrossRef]
- Paulson, V.; Chandler, G.; Rakheja, D.; Galindo, R.L.; Wilson, K.; Amatruda, J.F.; Cameron, S. High-Resolution Array CGH Identifies Common Mechanisms That Drive Embryonal Rhabdomyosarcoma Pathogenesis. Genes Chromosomes Cancer 2011, 50, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Ricker, C.A.; Crawford, K.; Matlock, K.; Lathara, M.; Seguin, B.; Rudzinski, E.R.; Berlow, N.E.; Keller, C. Defining an Embryonal Rhabdomyosarcoma Endotype. Mol. Case Stud. 2020, 6, a005066. [Google Scholar] [CrossRef]
- Sultan, I.; Qaddoumi, I.; Yaser, S.; Rodriguez-Galindo, C.; Ferrari, A. Comparing Adult and Pediatric Rhabdomyosarcoma in the Surveillance, Epidemiology and End Results Program, 1973 to 2005: An Analysis of 2600 Patients. J. Clin. Oncol. 2009, 27, 3391–3397. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, P.H.B.; Lynch, J.C.; Qualman, S.J.; Tirabosco, R.; Lim, J.F.; Maurer, H.M.; Bridge, J.A.; Crist, W.M.; Triche, T.J.; Barr, F.G. PAX3-FKHR and PAX7-FKHR Gene Fusions Are Prognostic Indicators in Alveolar Rhabdomyosarcoma: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2002, 20, 2672–2679. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C.R.; Zhang, L.; Shao, S.Y.; Mosquera, J.-M.; Weinreb, I.; Katabi, N.; Fletcher, C.D.M. Frequent PLAG1 Gene Rearrangements in Skin and Soft Tissue Myoepithelioma with Ductal Differentiation. Genes Chromosomes Cancer 2013, 52, 675–682. [Google Scholar] [CrossRef]
- Alaggio, R.; Zhang, L.; Sung, Y.-S.; Huang, S.-C.; Chen, C.-L.; Bisogno, G.; Zin, A.; Agaram, N.P.; LaQuaglia, M.P.; Wexler, L.H.; et al. A Molecular Study of Pediatric Spindle and Sclerosing Rhabdomyosarcoma. Am. J. Surg. Pathol. 2016, 40, 224–235. [Google Scholar] [CrossRef]
- Agaram, N.P.; Zhang, L.; Sung, Y.-S.; Cavalcanti, M.S.; Torrence, D.; Wexler, L.; Francis, G.; Sommerville, S.; Swanson, D.; Dickson, B.C.; et al. Expanding the Spectrum of Intraosseous Rhabdomyosarcoma. Am. J. Surg. Pathol. 2019, 43, 695–702. [Google Scholar] [CrossRef]
- Agaram, N.P.; LaQuaglia, M.P.; Alaggio, R.; Zhang, L.; Fujisawa, Y.; Ladanyi, M.; Wexler, L.H.; Antonescu, C.R. MYOD1-Mutant Spindle Cell and Sclerosing Rhabdomyosarcoma: An Aggressive Subtype Irrespective of Age. A Reappraisal for Molecular Classification and Risk Stratification. Mod. Pathol. 2019, 32, 27–36. [Google Scholar] [CrossRef]
- Agaram, N.P. Evolving Classification of Rhabdomyosarcoma. Histopathology 2022, 80, 98–108. [Google Scholar] [CrossRef]
- Xu, B.; Suurmeijer, A.J.H.; Agaram, N.P.; Zhang, L.; Antonescu, C.R. Head and Neck Rhabdomyosarcoma with TFCP2 Fusions and ALK Overexpression: A Clinicopathological and Molecular Analysis of 11 Cases. Histopathology 2021, 79, 347–357. [Google Scholar] [CrossRef]
- Watson, S.; Perrin, V.; Guillemot, D.; Reynaud, S.; Coindre, J.; Karanian, M.; Guinebretière, J.; Freneaux, P.; le Loarer, F.; Bouvet, M.; et al. Transcriptomic Definition of Molecular Subgroups of Small Round Cell Sarcomas. J. Pathol. 2018, 245, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Kaseb, H.; Kuhn, J.; Babiker, H.M. Rhabdomyosarcoma; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- PDQ Pediatric Treatment Editorial Board. Childhood Rhabdomyosarcoma Treatment (PDQ®): Health Professional Version; National Cancer Institute: Rockville, MD, USA, 2002. [Google Scholar]
- Gallego, S.; Bernabeu, D.; Garrido-Pontnou, M.; Guillen, G.; Hindi, N.; Juan-Ribelles, A.; Márquez, C.; Mata, C.; Orcajo, J.; Ramírez, G.; et al. GEIS-SEHOP Clinical Practice Guidelines for the Treatment of Rhabdomyosarcoma. Clin. Transl. Oncol. 2021, 23, 2460–2473. [Google Scholar] [CrossRef] [PubMed]
- Ruymann, F.B.; Grovas, A.C. Progress in the Diagnosis and Treatment of Rhabdomyosarcoma and Related Soft Tissue Sarcomas. Cancer Investig. 2000, 18, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Malempati, S.; Hawkins, D.S. Rhabdomyosarcoma: Review of the Children’s Oncology Group (COG) Soft-Tissue Sarcoma Committee Experience and Rationale for Current COG Studies. Pediatr. Blood Cancer 2012, 59, 5–10. [Google Scholar] [CrossRef]
- Bergeron, C.; Jenney, M.; de Corti, F.; Gallego, S.; Merks, H.; Glosli, H.; Ferrari, A.; Ranchère-Vince, D.; de Salvo, G.L.; Zanetti, I.; et al. Embryonal Rhabdomyosarcoma Completely Resected at Diagnosis: The European Paediatric Soft Tissue Sarcoma Study Group RMS2005 Experience. Eur. J. Cancer 2021, 146, 21–29. [Google Scholar] [CrossRef]
- Cecchetto, G.; Bisogno, G.; de Corti, F.; Dall’Igna, P.; Inserra, A.; Ferrari, A.; Garaventa, A.; Scagnellato, A.; Carli, M. Biopsy or Debulking Surgery as Initial Surgery for Locally Advanced Rhabdomyosarcomas in Children? Cancer 2007, 110, 2561–2567. [Google Scholar] [CrossRef]
- Rogers, T.N.; Seitz, G.; Fuchs, J.; Martelli, H.; Dasgupta, R.; Routh, J.C.; Hawkins, D.S.; Koscielniak, E.; Bisogno, G.; Rodeberg, D.A. Surgical Management of Paratesticular Rhabdomyosarcoma: A Consensus Opinion from the Children’s Oncology Group, European Paediatric Soft Tissue Sarcoma Study Group, and the Cooperative Weichteilsarkom Studiengruppe. Pediatr. Blood Cancer 2021, 68, e28938. [Google Scholar] [CrossRef]
- Ferrari, A.; van Noesel, M.M.; Brennan, B.; Zanetti, I.; Corradini, N.; Casanova, M.; Berlanga, P.; Merks, J.H.M.; Alaggio, R.; Schifflers, S.; et al. Paediatric Non-Rhabdomyosarcoma Soft Tissue Sarcomas: The Prospective NRSTS 2005 Study by the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG). Lancet Child Adolesc. Health 2021, 5, 546–558. [Google Scholar] [CrossRef]
- Von Mehren, M.; Kane, J.M.; Agulnik, M.; Bui, M.M.; Carr-Ascher, J.; Choy, E.; Connelly, M.; Dry, S.; Ganjoo, K.N.; Gonzalez, R.J.; et al. Soft Tissue Sarcoma, Version 2.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 815–833. [Google Scholar] [CrossRef]
- Hawkins, D.S.; Chi, Y.-Y.; Anderson, J.R.; Tian, J.; Arndt, C.A.S.; Bomgaars, L.; Donaldson, S.S.; Hayes-Jordan, A.; Mascarenhas, L.; McCarville, M.B.; et al. Addition of Vincristine and Irinotecan to Vincristine, Dactinomycin, and Cyclophosphamide Does Not Improve Outcome for Intermediate-Risk Rhabdomyosarcoma: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2018, 36, 2770–2777. [Google Scholar] [CrossRef]
- Terezakis, S.A.; Wharam, M.D. Radiotherapy for Rhabdomyosarcoma: Indications and Outcome. Clin. Oncol. 2013, 25, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Huang, D.; Zhang, W.; Zhang, Y.; Hu, H.; Li, J. Radiation Therapy Is an Important Factor to Improve Survival in Pediatric Patients with Head and Neck Rhabdomyosarcoma by Enhancing Local Control: A Historical Cohort Study from a Single Center. BMC Pediatr. 2020, 20, 265. [Google Scholar] [CrossRef] [PubMed]
- Million, L.; Anderson, J.; Breneman, J.; Hawkins, D.S.; Laurie, F.; Michalski, J.; Rodeberg, D.; Wharam, M.; Wolden, S.; Donaldson, S.S. Influence of Noncompliance with Radiation Therapy Protocol Guidelines and Operative Bed Recurrences for Children with Rhabdomyosarcoma and Microscopic Residual Disease: A Report from the Children’s Oncology Group. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Walterhouse, D.O.; Pappo, A.S.; Meza, J.L.; Breneman, J.C.; Hayes-Jordan, A.; Parham, D.M.; Cripe, T.P.; Anderson, J.R.; Meyer, W.H.; Hawkins, D.S. Reduction of Cyclophosphamide Dose for Patients with Subset 2 Low-Risk Rhabdomyosarcoma Is Associated with an Increased Risk of Recurrence: A Report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Cancer 2017, 123, 2368–2375. [Google Scholar] [CrossRef] [PubMed]
- Wolden, S.L.; Anderson, J.R.; Crist, W.M.; Breneman, J.C.; Wharam, J.M.D.; Wiener, E.S.; Qualman, S.J.; Donaldson, S.S. Indications for Radiotherapy and Chemotherapy After Complete Resection in Rhabdomyosarcoma: A Report from the Intergroup Rhabdomyosarcoma Studies I to III. J. Clin. Oncol. 1999, 17, 3468–3475. [Google Scholar] [CrossRef] [PubMed]
- Crist, W.M.; Anderson, J.R.; Meza, J.L.; Fryer, C.; Raney, R.B.; Ruymann, F.B.; Breneman, J.; Qualman, S.J.; Wiener, E.; Wharam, M.; et al. Intergroup Rhabdomyosarcoma Study-IV: Results for Patients with Nonmetastatic Disease. J. Clin. Oncol. 2001, 19, 3091–3102. [Google Scholar] [CrossRef]
- Walterhouse, D.O.; Pappo, A.S.; Meza, J.L.; Breneman, J.C.; Hayes-Jordan, A.A.; Parham, D.M.; Cripe, T.P.; Anderson, J.R.; Meyer, W.H.; Hawkins, D.S. Shorter-Duration Therapy Using Vincristine, Dactinomycin, and Lower-Dose Cyclophosphamide with or without Radiotherapy for Patients with Newly Diagnosed Low-Risk Rhabdomyosarcoma: A Report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. J. Clin. Oncol. 2014, 32, 3547–3552. [Google Scholar] [CrossRef]
- Ermoian, R.P.; Breneman, J.; Walterhouse, D.O.; Chi, Y.-Y.; Meza, J.; Anderson, J.; Hawkins, D.S.; Hayes-Jordan, A.A.; Parham, D.M.; Yock, T.I.; et al. 45 Gy Is Not Sufficient Radiotherapy Dose for Group III Orbital Embryonal Rhabdomyosarcoma after Less than Complete Response to 12 Weeks of ARST0331 Chemotherapy. Pediatr. Blood Cancer 2017, 64, e26540. [Google Scholar] [CrossRef]
- Casey, D.L.; Chi, Y.; Donaldson, S.S.; Hawkins, D.S.; Tian, J.; Arndt, C.A.; Rodeberg, D.A.; Routh, J.C.; Lautz, T.B.; Gupta, A.A.; et al. Increased Local Failure for Patients with Intermediate-risk Rhabdomyosarcoma on ARST0531: A Report from the Children’s Oncology Group. Cancer 2019, 125, 3242–3248. [Google Scholar] [CrossRef]
- Mohan, A.C.; Venkatramani, R.; Okcu, M.F.; Nuchtern, J.G.; Vasudevan, S.A.; Mahajan, A.; Rainusso, N.C.; Allen-Rhoades, W.; Chintagumpala, M.; Paulino, A.C. Local Therapy to Distant Metastatic Sites in Stage IV Rhabdomyosarcoma. Pediatr. Blood Cancer 2018, 65, e26859. [Google Scholar] [CrossRef]
- Wolden, S.L.; Lyden, E.R.; Arndt, C.A.; Hawkins, D.S.; Anderson, J.R.; Rodeberg, D.A.; Morris, C.D.; Donaldson, S.S. Local Control for Intermediate-Risk Rhabdomyosarcoma: Results from D9803 According to Histology, Group, Site, and Size: A Report from the Children’s Oncology Group. Int. J. Radiat. Oncol. Biol. Phys. 2015, 93, 1071–1076. [Google Scholar] [CrossRef]
- Mandeville, H.C. Radiotherapy in the Management of Childhood Rhabdomyosarcoma. Clin. Oncol. 2019, 31, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Casey, D.L.; Wexler, L.H.; LaQuaglia, M.P.; Meyers, P.A.; Wolden, S.L. Favorable Outcomes after Whole Abdominopelvic Radiation Therapy for Pediatric and Young Adult Sarcoma. Pediatr. Blood Cancer 2014, 61, 1565–1569. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.; Bergamaschi, L.; Chiaravalli, S.; Livellara, V.; Sironi, G.; Nigro, O.; Puma, N.; Gattuso, G.; Morosi, C.; Gasparini, P.; et al. Metastatic Rhabdomyosarcoma: Evidence of the Impact of Radiotherapy on Survival. A Retrospective Single-center Experience. Pediatr. Blood Cancer 2022, 69, e29853. [Google Scholar] [CrossRef] [PubMed]
- Gerber, N.K.; Wexler, L.H.; Singer, S.; Alektiar, K.M.; Keohan, M.L.; Shi, W.; Zhang, Z.; Wolden, S. Adult Rhabdomyosarcoma Survival Improved with Treatment on Multimodality Protocols. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 58–63. [Google Scholar] [CrossRef]
- Bergamaschi, L.; Bertulli, R.; Casanova, M.; Provenzano, S.; Chiaravalli, S.; Gasparini, P.; Collini, P.; Sangalli, C.; Gandola, L.; Diletto, B.; et al. Rhabdomyosarcoma in Adults: Analysis of Treatment Modalities in a Prospective Single-Center Series. Med. Oncol. 2019, 36, 59. [Google Scholar] [CrossRef]
- Zhao, R.; Yu, X.; Feng, Y.; Wang, J.; Chen, Y.; Mao, Y.; Yin, W.; Zhang, Z.; Guo, X.; Ma, S. The Survival Benefit of Radiotherapy in Localized Primary Adult Rhabdomyosarcoma. Asia Pac. J. Clin. Oncol. 2020, 16, 266–272. [Google Scholar] [CrossRef]
- Rodeberg, D.A.; Wharam, M.D.; Lyden, E.R.; Stoner, J.A.; Brown, K.; Wolden, S.L.; Paidas, C.N.; Donaldson, S.S.; Hawkins, D.S.; Spunt, S.L.; et al. Delayed Primary Excision with Subsequent Modification of Radiotherapy Dose for Intermediate-Risk Rhabdomyosarcoma: A Report from the Children’s Oncology Group Soft Tissue Sarcoma Committee. Int. J. Cancer 2015, 137, 204–211. [Google Scholar] [CrossRef]
- Donaldson, S.S.; Meza, J.; Breneman, J.C.; Crist, W.M.; Laurie, F.; Qualman, S.J.; Wharam, M. Results from the IRS-IV Randomized Trial of Hyperfractionated Radiotherapy in Children with Rhabdomyosarcoma—A Report from the IRSG 1 1For a Complete List of the Members of the Children’s Oncology Group Soft Tissue Sarcoma Committee (Formerly Intergroup Rhabdomyosarcoma Group) Representing the Children’s Oncology Group and the Quality Assurance Review Center, See the Appendix. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 718–728. [Google Scholar] [CrossRef]
- Soyfer, V.; Corn, B.W.; Kollender, Y.; Issakov, J.; Dadia, S.; Flusser, G.; Bickels, J.; Meller, I.; Merimsky, O. Hypofractionated Adjuvant Radiation Therapy of Soft-Tissue Sarcoma Achieves Excellent Results in Elderly Patients. Br. J. Radiol. 2013, 86, 20130258. [Google Scholar] [CrossRef]
- Spencer, R.M.; Aguiar Junior, S.; Ferreira, F.O.; Stevanato Filho, P.R.; Kupper, B.E.; Silva, M.L.; Mello, C.A.; Bezerra, T.S.; Lopes, A. Neoadjuvant Hypofractionated Radiotherapy and Chemotherapy in High-Grade Extremity Soft Tissue Sarcomas: Phase 2 Clinical Trial Protocol. JMIR Res. Protoc. 2017, 6, e97. [Google Scholar] [CrossRef] [PubMed]
- Kalbasi, A.; Kamrava, M.; Chu, F.-I.; Telesca, D.; van Dams, R.; Yang, Y.; Ruan, D.; Nelson, S.D.; Dry, S.M.; Hernandez, J.; et al. A Phase II Trial of 5-Day Neoadjuvant Radiotherapy for Patients with High-Risk Primary Soft Tissue Sarcoma. Clin. Cancer Res. 2020, 26, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Parsai, S.; Lawrenz, J.; Kilpatrick, S.; Rubin, B.; Hymes, C.; Gray, M.; Mesko, N.; Shah, C.; Nystrom, L.; Scott, J.G. Early Outcomes of Preoperative 5-Fraction Radiation Therapy for Soft Tissue Sarcoma Followed by Immediate Surgical Resection. Adv. Radiat. Oncol. 2020, 5, 1274–1279. [Google Scholar] [CrossRef] [PubMed]
- Spałek, M.; Koseła-Paterczyk, H.; Borkowska, A.; Wągrodzki, M.; Szumera-Ciećkiewicz, A.; Pałucki, J.; Cieszanowski, A.; Rutkowski, P. Hypofractionated Radiotherapy in Locally Advanced Myxoid Liposarcomas of Extremities or Trunk Wall: Results of a Single Arm Prospective Clinical Trial. Int. J. Radiat. Oncol. Biol. Phys. 2019, 105, S63. [Google Scholar] [CrossRef][Green Version]
- Pennington, J.D.; Eilber, F.C.; Eilber, F.R.; Singh, A.S.; Reed, J.P.; Chmielowski, B.; Eckardt, J.J.; Bukata, S.V.; Bernthal, N.M.; Federman, N.; et al. Long-Term Outcomes with Ifosfamide-Based Hypofractionated Preoperative Chemoradiotherapy for Extremity Soft Tissue Sarcomas. Am. J. Clin. Oncol. 2018, 41, 1154–1161. [Google Scholar] [CrossRef] [PubMed]
- Koseła-Paterczyk, H.; Szacht, M.; Morysiński, T.; Ługowska, I.; Dziewirski, W.; Falkowski, S.; Zdzienicki, M.; Pieńkowski, A.; Szamotulska, K.; Świtaj, T.; et al. Preoperative Hypofractionated Radiotherapy in the Treatment of Localized Soft Tissue Sarcomas. Eur. J. Surg. Oncol. 2014, 40, 1641–1647. [Google Scholar] [CrossRef]
- Meyer, J.M.; Perlewitz, K.S.; Hayden, J.B.; Doung, Y.-C.; Hung, A.Y.; Vetto, J.T.; Pommier, R.F.; Mansoor, A.; Beckett, B.R.; Tudorica, A.; et al. Phase I Trial of Preoperative Chemoradiation plus Sorafenib for High-Risk Extremity Soft Tissue Sarcomas with Dynamic Contrast-Enhanced MRI Correlates. Clin. Cancer Res. 2013, 19, 6902–6911. [Google Scholar] [CrossRef]
- MacDermed, D.M.; Miller, L.L.; Peabody, T.D.; Simon, M.A.; Luu, H.H.; Haydon, R.C.; Montag, A.G.; Undevia, S.D.; Connell, P.P. Primary Tumor Necrosis Predicts Distant Control in Locally Advanced Soft-Tissue Sarcomas After Preoperative Concurrent Chemoradiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2010, 76, 1147–1153. [Google Scholar] [CrossRef]
- Ryan, C.W.; Montag, A.G.; Hosenpud, J.R.; Samuels, B.; Hayden, J.B.; Hung, A.Y.; Mansoor, A.; Peabody, T.D.; Mundt, A.J.; Undevia, S. Histologic Response of Dose-Intense Chemotherapy with Preoperative Hypofractionated Radiotherapy for Patients with High-Risk Soft Tissue Sarcomas. Cancer 2008, 112, 2432–2439. [Google Scholar] [CrossRef]
- Temple, W.J.; Temple, C.L.F.; Arthur, K.; Schachar, N.S.; Paterson, A.H.G.; Crabtree, T.S. Prospective Cohort Study of Neoadjuvant Treatment in Conservative Surgery of Soft Tissue Sarcomas. Ann. Surg. Oncol. 1997, 4, 586–590. [Google Scholar] [CrossRef]
- Kılıç, L.; Ekenel, M.; Karabulut, S.; Ağaoğlu, F.; Darendeliler, E. Neoadjuvant Sequential Chemoradiotherapy versus Radiotherapy Alone for Treatment of High-Risk Extremity Soft Tissue Sarcoma: A Single-Institution Experience. Współczesna Onkol. 2017, 1, 60–65. [Google Scholar] [CrossRef]
- Spalek, M.; Koseła-Paterczyk, H.; Borkowska, A.; Wągrodzki, M.; Szumera-Ciećkiewicz, A.; Cieszanowski, A.; Castaneda-Wysocka, P.; Świtaj, T.; Dudzisz-Śledź, M.; Czarnecka, A.; et al. OC-0069 5x5 Gy with Chemotherapy in Borderline Resectable Soft Tissue Sarcomas: Early Results of a Trial. Radiother. Oncol. 2019, 133, S31–S32. [Google Scholar] [CrossRef]
- Gryder, B.E.; Wu, L.; Woldemichael, G.M.; Pomella, S.; Quinn, T.R.; Park, P.M.C.; Cleveland, A.; Stanton, B.Z.; Song, Y.; Rota, R.; et al. Chemical Genomics Reveals Histone Deacetylases Are Required for Core Regulatory Transcription. Nat. Commun. 2019, 10, 3004. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Pomella, S.; Sayers, C.; Wu, X.S.; Song, Y.; Chiarella, A.M.; Bagchi, S.; Chou, H.-C.; Sinniah, R.S.; Walton, A.; et al. Histone Hyperacetylation Disrupts Core Gene Regulatory Architecture in Rhabdomyosarcoma. Nat. Genet. 2019, 51, 1714–1722. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Yohe, M.E.; Chou, H.-C.; Zhang, X.; Marques, J.; Wachtel, M.; Schaefer, B.; Sen, N.; Song, Y.; Gualtieri, A.; et al. PAX3-FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov. 2017, 7, 884–899. [Google Scholar] [CrossRef]
- Schübeler, D. Function and Information Content of DNA Methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef]
- Laker, R.C.; Ryall, J.G. DNA Methylation in Skeletal Muscle Stem Cell Specification, Proliferation, and Differentiation. Stem Cells Int. 2016, 2016, 5725927. [Google Scholar] [CrossRef]
- Mattei, A.L.; Bailly, N.; Meissner, A. DNA Methylation: A Historical Perspective. Trends Genet. 2022, 38, 676–707. [Google Scholar] [CrossRef]
- Kim, J.K.; Samaranayake, M.; Pradhan, S. Epigenetic Mechanisms in Mammals. Cell. Mol. Life Sci. 2009, 66, 596–612. [Google Scholar] [CrossRef]
- Megiorni, F.; Camero, S.; Ceccarelli, S.; McDowell, H.P.; Mannarino, O.; Marampon, F.; Pizer, B.; Shukla, R.; Pizzuti, A.; Marchese, C.; et al. DNMT3B in Vitro Knocking-down Is Able to Reverse Embryonal Rhabdomyosarcoma Cell Phenotype through Inhibition of Proliferation and Induction of Myogenic Differentiation. Oncotarget 2016, 7, 79342–79356. [Google Scholar] [CrossRef]
- Camero, S.; Vitali, G.; Pontecorvi, P.; Ceccarelli, S.; Anastasiadou, E.; Cicchetti, F.; Flex, E.; Pomella, S.; Cassandri, M.; Rota, R.; et al. DNMT3A and DNMT3B Targeting as an Effective Radiosensitizing Strategy in Embryonal Rhabdomyosarcoma. Cells 2021, 10, 2956. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.D.; Grunstein, M. Functions of Site-Specific Histone Acetylation and Deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The Many Roles of Histone Deacetylases in Development and Physiology: Implications for Disease and Therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Federico, M.; Bagella, L. Histone Deacetylase Inhibitors in the Treatment of Hematological Malignancies and Solid Tumors. J. Biomed. Biotechnol. 2011, 2011, 475641. [Google Scholar] [CrossRef]
- Marampon, F.; di Nisio, V.; Pietrantoni, I.; Petragnano, F.; Fasciani, I.; Scicchitano, B.M.; Ciccarelli, C.; Gravina, G.L.; Festuccia, C.; del Fattore, A.; et al. Pro-Differentiating and Radiosensitizing Effects of Inhibiting HDACs by PXD-101 (Belinostat) in in Vitro and in Vivo Models of Human Rhabdomyosarcoma Cell Lines. Cancer Lett. 2019, 461, 90–101. [Google Scholar] [CrossRef]
- Bartek, J.; Lukas, J. Chk1 and Chk2 Kinases in Checkpoint Control and Cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef]
- Rossetti, A.; Petragnano, F.; Milazzo, L.; Vulcano, F.; Macioce, G.; Codenotti, S.; Cassandri, M.; Pomella, S.; Cicchetti, F.; Fasciani, I.; et al. Romidepsin (FK228) Fails in Counteracting the Transformed Phenotype of Rhabdomyosarcoma Cells but Efficiently Radiosensitizes, in Vitro and in Vivo, the Alveolar Phenotype Subtype. Int. J. Radiat. Biol. 2021, 97, 943–957. [Google Scholar] [CrossRef]
- Cassandri, M.; Pomella, S.; Rossetti, A.; Petragnano, F.; Milazzo, L.; Vulcano, F.; Camero, S.; Codenotti, S.; Cicchetti, F.; Maggio, R.; et al. MS-275 (Entinostat) Promotes Radio-Sensitivity in PAX3-FOXO1 Rhabdomyosarcoma Cells. Int. J. Mol. Sci. 2021, 22, 10671. [Google Scholar] [CrossRef]
- Stathis, A.; Bertoni, F. BET Proteins as Targets for Anticancer Treatment. Cancer Discov. 2018, 8, 24–36. [Google Scholar] [CrossRef]
- Bolden, J.E.; Tasdemir, N.; Dow, L.E.; van Es, J.H.; Wilkinson, J.E.; Zhao, Z.; Clevers, H.; Lowe, S.W. Inducible in Vivo Silencing of Brd4 Identifies Potential Toxicities of Sustained BET Protein Inhibition. Cell Rep. 2014, 8, 1919–1929. [Google Scholar] [CrossRef]
- Camero, S.; Camicia, L.; Marampon, F.; Ceccarelli, S.; Shukla, R.; Mannarino, O.; Pizer, B.; Schiavetti, A.; Pizzuti, A.; Tombolini, V.; et al. BET Inhibition Therapy Counteracts Cancer Cell Survival, Clonogenic Potential and Radioresistance Mechanisms in Rhabdomyosarcoma Cells. Cancer Lett. 2020, 479, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.R.; Morgan, W.F. Gene Expression Profiling after Irradiation: Clues to Understanding Acute and Persistent Responses? Cancer Metastasis Rev. 2004, 23, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Criswell, T.; Leskov, K.; Miyamoto, S.; Luo, G.; Boothman, D.A. Transcription Factors Activated in Mammalian Cells after Clinically Relevant Doses of Ionizing Radiation. Oncogene 2003, 22, 5813–5827. [Google Scholar] [CrossRef] [PubMed]
- Mistry, D.S.; Chen, Y.; Wang, Y.; Zhang, K.; Sen, G.L. SNAI2 Controls the Undifferentiated State of Human Epidermal Progenitor Cells. Stem Cells 2014, 32, 3209–3218. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Ng Eaton, E.; Weinberg, R.A. Distinct EMT Programs Control Normal Mammary Stem Cells and Tumour-Initiating Cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, Cell Plasticity and Metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef]
- Pérez-Mancera, P.A.; González-Herrero, I.; Pérez-Caro, M.; Gutiérrez-Cianca, N.; Flores, T.; Gutiérrez-Adán, A.; Pintado, B.; Sánchez-Martín, M.; Sánchez-García, I. SLUG in Cancer Development. Oncogene 2005, 24, 3073–3082. [Google Scholar] [CrossRef]
- Pomella, S.; Sreenivas, P.; Gryder, B.E.; Wang, L.; Milewski, D.; Cassandri, M.; Baxi, K.; Hensch, N.R.; Carcarino, E.; Song, Y.; et al. Interaction between SNAI2 and MYOD Enhances Oncogenesis and Suppresses Differentiation in Fusion Negative Rhabdomyosarcoma. Nat. Commun. 2021, 12, 192. [Google Scholar] [CrossRef]
- Pérez-Caro, M.; Bermejo-Rodríguez, C.; González-Herrero, I.; Sánchez-Beato, M.; Piris, M.A.; Sánchez-García, I. Transcriptomal Profiling of the Cellular Response to DNA Damage Mediated by Slug (Snai2). Br. J. Cancer 2008, 98, 480–488. [Google Scholar] [CrossRef]
- Wang, L.; Hensch, N.R.; Bondra, K.; Sreenivas, P.; Zhao, X.R.; Chen, J.; Moreno Campos, R.; Baxi, K.; Vaseva, A.V.; Sunkel, B.D.; et al. SNAI2-Mediated Repression of BIM Protects Rhabdomyosarcoma from Ionizing Radiation. Cancer Res. 2021, 81, 5451–5463. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Dias, P.; Kumar, P.; Marsden, H.B.; Gattamaneni, H.R.; Kumar, S. N- and c-Myc Oncogenes in Childhood Rhabdomyosarcoma. J. Natl. Cancer Inst. 1990, 82, 151. [Google Scholar] [CrossRef]
- Yoshida, G.J. Emerging Roles of Myc in Stem Cell Biology and Novel Tumor Therapies. J. Exp. Clin. Cancer Res. 2018, 37, 173. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-J.; Wu, S.-P.; Liu, J.-B.; Shi, Y.-S.; Huang, X.; Zhang, Q.-B.; Yao, K.-T. MYC Regulation of CHK1 and CHK2 Promotes Radioresistance in a Stem Cell-like Population of Nasopharyngeal Carcinoma Cells. Cancer Res. 2013, 73, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Festuccia, C.; Popov, V.M.; di Rocco, A.; Colapietro, A.; Sanità, P.; Monache, S.D.; Musio, D.; de Felice, F.; di Cesare, E.; et al. C-Myc Sustains Transformed Phenotype and Promotes Radioresistance of Embryonal Rhabdomyosarcoma Cell Lines. Radiat. Res. 2016, 185, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, M.C.; Zhang, D.D. The Emerging Role of the Nrf2-Keap1 Signaling Pathway in Cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef]
- Marampon, F.; Codenotti, S.; Megiorni, F.; del Fattore, A.; Camero, S.; Gravina, G.L.; Festuccia, C.; Musio, D.; de Felice, F.; Nardone, V.; et al. NRF2 Orchestrates the Redox Regulation Induced by Radiation Therapy, Sustaining Embryonal and Alveolar Rhabdomyosarcoma Cells Radioresistance. J. Cancer Res. Clin. Oncol. 2019, 145, 881–893. [Google Scholar] [CrossRef]
- Madoz-Gúrpide, J.; Cañamero, M.; Sanchez, L.; Solano, J.; Alfonso, P.; Casal, J.I. A Proteomics Analysis of Cell Signaling Alterations in Colorectal Cancer. Mol. Cell. Proteom. 2007, 6, 2150–2164. [Google Scholar] [CrossRef]
- Verlinden, L.; vanden Bempt, I.; Eelen, G.; Drijkoningen, M.; Verlinden, I.; Marchal, K.; de Wolf-Peeters, C.; Christiaens, M.-R.; Michiels, L.; Bouillon, R.; et al. The E2F-Regulated Gene Chk1 Is Highly Expressed in Triple-Negative Estrogen Receptor/Progesterone Receptor /HER-2 Breast Carcinomas. Cancer Res. 2007, 67, 6574–6581. [Google Scholar] [CrossRef]
- Amé, J.-C.; Spenlehauer, C.; de Murcia, G. The PARP Superfamily. Bioessays 2004, 26, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Sousa, F.G.; Matuo, R.; Soares, D.G.; Escargueil, A.E.; Henriques, J.A.P.; Larsen, A.K.; Saffi, J. PARPs and the DNA Damage Response. Carcinogenesis 2012, 33, 1433–1440. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.; Sottili, M.; Salvatore, G.; Meattini, I.; Desideri, I.; Greto, D.; Loi, M.; Becherini, C.; Garlatti, P.; Delli Paoli, C.; et al. Enhancement of Soft Tissue Sarcoma Cell Radiosensitivity by Poly(ADP-Ribose) Polymerase-1 Inhibitors. Radiat. Res. 2018, 190, 464–472. [Google Scholar] [CrossRef]
- Camero, S.; Ceccarelli, S.; de Felice, F.; Marampon, F.; Mannarino, O.; Camicia, L.; Vescarelli, E.; Pontecorvi, P.; Pizer, B.; Shukla, R.; et al. PARP Inhibitors Affect Growth, Survival and Radiation Susceptibility of Human Alveolar and Embryonal Rhabdomyosarcoma Cell Lines. J. Cancer Res. Clin. Oncol. 2019, 145, 137–152. [Google Scholar] [CrossRef]
- Ohi, M.D.; Kenworthy, A.K. Emerging Insights into the Molecular Architecture of Caveolin-1. J. Membr. Biol. 2022, 255, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Poliani, P.L.; Cominelli, M.; Bozzato, A.; Vescovi, R.; Monti, E.; Fanzani, A. Caveolin 1 Is a Marker of Poor Differentiation in Rhabdomyosarcoma. Eur. J. Cancer 2011, 47, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Faggi, F.; Mitola, S.; Sorci, G.; Riuzzi, F.; Donato, R.; Codenotti, S.; Poliani, P.L.; Cominelli, M.; Vescovi, R.; Rossi, S.; et al. Phosphocaveolin-1 Enforces Tumor Growth and Chemoresistance in Rhabdomyosarcoma. PLoS ONE 2014, 9, e84618. [Google Scholar] [CrossRef]
- Codenotti, S.; Faggi, F.; Ronca, R.; Chiodelli, P.; Grillo, E.; Guescini, M.; Megiorni, F.; Marampon, F.; Fanzani, A. Caveolin-1 Enhances Metastasis Formation in a Human Model of Embryonal Rhabdomyosarcoma through Erk Signaling Cooperation. Cancer Lett. 2019, 449, 135–144. [Google Scholar] [CrossRef]
- Pavlides, S.; Tsirigos, A.; Vera, I.; Flomenberg, N.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; Pestell, R.G.; et al. Loss of Stromal Caveolin-1 Leads to Oxidative Stress, Mimics Hypoxia and Drives Inflammation in the Tumor Microenvironment, Conferring the “Reverse Warburg Effect”: A Transcriptional Informatics Analysis with Validation. Cell Cycle 2010, 9, 2201–2219. [Google Scholar] [CrossRef]
- Petragnano, F.; Pietrantoni, I.; Camero, S.; Codenotti, S.; Milazzo, L.; Vulcano, F.; Macioce, G.; Giordani, I.; Tini, P.; Cheleschi, S.; et al. Clinically Relevant Radioresistant Rhabdomyosarcoma Cell Lines: Functional, Molecular and Immune-Related Characterization. J. Biomed. Sci. 2020, 27, 90. [Google Scholar] [CrossRef]
- Codenotti, S.; Marampon, F.; Triggiani, L.; Bonù, M.L.; Magrini, S.M.; Ceccaroli, P.; Guescini, M.; Gastaldello, S.; Tombolini, V.; Poliani, P.L.; et al. Caveolin-1 Promotes Radioresistance in Rhabdomyosarcoma through Increased Oxidative Stress Protection and DNA Repair. Cancer Lett. 2021, 505, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The First 30 Years of P53: Growing Ever More Complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Petrenko, O. The MDM2-P53 Interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar] [PubMed]
- Werbrouck, C.; Evangelista, C.C.S.; Lobón-Iglesias, M.-J.; Barret, E.; le Teuff, G.; Merlevede, J.; Brusini, R.; Kergrohen, T.; Mondini, M.; Bolle, S.; et al. TP53 Pathway Alterations Drive Radioresistance in Diffuse Intrinsic Pontine Gliomas (DIPG). Clin. Cancer Res. 2019, 25, 6788–6800. [Google Scholar] [CrossRef] [PubMed]
- Yogev, O.; Barker, K.; Sikka, A.; Almeida, G.S.; Hallsworth, A.; Smith, L.M.; Jamin, Y.; Ruddle, R.; Koers, A.; Webber, H.T.; et al. P53 Loss in MYC-Driven Neuroblastoma Leads to Metabolic Adaptations Supporting Radioresistance. Cancer Res. 2016, 76, 3025–3035. [Google Scholar] [CrossRef] [PubMed]
- Casey, D.L.; Pitter, K.L.; Wexler, L.H.; Slotkin, E.K.; Gupta, G.P.; Wolden, S.L. TP53 Mutations Increase Radioresistance in Rhabdomyosarcoma and Ewing Sarcoma. Br. J. Cancer 2021, 125, 576–581. [Google Scholar] [CrossRef]
- Phelps, D.; Bondra, K.; Seum, S.; Chronowski, C.; Leasure, J.; Kurmasheva, R.T.; Middleton, S.; Wang, D.; Mo, X.; Houghton, P.J. Inhibition of MDM2 by RG7388 Confers Hypersensitivity to X-Radiation in Xenograft Models of Childhood Sarcoma. Pediatr. Blood Cancer 2015, 62, 1345–1352. [Google Scholar] [CrossRef]
- O’Neill, E.; Kolch, W. Conferring Specificity on the Ubiquitous Raf/MEK Signalling Pathway. Br. J. Cancer 2004, 90, 283–288. [Google Scholar] [CrossRef]
- Caunt, C.J.; Sale, M.J.; Smith, P.D.; Cook, S.J. MEK1 and MEK2 Inhibitors and Cancer Therapy: The Long and Winding Road. Nat. Rev. Cancer 2015, 15, 577–592. [Google Scholar] [CrossRef]
- Shern, J.F.; Selfe, J.; Izquierdo, E.; Patidar, R.; Chou, H.-C.; Song, Y.K.; Yohe, M.E.; Sindiri, S.; Wei, J.; Wen, X.; et al. Genomic Classification and Clinical Outcome in Rhabdomyosarcoma: A Report from an International Consortium. J. Clin. Oncol. 2021, 39, 2859–2871. [Google Scholar] [CrossRef]
- Gupta, A.K.; Bakanauskas, V.J.; Cerniglia, G.J.; Cheng, Y.; Bernhard, E.J.; Muschel, R.J.; McKenna, W.G. The Ras Radiation Resistance Pathway. Cancer Res. 2001, 61, 4278–4282. [Google Scholar] [PubMed]
- Toulany, M.; Baumann, M.; Rodemann, H.P. Stimulated PI3K-AKT Signaling Mediated through Ligand or Radiation-Induced EGFR Depends Indirectly, but Not Directly, on Constitutive K-Ras Activity. Mol. Cancer Res. 2007, 5, 863–872. [Google Scholar] [CrossRef]
- Bachireddy, P.; Bendapudi, P.K.; Felsher, D.W. Getting at MYC through RAS. Clin. Cancer Res. 2005, 11, 4278–4281. [Google Scholar] [CrossRef] [PubMed]
- McKenna, W.G.; Weiss, M.C.; Endlich, B.; Ling, C.C.; Bakanauskas, V.J.; Kelsten, M.L.; Muschel, R.J. Synergistic Effect of the V-Myc Oncogene with H-Ras on Radioresistance. Cancer Res. 1990, 50, 97–102. [Google Scholar]
- Marampon, F.; Gravina, G.L.; di Rocco, A.; Bonfili, P.; di Staso, M.; Fardella, C.; Polidoro, L.; Ciccarelli, C.; Festuccia, C.; Popov, V.M.; et al. MEK/ERK Inhibitor U0126 Increases the Radiosensitivity of Rhabdomyosarcoma Cells in Vitro and in Vivo by Downregulating Growth and DNA Repair Signals. Mol. Cancer Ther. 2011, 10, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, C.; Vulcano, F.; Milazzo, L.; Gravina, G.L.; Marampon, F.; Macioce, G.; Giampaolo, A.; Tombolini, V.; di Paolo, V.; Hassan, H.J.; et al. Key Role of MEK/ERK Pathway in Sustaining Tumorigenicity and in Vitro Radioresistance of Embryonal Rhabdomyosarcoma Stem-like Cell Population. Mol. Cancer 2016, 15, 16. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and Saturation Analysis of Cancer Genes across 21 Tumour Types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef]
- Cen, L.; Hsieh, F.-C.; Lin, H.-J.; Chen, C.-S.; Qualman, S.J.; Lin, J. PDK-1/AKT Pathway as a Novel Therapeutic Target in Rhabdomyosarcoma Cells Using OSU-03012 Compound. Br. J. Cancer 2007, 97, 785–791. [Google Scholar] [CrossRef]
- Kohsaka, S.; Shukla, N.; Ameur, N.; Ito, T.; Ng, C.K.Y.; Wang, L.; Lim, D.; Marchetti, A.; Viale, A.; Pirun, M.; et al. A Recurrent Neomorphic Mutation in MYOD1 Defines a Clinically Aggressive Subset of Embryonal Rhabdomyosarcoma Associated with PI3K-AKT Pathway Mutations. Nat. Genet. 2014, 46, 595–600. [Google Scholar] [CrossRef]
- Granados, V.A.; Avirneni-Vadlamudi, U.; Dalal, P.; Scarborough, S.R.; Galindo, K.A.; Mahajan, P.; Galindo, R.L. Selective Targeting of Myoblast Fusogenic Signaling and Differentiation-Arrest Antagonizes Rhabdomyosarcoma Cells. Cancer Res. 2019, 79, 4585–4591. [Google Scholar] [CrossRef]
- Codenotti, S.; Zizioli, D.; Mignani, L.; Rezzola, S.; Tabellini, G.; Parolini, S.; Giacomini, A.; Asperti, M.; Poli, M.; Mandracchia, D.; et al. Hyperactive Akt1 Signaling Increases Tumor Progression and DNA Repair in Embryonal Rhabdomyosarcoma RD Line and Confers Susceptibility to Glycolysis and Mevalonate Pathway Inhibitors. Cells 2022, 11, 2859. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; English, C.A.; Ohi, R. The Kinesin-8 Kif18A Dampens Microtubule plus-End Dynamics. Curr. Biol. 2010, 20, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Yu, L.; Fang, Z.; Liu, J.; Bai, C.; Li, S.; Xue, R.; Zhang, L.; Tan, Z.; Fan, Z. KIF18B Promotes Tumor Progression in Osteosarcoma by Activating β-Catenin. Cancer Biol. Med. 2020, 17, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yu, Z.; Tang, H.; Wang, X.; Zhang, B.; Zhao, J.; Liu, X.; Zhang, J.; Wei, M. Silencing KIF18B Enhances Radiosensitivity: Identification of a Promising Therapeutic Target in Sarcoma. eBioMedicine 2020, 61, 103056. [Google Scholar] [CrossRef]
- Peake, J.D.; Noguchi, E. Fanconi Anemia: Current Insights Regarding Epidemiology, Cancer, and DNA Repair. Hum. Genet. 2022, 141, 1–26. [Google Scholar] [CrossRef]
- Dextraze, M.-E.; Gantchev, T.; Girouard, S.; Hunting, D. DNA Interstrand Cross-Links Induced by Ionizing Radiation: An Unsung Lesion. Mutat. Res. 2010, 704, 101–107. [Google Scholar] [CrossRef]
- Singh, M.; Leasure, J.M.; Chronowski, C.; Geier, B.; Bondra, K.; Duan, W.; Hensley, L.A.; Villalona-Calero, M.; Li, N.; Vergis, A.M.; et al. FANCD2 Is a Potential Therapeutic Target and Biomarker in Alveolar Rhabdomyosarcoma Harboring the PAX3-FOXO1 Fusion Gene. Clin. Cancer Res. 2014, 20, 3884–3895. [Google Scholar] [CrossRef]
- Shen, C.; Oswald, D.; Phelps, D.; Cam, H.; Pelloski, C.E.; Pang, Q.; Houghton, P.J. Regulation of FANCD2 by the MTOR Pathway Contributes to the Resistance of Cancer Cells to DNA Double-Strand Breaks. Cancer Res. 2013, 73, 3393–3401. [Google Scholar] [CrossRef]
- Houghton, P.J.; Gorlick, R.; Kolb, E.A.; Lock, R.; Carol, H.; Morton, C.L.; Keir, S.T.; Reynolds, C.P.; Kang, M.H.; Phelps, D.; et al. Initial Testing (Stage 1) of the MTOR Kinase Inhibitor AZD8055 by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2012, 58, 191–199. [Google Scholar] [CrossRef]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in Clinical Cancer Immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef]
- di Maggio, F.M.; Minafra, L.; Forte, G.I.; Cammarata, F.P.; Lio, D.; Messa, C.; Gilardi, M.C.; Bravatà, V. Portrait of Inflammatory Response to Ionizing Radiation Treatment. J. Inflamm. 2015, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Sakai, K.; Nakamura, T.; Matsumoto, K. Hepatocyte Growth Factor Twenty Years on: Much More than a Growth Factor. J. Gastroenterol. Hepatol. 2011, 26 (Suppl. S1), 188–202. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, K.; Kucia, M.; Wysoczynski, M.; Reca, R.; Zhao, D.; Trzyna, E.; Trent, J.; Peiper, S.; Zembala, M.; Ratajczak, J.; et al. Both Hepatocyte Growth Factor (HGF) and Stromal-Derived Factor-1 Regulate the Metastatic Behavior of Human Rhabdomyosarcoma Cells, but Only HGF Enhances Their Resistance to Radiochemotherapy. Cancer Res. 2003, 63, 7926–7935. [Google Scholar] [PubMed]
- Teicher, B.A.; Holden, S.A.; Ara, G.; Sotomayor, E.A.; Huang, Z.D.; Chen, Y.N.; Brem, H. Potentiation of Cytotoxic Cancer Therapies by TNP-470 Alone and with Other Anti-Angiogenic Agents. Int. J. Cancer 1994, 57, 920–925. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour Actions of Interferons: Implications for Cancer Therapy. Nat. Rev. Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef]
- Sims, T.L.; McGee, M.; Williams, R.F.; Myers, A.L.; Tracey, L.; Hamner, J.B.; Ng, C.; Wu, J.; Gaber, M.W.; McCarville, B.; et al. IFN-Beta Restricts Tumor Growth and Sensitizes Alveolar Rhabdomyosarcoma to Ionizing Radiation. Mol. Cancer Ther. 2010, 9, 761–771. [Google Scholar] [CrossRef]
- Brantley-Sieders, D.; Schmidt, S.; Parker, M.; Chen, J. Eph Receptor Tyrosine Kinases in Tumor and Tumor Microenvironment. Curr. Pharm. Des. 2004, 10, 3431–3442. [Google Scholar] [CrossRef]
- Berardi, A.C.; Marsilio, S.; Rofani, C.; Salvucci, O.; Altavista, P.; Perla, F.M.; Diomedi-Camassei, F.; Uccini, S.; Kokai, G.; Landuzzi, L.; et al. Up-Regulation of EphB and Ephrin-B Expression in Rhabdomyosarcoma. Anticancer Res. 2008, 28, 763–769. [Google Scholar]
- Megiorni, F.; Gravina, G.L.; Camero, S.; Ceccarelli, S.; del Fattore, A.; Desiderio, V.; Papaccio, F.; McDowell, H.P.; Shukla, R.; Pizzuti, A.; et al. Pharmacological Targeting of the Ephrin Receptor Kinase Signalling by GLPG1790 in Vitro and in Vivo Reverts Oncophenotype, Induces Myogenic Differentiation and Radiosensitizes Embryonal Rhabdomyosarcoma Cells. J. Hematol. Oncol. 2017, 10, 161. [Google Scholar] [CrossRef]
- Farhood, B.; Mortezaee, K.; Motevaseli, E.; Mirtavoos-Mahyari, H.; Shabeeb, D.; Eleojo Musa, A.; Sanikhani, N.S.; Najafi, M.; Ahmadi, A. Selenium as an Adjuvant for Modification of Radiation Response. J. Cell Biochem. 2019, 120, 18559–18571. [Google Scholar] [CrossRef]
- Battin, E.E.; Brumaghim, J.L. Antioxidant Activity of Sulfur and Selenium: A Review of Reactive Oxygen Species Scavenging, Glutathione Peroxidase, and Metal-Binding Antioxidant Mechanisms. Cell Biochem. Biophys. 2009, 55, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Gehrisch, A.; Dörr, W. Effects of Systemic or Topical Administration of Sodium Selenite on Early Radiation Effects in Mouse Oral Mucosa. Strahlenther. Onkol. 2007, 183, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Dörr, W. Effects of Selenium on Radiation Responses of Tumor Cells and Tissue. Strahlenther. Onkol. 2006, 182, 693–695. [Google Scholar] [CrossRef] [PubMed]
- Beck-Bornholdt, H.P.; Omniczynski, M.; Theis, E.; Vogler, H.; Würschmidt, F. Influence of Treatment Time on the Response of Rat Rhabdomyosarcoma R1H to Fractionated Irradiation. Acta Oncol. 1991, 30, 57–63. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Mattson, M.P.; Calabrese, V. Resveratrol Commonly Displays Hormesis: Occurrence and Biomedical Significance. Hum. Exp. Toxicol. 2010, 29, 980–1015. [Google Scholar] [CrossRef]
- Magalhães, V.D.; Rogero, S.O.; Cruz, A.S.; Vieira, D.P.; Okazaki, K.; Rogero, J.R. In Vitro Tests of Resveratrol Radiomodifying Effect on Rhabdomyosarcoma Cells by Comet Assay. Toxicol. In Vitro 2014, 28, 1436–1442. [Google Scholar] [CrossRef]
- Chow, A.W.; Murillo, G.; Yu, C.; van Breemen, R.B.; Boddie, A.W.; Pezzuto, J.M.; das Gupta, T.K.; Mehta, R.G. Resveratrol Inhibits Rhabdomyosarcoma Cell Proliferation. Eur. J. Cancer Prev. 2005, 14, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Mohrbacher, A.M.; Yang, A.S.; Groshen, S.; Kummar, S.; Gutierrez, M.E.; Kang, M.H.; Tsao-Wei, D.; Reynolds, C.P.; Newman, E.M.; Maurer, B.J. Phase I Study of Fenretinide Delivered Intravenously in Patients with Relapsed or Refractory Hematologic Malignancies: A California Cancer Consortium Trial. Clin. Cancer Res. 2017, 23, 4550–4555. [Google Scholar] [CrossRef]
- Hail, N.; Kim, H.J.; Lotan, R. Mechanisms of Fenretinide-Induced Apoptosis. Apoptosis 2006, 11, 1677–1694. [Google Scholar] [CrossRef]
- Tsou, C.T.; Chen, C.F.; Chow, S.Y. Interactions between Ketamine and Other Drugs. Taiwan Yi Xue Hui Za Zhi 1975, 74, 244–250. [Google Scholar]
- Chen, N.E.; Maldonado, N.V.; Khankaldyyan, V.; Shimada, H.; Song, M.M.; Maurer, B.J.; Reynolds, C.P. Reactive Oxygen Species Mediates the Synergistic Activity of Fenretinide Combined with the Microtubule Inhibitor ABT-751 against Multidrug-Resistant Recurrent Neuroblastoma Xenografts. Mol. Cancer Ther. 2016, 15, 2653–2664. [Google Scholar] [CrossRef] [PubMed]
- Brack, E.; Bender, S.; Wachtel, M.; Pruschy, M.; Schäfer, B.W. Fenretinide Acts as Potent Radiosensitizer for Treatment of Rhabdomyosarcoma Cells. Front. Oncol. 2021, 11, 664462. [Google Scholar] [CrossRef] [PubMed]
- Brack, E.; Wachtel, M.; Wolf, A.; Kaech, A.; Ziegler, U.; Schäfer, B.W. Fenretinide Induces a New Form of Dynamin-Dependent Cell Death in Pediatric Sarcoma. Cell Death Differ. 2020, 27, 2500–2516. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Stage | Sites | Tumor Stage | N | M | |
|---|---|---|---|---|---|
| Invasiveness | Size | N0 → No Nodes | |||
| T1 → Confined | a → < 5 cm. | N1 → Nodes Positive | |||
| T2 → Extended | b → >5 cm. | NX → Unknown | |||
| 1 |
| T1 or T2 | a or b | Any N | M0 |
| |||||
| |||||
| |||||
| 2 |
| T1 or T2 | a or b | N0 or NX | M0 |
| |||||
| |||||
| |||||
| 3 |
| T1 or T2 | a b | N1 Any N | M0 |
| |||||
| |||||
| |||||
| 4 | All | T1 or T2 | a or b | N0 or N1 | M1 |
| Children’s Oncology Group (COG) Soft-Tissue Sarcoma Committee | ||||||
|---|---|---|---|---|---|---|
| Risk Group | Stage (tnm) | Clinical Group (IRS) | FN-FP | Radiation Dose and Specific Indications | Ref. | |
| I → R0 + N0 | ||||||
| II → R0 + N1 → R1 + N0 → R1 + N1 | ||||||
| II → R2 → Only Biopsy | ||||||
| IV → Metastatic | ||||||
| Low—Subset 1 | 1–2 | I | FN | 0 Gy | [37] | |
| 1–2 | II | 36–41.4 Gy | [39] | |||
| 1 | III Orbit | 45 Gy | Complete Response after CHT | [40] | ||
| 50.4 Gy | Partial Response after CHT | |||||
| Low—Subset 2 | 1 | III Non-Orbit | FN | 50.4 Gy | ≤5 cm | [41] |
| 59.4 Gy | >5 cm | |||||
| 3 | I | 0 Gy | [37] | |||
| 3 | II | 36–41.4 Gy | [39] | |||
| Intermediate | 2–3 | III | FN | 50.4 Gy | ≤5 cm | [41] |
| 59.4 Gy | >5 cm | |||||
| 4 | IV ≤ 10 years old | Sites M+ and | See Risk Group | [42] | ||
| 15 Gy Whole Lung | ||||||
| 1–3 | I | FP | 36 Gy | [43] | ||
| 1–3 | II | 36–41.4 Gy | [39] | |||
| 1–3 | III | 50.4 Gy | ≤5 cm | [41] | ||
| 59.4 Gy | >5 cm | |||||
| High | 4 | IV | FN-FP | Sites M+ | See Risk Group | [42] |
| European Pediatric Soft Tissue Sarcoma Group (EpSSG) RMS2005 | ||||||
|---|---|---|---|---|---|---|
| Risk Group | Site | CLINICAL GROUP (IRS) | Nodal | FN-FP | Age/Size | Dose |
| I → R0 + N0 | ||||||
| II → R0 + N1 → R1 + N0 → R1 + N1 | ||||||
| II → R2 → Only Biopsy | ||||||
| IV → Metastatic | ||||||
| Low | Any | I | N0 | FN | ≤10 years | 41.4 Gy |
| ≤5 cm | ||||||
| Standard | Any | I | N0 | FN | >10 years | 41.4 Gy |
| >5 cm | ||||||
| Favorable | II–III | N0 | FN | Any | 50.4 Gy | |
| Unfavorable | II–III | N0 | FN | ≤10 years | 50.4 Gy | |
| ≤5 cm | ||||||
| High | Unfavorable | II–III | N0 | FN | >10 years | 50.4 Gy |
| >5 cm | ||||||
| Any | II–III | N1 | FN | Any | 50.4 Gy | |
| Any | 1-II-III | N0 | FP | Any | 50.4 Gy | |
| Very high | Any | 1-II-III | N1 | FP | Any | 50.4 Gy |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pomella, S.; Porrazzo, A.; Cassandri, M.; Camero, S.; Codenotti, S.; Milazzo, L.; Vulcano, F.; Barillari, G.; Cenci, G.; Marchese, C.; et al. Translational Implications for Radiosensitizing Strategies in Rhabdomyosarcoma. Int. J. Mol. Sci. 2022, 23, 13281. https://doi.org/10.3390/ijms232113281
Pomella S, Porrazzo A, Cassandri M, Camero S, Codenotti S, Milazzo L, Vulcano F, Barillari G, Cenci G, Marchese C, et al. Translational Implications for Radiosensitizing Strategies in Rhabdomyosarcoma. International Journal of Molecular Sciences. 2022; 23(21):13281. https://doi.org/10.3390/ijms232113281
Chicago/Turabian StylePomella, Silvia, Antonella Porrazzo, Matteo Cassandri, Simona Camero, Silvia Codenotti, Luisa Milazzo, Francesca Vulcano, Giovanni Barillari, Giovanni Cenci, Cinzia Marchese, and et al. 2022. "Translational Implications for Radiosensitizing Strategies in Rhabdomyosarcoma" International Journal of Molecular Sciences 23, no. 21: 13281. https://doi.org/10.3390/ijms232113281
APA StylePomella, S., Porrazzo, A., Cassandri, M., Camero, S., Codenotti, S., Milazzo, L., Vulcano, F., Barillari, G., Cenci, G., Marchese, C., Fanzani, A., Megiorni, F., Rota, R., & Marampon, F. (2022). Translational Implications for Radiosensitizing Strategies in Rhabdomyosarcoma. International Journal of Molecular Sciences, 23(21), 13281. https://doi.org/10.3390/ijms232113281