
Isolating Linum usitatissimum L. Nuclear DNA Enabled Assembling High-Quality Genome

 , ,
, , 

Abstract
1. Introduction
2. Results
3. Discussion
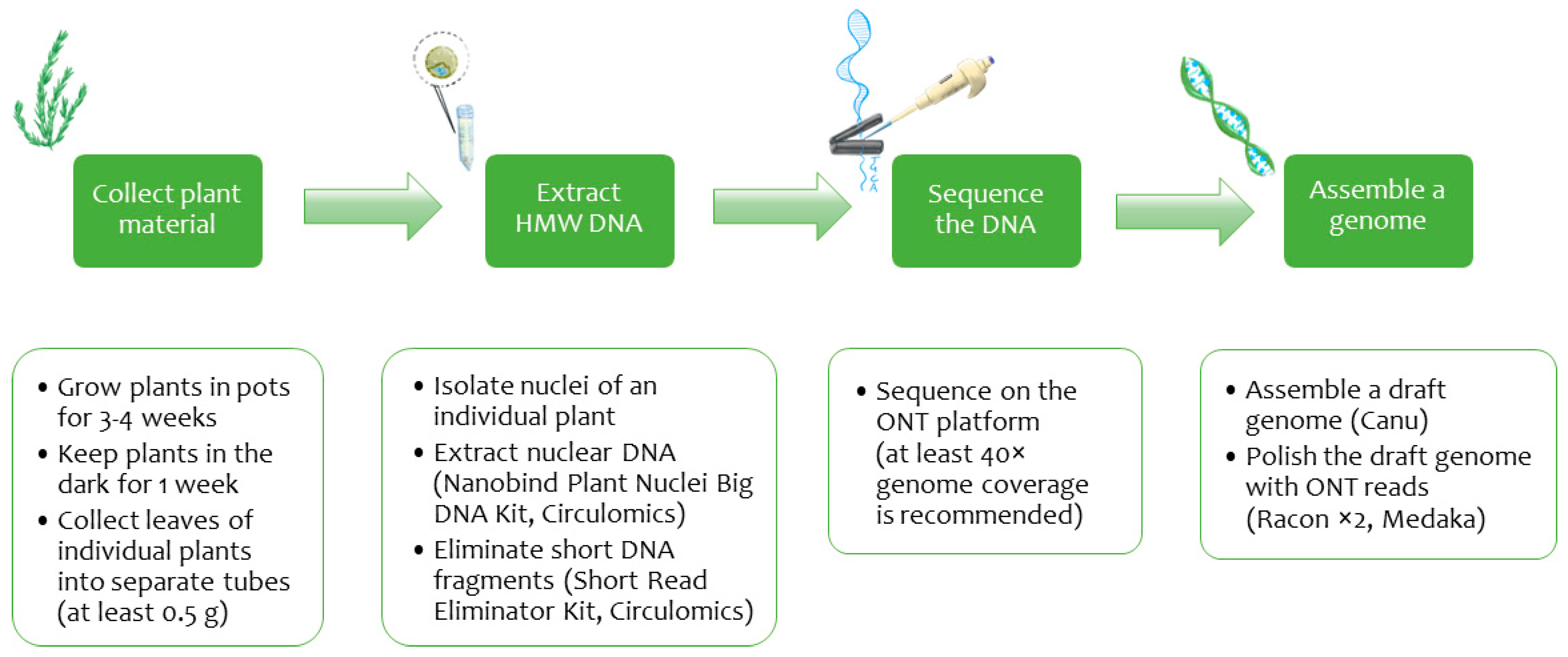
4. Materials and Methods
4.1. Growing Plant Material
4.2. Nuclei Isolation and DNA Extraction
4.3. ONT Library Preparation and Sequencing
4.4. Genome Assembly
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goyal, A.; Sharma, V.; Upadhyay, N.; Gill, S.; Sihag, M. Flax and flaxseed oil: An ancient medicine & modern functional food. J. Food Sci. Technol. 2014, 51, 1633–1653. [Google Scholar] [PubMed]
- Preisner, M.; Wojtasik, W.; Kulma, A.; Żuk, M.; Szopa, J. Flax fiber. In Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley & Sons: Hoboken, NJ, USA, 2014; pp. 1–32. [Google Scholar]
- Teeter, H.M. Some Industrial Outlets for Seed Flax. In The Yearbook of Agriculture 1950–1951; Department of Agriculture: Washington, DC, USA, 1950. [Google Scholar]
- Zhang, Y.; Mao, T.; Wu, H.; Cheng, L.; Zheng, L. Carbon Nanotubes Grown on Flax Fabric as Hierarchical All-Carbon Flexible Electrodes for Supercapacitors. Adv. Mater. Interfaces 2017, 4, 1601123. [Google Scholar] [CrossRef]
- Singh, K.; Mridula, D.; Rehal, J.; Barnwal, P. Flaxseed: A potential source of food, feed and fiber. Crit. Rev. Food Sci. Nutr. 2011, 51, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Caroprese, M.; Marzano, A.; Marino, R.; Gliatta, G.; Muscio, A.; Sevi, A. Flaxseed supplementation improves fatty acid profile of cow milk. J. Dairy Sci. 2010, 93, 2580–2588. [Google Scholar] [CrossRef]
- Jhala, A.J.; Hall, L.M. Flax (Linum usitatissimum L.): Current uses and future applications. Aust. J. Basic Appl. Sci 2010, 4, 4304–4312. [Google Scholar]
- Asyraf, M.R.M.; Ishak, M.R.; Norrrahim, M.N.F.; Amir, A.L.; Nurazzi, N.M.; Ilyas, R.A.; Asrofi, M.; Rafidah, M.; Razman, M.R. Potential of Flax Fiber Reinforced Biopolymer Composites for Cross-Arm Application in Transmission Tower: A Review. Fibers Polym. 2022, 23, 853–877. [Google Scholar] [CrossRef]
- Royle, J.F. The Fibrous Plants of India Fitted for Cordage, Clothing, and Paper: With an Account of the Cultivation and Preparation of Flax, Hemp, and Their Substitutes; Smith, Elder, and Company: London, UK, 1855. [Google Scholar]
- Bakowska-Barczak, A.; de Larminat, M.-A.; Kolodziejczyk, P.P. The application of flax and hempseed in food, nutraceutical and personal care products. In Handbook of Natural Fibres; Elsevier: Amsterdam, The Netherlands, 2020; pp. 557–590. [Google Scholar]
- Gomez-Campos, A.; Vialle, C.; Rouilly, A.; Sablayrolles, C.; Hamelin, L. Flax fiber for technical textile: A life cycle inventory. J. Clean. Prod. 2021, 281, 125177. [Google Scholar] [CrossRef]
- Hall, L.M.; Booker, H.; Siloto, R.M.; Jhala, A.J.; Weselake, R.J. Flax (Linum usitatissimum L.). In Industrial Oil Crops; Elsevier: Amsterdam, The Netherlands, 2016; pp. 157–194. [Google Scholar]
- Kulma, A.; Skórkowska-Telichowska, K.; Kostyn, K.; Szatkowski, M.; Skała, J.; Drulis-Kawa, Z.; Preisner, M.; Żuk, M.; Szperlik, J.; Wang, Y. New flax producing bioplastic fibers for medical purposes. Ind. Crops Prod. 2015, 68, 80–89. [Google Scholar] [CrossRef]
- Allaby, R.G.; Peterson, G.W.; Merriwether, D.A.; Fu, Y.-B. Evidence of the domestication history of flax (Linum usitatissimum L.) from genetic diversity of the sad2 locus. Theor. Appl. Genet. 2005, 112, 58–65. [Google Scholar] [CrossRef]
- Diederichsen, A. Comparison of genetic diversity of flax (Linum usitatissimum L.) between Canadian cultivars and a world collection. Plant Breed. 2001, 120, 360–362. [Google Scholar] [CrossRef]
- Nag, S.; Mitra, J.; Karmakar, P. An overview on flax (Linum usitatissimum L.) and its genetic diversity. Int. J. Agric. Environ. Biotechnol. 2015, 8, 805. [Google Scholar] [CrossRef]
- Hoque, A.; Fiedler, J.D.; Rahman, M. Genetic diversity analysis of a flax (Linum usitatissimum L.) global collection. BMC Genom. 2020, 21, 557. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, P.; Verma, G.; Sidhu, G.; Duhan, N. Epigenomics: Role, approaches and applications in plants. J. Anim. Plant Sci. 2020, 30, 1071–1081. [Google Scholar]
- Shivaraj, S.; Dhakate, P.; Sonah, H.; Vuong, T.; Nguyen, H.T.; Deshmukh, R. Progress toward development of climate-smart flax: A perspective on omics-assisted breeding. In Genomic Designing of Climate-Smart Oilseed Crops; Springer: Cham, Switzerland, 2019; pp. 239–274. [Google Scholar]
- Kyriakidou, M.; Achakkagari, S.R.; Gálvez López, J.H.; Zhu, X.; Tang, C.Y.; Tai, H.H.; Anglin, N.L.; Ellis, D.; Strömvik, M.V. Structural genome analysis in cultivated potato taxa. Theor. Appl. Genet. 2020, 133, 951–966. [Google Scholar] [CrossRef]
- Hibrand Saint-Oyant, L.; Ruttink, T.; Hamama, L.; Kirov, I.; Lakhwani, D.; Zhou, N.N.; Bourke, P.M.; Daccord, N.; Leus, L.; Schulz, D.; et al. A high-quality genome sequence of Rosa chinensis to elucidate ornamental traits. Nat. Plants 2018, 4, 473–484. [Google Scholar] [CrossRef]
- Li, C.; Xiang, X.; Huang, Y.; Zhou, Y.; An, D.; Dong, J.; Zhao, C.; Liu, H.; Li, Y.; Wang, Q.; et al. Long-read sequencing reveals genomic structural variations that underlie creation of quality protein maize. Nat. Commun. 2020, 11, 17. [Google Scholar] [CrossRef]
- Bolger, M.E.; Weisshaar, B.; Scholz, U.; Stein, N.; Usadel, B.; Mayer, K.F.X. Plant genome sequencing—Applications for crop improvement. Curr. Opin. Biotechnol. 2014, 26, 31–37. [Google Scholar] [CrossRef]
- Kelly, L.J.; Leitch, I.J. Exploring giant plant genomes with next-generation sequencing technology. Chromosome Res. 2011, 19, 939–953. [Google Scholar] [CrossRef]
- Weigel, D.; Mott, R. The 1001 Genomes Project for Arabidopsis thaliana. Genome Biol. 2009, 10, 107. [Google Scholar] [CrossRef]
- Hodzic, J.; Gurbeta, L.; Omanovic-Miklicanin, E.; Badnjevic, A. Overview of Next-generation Sequencing Platforms Used in Published Draft Plant Genomes in Light of Genotypization of Immortelle Plant (Helichrysium arenarium). Med. Arch. 2017, 71, 288–292. [Google Scholar] [CrossRef]
- Kubis, S.; Schmidt, T.; Heslop-Harrison, J.S. Repetitive DNA Elements as a Major Component of Plant Genomes. Ann. Bot. 1998, 82, 45–55. [Google Scholar] [CrossRef]
- Ossowski, S.; Schneeberger, K.; Clark, R.M.; Lanz, C.; Warthmann, N.; Weigel, D. Sequencing of natural strains of Arabidopsis thaliana with short reads. Genome Res. 2008, 18, 2024–2033. [Google Scholar] [CrossRef] [PubMed]
- Weiss-Schneeweiss, H.; Leitch, A.R.; McCann, J.; Jang, T.-S.; Macas, J. Employing next generation sequencing to explore the repeat landscape of the plant genome. Next Gener. Seq. Plant Syst. Regnum Veg. 2015, 157, 155–179. [Google Scholar]
- Kyriakidou, M.; Tai, H.H.; Anglin, N.L.; Ellis, D.; Strömvik, M.V. Current Strategies of Polyploid Plant Genome Sequence Assembly. Front. Plant Sci. 2018, 9, 1660. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Shang, L.; Zhu, Q.-H.; Fan, L.; Guo, L. Twenty years of plant genome sequencing: Achievements and challenges. Trends Plant Sci. 2022, 27, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Wang, X.; Li, X.; Xu, M.; Li, H.; Abla, M.; Sun, H.; Wei, S.; Feng, J.; Zhou, Y. Long-read sequencing and de novo genome assembly of Ammopiptanthus nanus, a desert shrub. GigaScience 2018, 7, giy074. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Zhou, Y.; Li, C.; Xiao, Q.; Wang, T.; Zhang, Y.; Wu, Y.; Li, Y.; Chao, D.-Y.; Messing, J. Plant evolution and environmental adaptation unveiled by long-read whole-genome sequencing of Spirodela. Proc. Natl. Acad. Sci. USA 2019, 116, 18893–18899. [Google Scholar] [CrossRef]
- Wei, C.; Yang, H.; Wang, S.; Zhao, J.; Liu, C.; Gao, L.; Xia, E.; Lu, Y.; Tai, Y.; She, G. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. USA 2018, 115, E4151–E4158. [Google Scholar] [CrossRef]
- Lan, T.; Renner, T.; Ibarra-Laclette, E.; Farr, K.M.; Chang, T.-H.; Cervantes-Pérez, S.A.; Zheng, C.; Sankoff, D.; Tang, H.; Purbojati, R.W. Long-read sequencing uncovers the adaptive topography of a carnivorous plant genome. Proc. Natl. Acad. Sci. USA 2017, 114, E4435–E4441. [Google Scholar] [CrossRef]
- Lee, H.; Chawla, H.S.; Obermeier, C.; Dreyer, F.; Abbadi, A.; Snowdon, R. Chromosome-Scale Assembly of Winter Oilseed Rape Brassica napus. Front. Plant Sci. 2020, 11, 496. [Google Scholar] [CrossRef]
- Danilevicz, M.F.; Fernandez, C.G.T.; Marsh, J.I.; Bayer, P.E.; Edwards, D. Plant pangenomics: Approaches, applications and advancements. Curr. Opin. Plant Biol. 2020, 54, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Belser, C.; Baurens, F.-C.; Noel, B.; Martin, G.; Cruaud, C.; Istace, B.; Yahiaoui, N.; Labadie, K.; Hřibová, E.; Doležel, J. Telomere-to-telomere gapless chromosomes of banana using nanopore sequencing. Commun. Biol. 2021, 4, 1047. [Google Scholar] [CrossRef]
- Marrano, A.; Britton, M.; Zaini, P.A.; Zimin, A.V.; Workman, R.E.; Puiu, D.; Bianco, L.; Pierro, E.A.D.; Allen, B.J.; Chakraborty, S.; et al. High-quality chromosome-scale assembly of the walnut (Juglans regia L.) reference genome. GigaScience 2020, 9, giaa050. [Google Scholar] [CrossRef]
- Dmitriev, A.A.; Pushkova, E.N.; Melnikova, N.V. Plant Genome Sequencing: Modern Technologies and Novel Opportunities for Breeding. Mol. Biol. 2022, 56, 495–507. [Google Scholar] [CrossRef]
- Sa, R.; Yi, L.; Siqin, B.; An, M.; Bao, H.; Song, X.; Wang, S.; Li, Z.; Zhang, Z.; Hazaisi, H.; et al. Chromosome-Level Genome Assembly and Annotation of the Fiber Flax (Linum usitatissimum) Genome. Front Genet 2021, 12, 735690. [Google Scholar] [CrossRef] [PubMed]
- You, F.M.; Xiao, J.; Li, P.; Yao, Z.; Jia, G.; He, L.; Zhu, T.; Luo, M.-C.; Wang, X.; Deyholos, M.K.; et al. Chromosome-scale pseudomolecules refined by optical, physical and genetic maps in flax. Plant J. 2018, 95, 371–384. [Google Scholar] [CrossRef]
- Dmitriev, A.A.; Pushkova, E.N.; Novakovskiy, R.O.; Beniaminov, A.D.; Rozhmina, T.A.; Zhuchenko, A.A.; Bolsheva, N.L.; Muravenko, O.V.; Povkhova, L.V.; Dvorianinova, E.M.; et al. Genome Sequencing of Fiber Flax Cultivar Atlant Using Oxford Nanopore and Illumina Platforms. Front. Genet. 2021, 11, 590282. [Google Scholar] [CrossRef]
- Zhang, J.; Qi, Y.; Wang, L.; Wang, L.; Yan, X.; Dang, Z.; Li, W.; Zhao, W.; Pei, X.; Li, X.; et al. Genomic Comparison and Population Diversity Analysis Provide Insights into the Domestication and Improvement of Flax. iScience 2020, 23, 100967. [Google Scholar] [CrossRef]
- Clarke, J.D. Cetyltrimethyl ammonium bromide (CTAB) DNA miniprep for plant DNA isolation. Cold Spring Harb. Protoc. 2009. [Google Scholar] [CrossRef]
- Dumschott, K.; Schmidt, M.H.-W.; Chawla, H.S.; Snowdon, R.; Usadel, B. Oxford Nanopore sequencing: New opportunities for plant genomics? J. Exp. Bot. 2020, 71, 5313–5322. [Google Scholar] [CrossRef]
- Kidwell, K.K.; Osborn, T.C. Simple plant DNA isolation procedures. In Plant Genomes: Methods for Genetic and Physical Mapping; Springer: Dordrecht, The Netherlands, 1992; pp. 1–13. [Google Scholar]
- Križman, M.; Jakše, J.; Baričevič, D.; Javornik, B.; Prošek, M. Robust CTAB-activated charcoal protocol for plant DNA extraction. Acta Agric. Slov. 2006, 87, 427–433. [Google Scholar]
- Li, J.; Wang, S.; Yu, J.; Wang, L.; Zhou, S. A modified CTAB protocol for plant DNA extraction. Chin. Bull. Bot. 2013, 48, 72. [Google Scholar]
- Sharma, R.; Mahla, H.R.; Mohapatra, T.; Bhargava, S.C.; Sharma, M.M. Isolating plant genomic DNA without liquid nitrogen. Plant Mol. Biol. Report. 2003, 21, 43–50. [Google Scholar] [CrossRef]
- Vondrak, T.; Ávila Robledillo, L.; Novák, P.; Koblížková, A.; Neumann, P.; Macas, J. Characterization of repeat arrays in ultra-long nanopore reads reveals frequent origin of satellite DNA from retrotransposon-derived tandem repeats. Plant J. 2020, 101, 484–500. [Google Scholar] [CrossRef] [PubMed]
- Luro, F.; Laigret, F. Preparation of high molecular weight genomic DNA from nuclei of woody plants. BioTechniques 1995, 19, 388–392. [Google Scholar]
- Frei, D.; Veekman, E.; Grogg, D.; Stoffel-Studer, I.; Morishima, A.; Shimizu-Inatsugi, R.; Yates, S.; Shimizu, K.K.; Frey, J.E.; Studer, B.; et al. Ultralong Oxford Nanopore Reads Enable the Development of a Reference-Grade Perennial Ryegrass Genome Assembly. Genome Biol. Evol. 2021, 13, evab159. [Google Scholar] [CrossRef]
- Workman, R.; Fedak, R.; Kilburn, D.; Hao, S.; Liu, K.; Timp, W. High Molecular Weight DNA Extraction from Recalcitrant Plant Species for Third Generation Sequencing. Protocols.io 2019. [Google Scholar] [CrossRef]
- Ma, L.; Dong, C.; Song, C.; Wang, X.; Zheng, X.; Niu, Y.; Chen, S.; Feng, W. De novo genome assembly of the potent medicinal plant Rehmannia glutinosa using nanopore technology. Comput. Struct. Biotechnol. J. 2021, 19, 3954–3963. [Google Scholar] [CrossRef]
- Driguez, P.; Bougouffa, S.; Carty, K.; Putra, A.; Jabbari, K.; Reddy, M.; Soppe, R.; Cheung, M.S.; Fukasawa, Y.; Ermini, L. LeafGo: Leaf to Genome, a quick workflow to produce high-quality de novo plant genomes using long-read sequencing technology. Genome Biol. 2021, 22, 256. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Hu, J.; Fan, J.; Sun, Z.; Liu, S. NextPolish: A fast and efficient genome polishing tool for long-read assembly. Bioinformatics 2020, 36, 2253–2255. [Google Scholar] [CrossRef] [PubMed]
- Shafin, K.; Pesout, T.; Chang, P.-C.; Nattestad, M.; Kolesnikov, A.; Goel, S.; Baid, G.; Kolmogorov, M.; Eizenga, J.M.; Miga, K.H. Haplotype-aware variant calling with PEPPER-Margin-DeepVariant enables high accuracy in nanopore long-reads. Nat. Methods 2021, 18, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.; Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol. 2018, 176, 1410–1422. [Google Scholar] [CrossRef]
- Belser, C.; Istace, B.; Denis, E.; Dubarry, M.; Baurens, F.-C.; Falentin, C.; Genete, M.; Berrabah, W.; Chèvre, A.-M.; Delourme, R.; et al. Chromosome-scale assemblies of plant genomes using nanopore long reads and optical maps. Nat. Plants 2018, 4, 879–887. [Google Scholar] [CrossRef]
- Hamilton, J.P.; Robin Buell, C. Advances in plant genome sequencing. Plant J. 2012, 70, 177–190. [Google Scholar] [CrossRef]
- Istace, B.; Belser, C.; Falentin, C.; Labadie, K.; Boideau, F.; Deniot, G.; Maillet, L.; Cruaud, C.; Bertrand, L.; Chèvre, A.-M. Sequencing and chromosome-scale assembly of plant genomes, Brassica rapa as a use case. Biology 2021, 10, 732. [Google Scholar] [CrossRef]
- Dvorianinova, E.M.; Pushkova, E.N.; Novakovskiy, R.O.; Povkhova, L.V.; Bolsheva, N.L.; Kudryavtseva, L.P.; Rozhmina, T.A.; Melnikova, N.V.; Dmitriev, A.A. Nanopore and Illumina genome sequencing of Fusarium oxysporum f. sp. lini strains of different virulence. Front. Genet. 2021, 12, 662928. [Google Scholar] [CrossRef]
- Fu, Z.-Y.; Song, J.-C.; Jameson, P.E. A rapid and cost effective protocol for plant genomic DNA isolation using regenerated silica columns in combination with CTAB extraction. J. Integr. Agric. 2017, 16, 1682–1688. [Google Scholar] [CrossRef]
- Healey, A.; Furtado, A.; Cooper, T.; Henry, R.J. Protocol: A simple method for extracting next-generation sequencing quality genomic DNA from recalcitrant plant species. Plant Methods 2014, 10, 21. [Google Scholar] [CrossRef]
- Manen, J.-F.; Sinitsyna, O.; Aeschbach, L.; Markov, A.V.; Sinitsyn, A. A fully automatable enzymatic method for DNA extraction from plant tissues. BMC Plant Biol. 2005, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Pushkova, E.N.; Beniaminov, A.D.; Krasnov, G.S.; Novakovskiy, R.O.; Povkhova, L.V.; Melnikova, N.V.; Dmitriev, A.A. Extraction of high-molecular-weight DNA from poplar plants for Nanopore sequencing. In Current Challenges in Plant Genetics, Genomics, Bioinformatics, and Biotechnology: Proceedings of the Fifth International Scientific Conference PlantGen2019 (June 24–29, 2019, Novosibirsk, Russia); ICG SB RAS: Novosibirsk, Russia, 2019; pp. 158–160. [Google Scholar] [CrossRef]
- Xin, Z.; Chen, J. A high throughput DNA extraction method with high yield and quality. Plant Methods 2012, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Stewart, J. Economical and rapid method for extracting cotton genomic DNA. J. Cotton Sci. 2000, 4, 193–201. [Google Scholar]
- Zhou, L.; Shen, R.; Ma, X.; Li, H.; Li, G.; Liu, Y.G. Preparation of rice plant genomic DNA for various applications. Curr. Protoc. Plant Biol. 2016, 1, 29–42. [Google Scholar] [CrossRef]
- Krasnov, G.S.; Pushkova, E.N.; Novakovskiy, R.O.; Kudryavtseva, L.P.; Rozhmina, T.A.; Dvorianinova, E.M.; Povkhova, L.V.; Kudryavtseva, A.V.; Dmitriev, A.A.; Melnikova, N.V. High-quality genome assembly of Fusarium oxysporum f. sp. lini. Front. Genet. 2020, 11, 959. [Google Scholar] [CrossRef]
- Melnikova, N.V.; Pushkova, E.N.; Dvorianinova, E.M.; Beniaminov, A.D.; Novakovskiy, R.O.; Povkhova, L.V.; Bolsheva, N.L.; Snezhkina, A.V.; Kudryavtseva, A.V.; Krasnov, G.S.; et al. Genome Assembly and Sex-Determining Region of Male and Female Populus × sibirica. Front. Plant Sci. 2021, 12, 625416. [Google Scholar] [CrossRef]
- Wang, Z.; Hobson, N.; Galindo, L.; Zhu, S.; Shi, D.; McDill, J.; Yang, L.; Hawkins, S.; Neutelings, G.; Datla, R. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 2012, 72, 461–473. [Google Scholar] [CrossRef]
- Bolsheva, N.L.; Melnikova, N.V.; Kirov, I.V.; Speranskaya, A.S.; Krinitsina, A.A.; Dmitriev, A.A.; Belenikin, M.S.; Krasnov, G.S.; Lakunina, V.A.; Snezhkina, A.V.; et al. Evolution of blue-flowered species of genus Linum based on high-throughput sequencing of ribosomal RNA genes. BMC Evol. Biol. 2017, 17, 253. [Google Scholar] [CrossRef]
- Wu, X.; Liu, Y.; Zhang, Y.; Gu, R. Advances in Research on the Mechanism of Heterosis in Plants. Front. Plant Sci. 2021, 12, 2124. [Google Scholar] [CrossRef]
- Yang, F.X.; Gao, J. The genome of Cymbidium sinense revealed the evolution of orchid traits. Plant Biotechnol. J. 2021, 19, 2501–2516. [Google Scholar] [CrossRef]
- Miga, K.H.; Koren, S.; Rhie, A.; Vollger, M.R.; Gershman, A.; Bzikadze, A.; Brooks, S.; Howe, E.; Porubsky, D.; Logsdon, G.A.; et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature 2020, 585, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Wu, H.; Yang, Q.; Huang, L.; Hu, Q.; Ma, T.; Li, Z.; Liu, J. A chromosome-scale genome assembly of Isatis indigotica, an important medicinal plant used in traditional Chinese medicine: An Isatis genome. Hortic. Res. 2020, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, H.; Huang, W.; Xu, Y.; Zhou, Q.; Wang, S.; Ruan, J.; Huang, S.; Zhang, Z. A chromosome-scale genome assembly of cucumber (Cucumis sativus L.). GigaScience 2019, 8, giz072. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, X.; Jia, Y.; Xu, Y.; Jia, P.; Dang, N.; Wang, S.; Xu, T.; Zhao, X.; Gao, S.; et al. High-quality Arabidopsis thaliana Genome Assembly with Nanopore and HiFi Long Reads. Genom. Proteom. Bioinform. 2021, 20, 4–13. [Google Scholar] [CrossRef]
- Van Rengs, W.M.; Schmidt, M.H.W.; Effgen, S.; Le, D.B.; Wang, Y.; Zaidan, M.W.A.M.; Huettel, B.; Schouten, H.J.; Usadel, B.; Underwood, C.J. A chromosome scale tomato genome built from complementary PacBio and Nanopore sequences alone reveals extensive linkage drag during breeding. Plant J. 2022, 110, 572–588. [Google Scholar] [CrossRef]
- Deschamps, S.; Zhang, Y.; Llaca, V.; Ye, L.; Sanyal, A.; King, M.; May, G.; Lin, H. A chromosome-scale assembly of the sorghum genome using nanopore sequencing and optical mapping. Nat. Commun. 2018, 9, 4844. [Google Scholar] [CrossRef]
- Rozhmina, T.; Zhuchenko, A., Jr.; Melnikova, N.; Gerasimova, E. Identification of genes and creation of resistance donors to Fusarium wilt disease for breeding fibre-flax and dual use flax. Bull. Agrar. Sci. 2019, 1, 3–10. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Liu, H.; Wu, S.; Li, A.; Ruan, J. SMARTdenovo: A de novo assembler using long noisy reads. Gigabyte 2021. [Google Scholar] [CrossRef]
- Ruan, J.; Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 2020, 17, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Šikić, M. Time- and memory-efficient genome assembly with Raven. Nat. Comput. Sci. 2021, 1, 332–336. [Google Scholar] [CrossRef]
- Li, H. Minimap and miniasm: Fast mapping and de novo assembly for noisy long sequences. Bioinformatics 2016, 32, 2103–2110. [Google Scholar] [CrossRef] [PubMed]
- Shafin, K.; Pesout, T.; Lorig-Roach, R.; Haukness, M.; Olsen, H.E.; Bosworth, C.; Armstrong, J.; Tigyi, K.; Maurer, N.; Koren, S.; et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotechnol. 2020, 38, 1044–1053. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Assembler | QUAST | BUSCO | QUAST (Reference = CDC Bethune, GCA_000224295.1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Length, Mb | Contigs | N50, Mb | L50 | GC, % | C, % | D, % | F, % | Genome Fraction, % | Duplication Ratio | Genomic Features | |
| Canu | 447.4 | 1728 | 6.2 | 26 | 38.8 | 93.3 | 59.8 | 0.8 | 93.9 | 1.11 | 8123 + 20,506 |
| Flye | 345.1 | 7720 | 0.3 | 179 | 39.3 | 91.8 | 48.0 | 1.8 | 91.5 | 1.20 | 7997 + 20,858 |
| Flye (from Canu-corrected reads) | 335.9 | 1571 | 5.8 | 21 | 39.1 | 93.8 | 61.8 | 0.9 | 93.9 | 1.06 | 8219 + 20,309 |
| Miniasm | 337.6 | 1104 | 0.6 | 108 | 39.0 | 21.7 | 20.7 | 11.2 | 57.8 | 1.07 | 1957 + 22,130 |
| NextDenovo | 289.5 | 248 | 3.1 | 26 | 39.4 | 91.1 | 44.5 | 1.3 | 83.3 | 1.05 | 7117 + 18,732 |
| Raven | 269.3 | 1722 | 0.2 | 328 | 39.4 | 89.7 | 29.8 | 1.5 | 72.4 | 1.06 | 4860 + 19,855 |
| Shasta | 372.5 | 6952 | 1.5 | 67 | 38.7 | 93.2 | 57.9 | 0.8 | 91.0 | 1.04 | 7212 + 20,962 |
| SMARTdenovo | 163.0 | 110 | 2.8 | 21 | 39.2 | 62.9 | 16.6 | 1.9 | 48.9 | 1.03 | 4027 + 11,827 |
| Wtdbg2 | 243.7 | 3678 | 0.2 | 229 | 40.0 | 74.5 | 6.9 | 4.0 | 61.7 | 1.10 | 214 + 2633 |
| Assembler + (Polisher) | Polisher | QUAST | BUSCO | QUAST (Reference = CDC Bethune, GCA_000224295.1) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Length, Mb | Contigs | N50, Mb | C, % | D, % | F, % | Genomic Features | Mismatches per 100 kbp | Indels per 100 kbp | ||
| Canu | - | 447.4 | 1728 | 6.2 | 93.3 | 59.8 | 0.8 | 8123 + 20,506 part | 1210.3 | 366.4 |
| Medaka | 448.5 | 1728 | 6.2 | 93.8 | 62.3 | 0.8 | 8403 + 20,441 part | 1110.3 | 268.9 | |
| Medaka ×2 | 448.6 | 1728 | 6.2 | 93.8 | 62.3 | 0.7 | 8448 + 20,437 part | 1094.1 | 264.8 | |
| NextPolish | 450.2 | 1728 | 6.2 | 93.0 | 54.1 | 1.0 | 8150 + 20,705 part | 1201.9 | 342.6 | |
| NextPolish ×2 | 449.6 | 1728 | 6.2 | 93.7 | 60.3 | 0.8 | 8342 + 20,548 part | 1163.5 | 257.4 | |
| Pepper | 424.4 | 1720 | 6.3 | 93.7 | 62.0 | 0.9 | 8446 + 20,402 part | 1073.4 | 229.8 | |
| Pepper ×2 | 410.9 | 1661 | 6.4 | 93.8 | 62.2 | 0.9 | 8472 + 20,404 part | 1074.5 | 213.9 | |
| Racon | 447.6 | 1701 | 6.2 | 93.8 | 61.1 | 0.7 | 8349 + 20,479 part | 1156.2 | 253.5 | |
| Racon ×2 | 446.7 | 1695 | 6.2 | 93.6 | 61.1 | 0.7 | 8384 + 20,499 part | 1143.3 | 247.4 | |
| Canu, Pepper | Medaka | 425.0 | 1715 | 6.3 | 93.8 | 62.2 | 0.9 | 8508 + 20,382 part | 1061.8 | 214.3 |
| Canu, Pepper ×2 | 411.3 | 1661 | 6.4 | 93.8 | 62.5 | 0.9 | 8502 + 20,392 part | 1061.0 | 203.4 | |
| Canu, Racon | 447.9 | 1701 | 6.2 | 93.8 | 62.2 | 0.7 | 8448 + 20,436 part | 1122.0 | 225.3 | |
| Canu, Racon ×2 | 447.1 | 1695 | 6.2 | 93.8 | 62.3 | 0.7 | 8483 + 20,419 part | 1115.3 | 222.5 | |
| Assembly | Length, Mb | Contigs | N50, Mb | BUSCO | Total Interspersed Repeats, % | ||
|---|---|---|---|---|---|---|---|
| C, % | D, % | F, % | |||||
| 3896 | 447.1 | 1695 | 6.2 | 93.8 | 62.3 | 0.7 | 49.3 |
| Atlant (GCA_014858635.1) | 361.8 | 2458 | 0.4 | 94.4 | 63.4 | 0.7 | 44.7 |
| CDC Bethune v.1 (GCA_000224295.1) | 282.2 | 48,397 | 0.02 | 93.9 | 60.4 | 1.3 | 33.3 |
| CDC Bethune v.2 (GCA_000224295.2) | 316.2 | 24,829 | 0.02 | 93.7 | 57.4 | 0.9 | 27.7 |
| longya 10 (GCA_010665275.2) | 306.4 | 4419 | 0.2 | 94.4 | 60.5 | 0.9 | 36.0 |
| Heiya 14 (GCA_010665265.1) | 303.7 | 4581 | 0.3 | 94.5 | 62.6 | 0.9 | 36.1 |
| YY5 v.2 (https://zenodo.org/record/4872894) | 455.0 | 336 | 9.6 | 94.5 | 63.1 | 0.7 | 50.1 |
| L. bienne (GCA_010665285.1) | 293.6 | 6369 | 0.1 | 93.3 | 50.4 | 1.3 | 36.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dvorianinova, E.M.; Bolsheva, N.L.; Pushkova, E.N.; Rozhmina, T.A.; Zhuchenko, A.A.; Novakovskiy, R.O.; Povkhova, L.V.; Sigova, E.A.; Zhernova, D.A.; Borkhert, E.V.; et al. Isolating Linum usitatissimum L. Nuclear DNA Enabled Assembling High-Quality Genome. Int. J. Mol. Sci. 2022, 23, 13244. https://doi.org/10.3390/ijms232113244
Dvorianinova EM, Bolsheva NL, Pushkova EN, Rozhmina TA, Zhuchenko AA, Novakovskiy RO, Povkhova LV, Sigova EA, Zhernova DA, Borkhert EV, et al. Isolating Linum usitatissimum L. Nuclear DNA Enabled Assembling High-Quality Genome. International Journal of Molecular Sciences. 2022; 23(21):13244. https://doi.org/10.3390/ijms232113244
Chicago/Turabian StyleDvorianinova, Ekaterina M., Nadezhda L. Bolsheva, Elena N. Pushkova, Tatiana A. Rozhmina, Alexander A. Zhuchenko, Roman O. Novakovskiy, Liubov V. Povkhova, Elizaveta A. Sigova, Daiana A. Zhernova, Elena V. Borkhert, and et al. 2022. "Isolating Linum usitatissimum L. Nuclear DNA Enabled Assembling High-Quality Genome" International Journal of Molecular Sciences 23, no. 21: 13244. https://doi.org/10.3390/ijms232113244
APA StyleDvorianinova, E. M., Bolsheva, N. L., Pushkova, E. N., Rozhmina, T. A., Zhuchenko, A. A., Novakovskiy, R. O., Povkhova, L. V., Sigova, E. A., Zhernova, D. A., Borkhert, E. V., Kaluzhny, D. N., Melnikova, N. V., & Dmitriev, A. A. (2022). Isolating Linum usitatissimum L. Nuclear DNA Enabled Assembling High-Quality Genome. International Journal of Molecular Sciences, 23(21), 13244. https://doi.org/10.3390/ijms232113244