

Solutions to a Radical Problem: Overview of Current and Future Treatment Strategies in Leber’s Hereditary Opic Neuropathy


Abstract
1. Introduction
2. Natural Disease Course
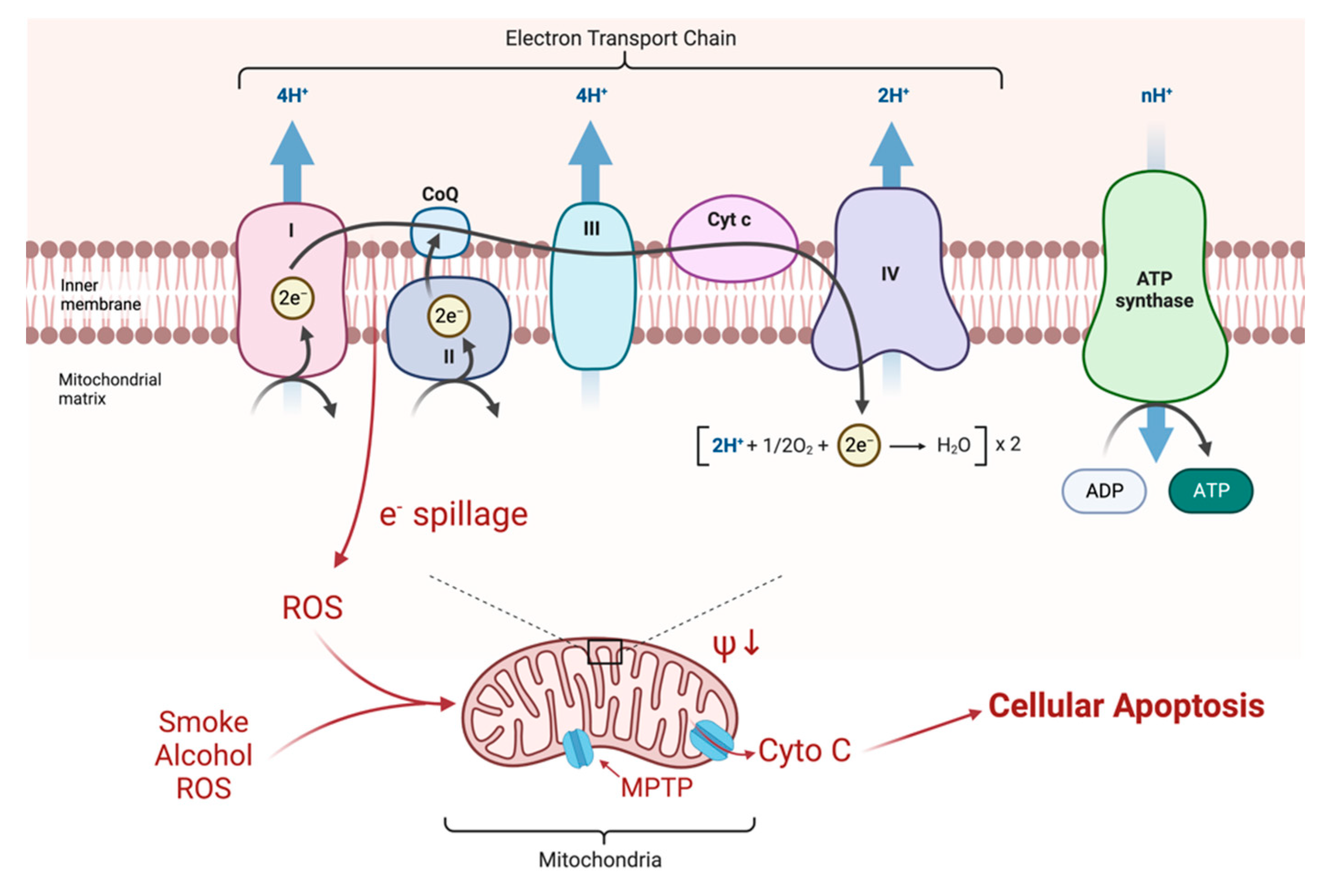
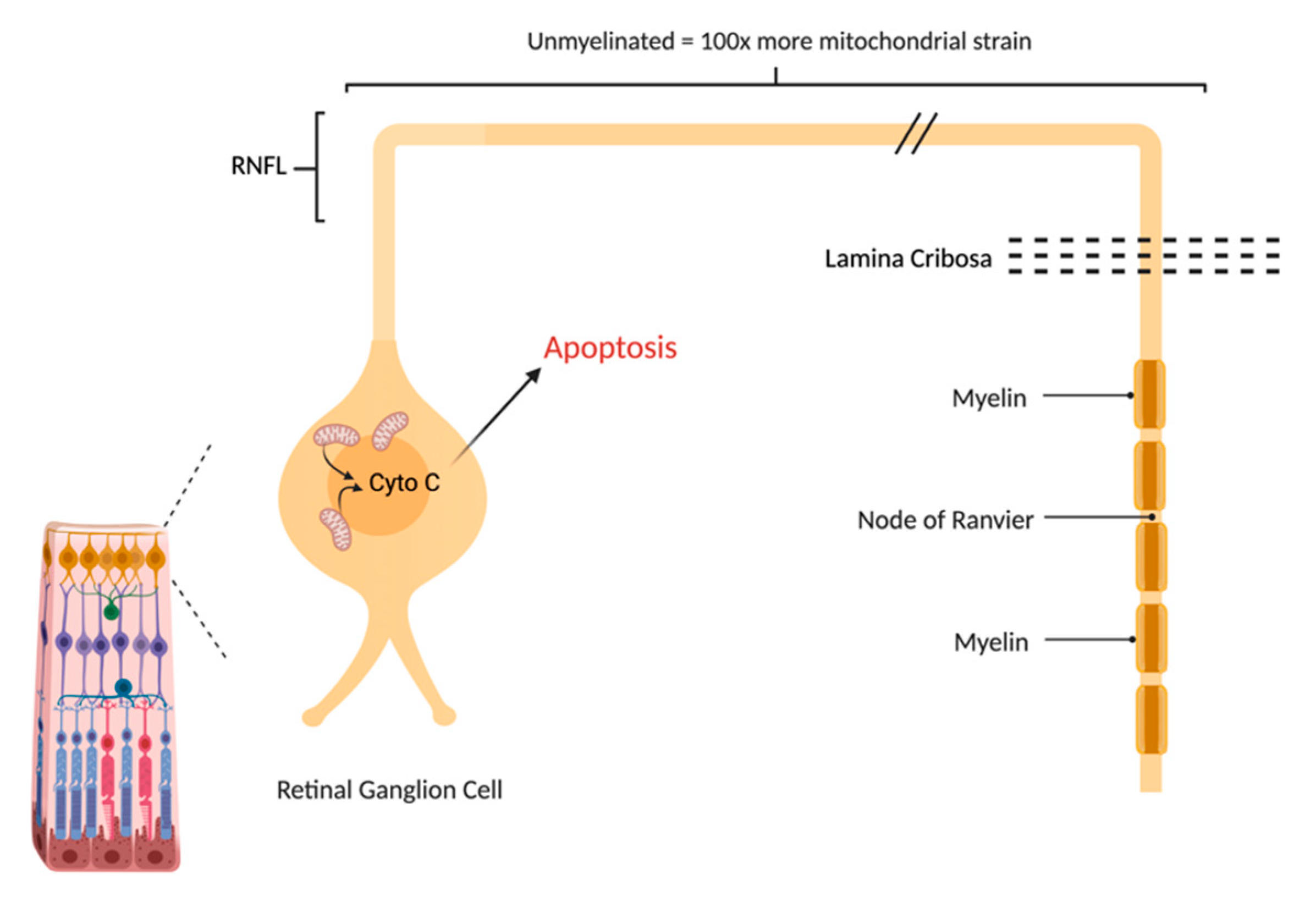
3. Molecular Background and Pathophysiology
4. Unaffected Carriers and Pre-Symptomatic/Pre-Clinical
4.1. Genetics and Counselling
4.2. Neurotoxins
4.3. Mitochondrial Biogenesis, Autophagy, and Transmitophagy
4.4. Estrogens
5. Symptomatic Phase
5.1. Antioxidants
5.2. Coenzyme Q10 (Coq10)
5.3. Idebenone
5.4. Elampritide
5.5. EPI-743
5.6. Cyclosporine
5.7. Light Therapy and Electrical Stimulation
5.8. Gene Therapy
6. Chronic Phase
6.1. Supportive Therapy
6.2. Psychological Counseling
6.3. Induced Pluripotent Stem Cells
6.4. Mitochondrial Gene Editing
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Man, P.; Griffiths, P.; Brown, D.; Howell, N.; Turnbull, D.; Chinnery, P. The Epidemiology of Leber Hereditary Optic Neuropathy in the North East of England. Am. J. Hum. Genet. 2003, 72, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; Carbonelli, M.; Irenaeus, F.; Kawasaki, A.; Klopstock, T.; Lagrèze, W.A.; La Morgia, C.; Newman, N.J.; Orssaud, C.; Pott, J.W.R. International Consensus Statement on the Clinical and Therapeutic Management of Leber Hereditary Optic Neuropathy. J. Neuro-Ophthalmol. 2017, 37, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Huoponen, K.; Vilkki, J.; Aula, P.; Nikoskelainen, E.K.; Savontaus, M. A New MtDNA Mutation Associated with Leber Hereditary Optic Neuroretinopathy. Am. J. Hum. Genet. 1991, 48, 1147. [Google Scholar] [PubMed]
- Johns, D.R.; Neufeld, M.J.; Park, R.D. An ND-6 Mitochondrial DNA Mutation Associated with Leber Hereditary Optic Neuropathy. Biochem. Biophys. Res. Commun. 1992, 187, 1551–1557. [Google Scholar] [CrossRef]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J.; Nikoskelainen, E.K. Mitochondrial DNA Mutation Associated with Leber’s Hereditary Optic Neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef]
- Sadun, A.A.; Salomao, S.R.; Berezovsky, A.; Sadun, F.; DeNegri, A.M.; Quiros, P.A.; Chicani, F.; Ventura, D.; Barboni, P.; Sherman, J. Subclinical Carriers and Conversions in Leber Hereditary Optic Neuropathy: A Prospective Psychophysical Study. Trans. Am. Ophthalmol. Soc. 2006, 104, 51. [Google Scholar]
- Savini, G.; Barboni, P.; Valentino, M.L.; Montagna, P.; Cortelli, P.; De Negri, A.M.; Sadun, F.; Bianchi, S.; Longanesi, L.; Zanini, M. Retinal Nerve Fiber Layer Evaluation by Optical Coherence Tomography in Unaffected Carriers with Leber’s Hereditary Optic Neuropathy Mutations. Ophthalmology 2005, 112, 127–131. [Google Scholar] [CrossRef]
- Balducci, N.; Cascavilla, M.L.; Ciardella, A.; La Morgia, C.; Triolo, G.; Parisi, V.; Bandello, F.; Sadun, A.A.; Carelli, V.; Barboni, P. Peripapillary Vessel Density Changes in Leber’s Hereditary Optic Neuropathy: A New Biomarker. Clin. Exp. Ophthalmol. 2018, 46, 1055–1062. [Google Scholar] [CrossRef]
- Jia, Y.; Simonett, J.M.; Wang, J.; Hua, X.; Liu, L.; Hwang, T.S.; Huang, D. Wide-Field OCT Angiography Investigation of the Relationship between Radial Peripapillary Capillary Plexus Density and Nerve Fiber Layer Thickness. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5188–5194. [Google Scholar] [CrossRef]
- Silva, M.; Llòria, X.; Catarino, C.; Klopstock, T. Natural History Findings from a Large Cohort of Patients with Lebers Hereditary Optic Neuropathy (LHON): New Insights into the Natural Disease Course. Acta Ophthalmol. 2018, 96, 117. [Google Scholar]
- Man, P.Y.W.; Turnbull, D.; Chinnery, P. Leber Hereditary Optic Neuropathy. J. Med. Genet. 2002, 39, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; d’Adamo, P.; Valentino, M.L.; La Morgia, C.; Ross-Cisneros, F.N.; Caporali, L.; Maresca, A.; Loguercio Polosa, P.; Barboni, P.; De Negri, A. Parsing the Differences in Affected with LHON: Genetic versus Environmental Triggers of Disease Conversion. Brain 2016, 139, e17. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.J.; Carelli, V.; Taiel, M.; Yu-Wai-Man, P. Visual Outcomes in Leber Hereditary Optic Neuropathy Patients with the m. 11778G>A (MTND4) Mitochondrial DNA Mutation. J. Neuro-Ophthalmol. 2020, 40, 547–557. [Google Scholar] [CrossRef] [PubMed]
- do VF Ramos, C.; Bellusci, C.; Savini, G.; Carbonelli, M.; Berezovsky, A.; Tamaki, C.; Cinoto, R.; Sacai, P.Y.; Moraes-Filho, M.N.; Miura, H.M. Association of Optic Disc Size with Development and Prognosis of Leber’s Hereditary Optic Neuropathy. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1666–1674. [Google Scholar] [CrossRef] [PubMed]
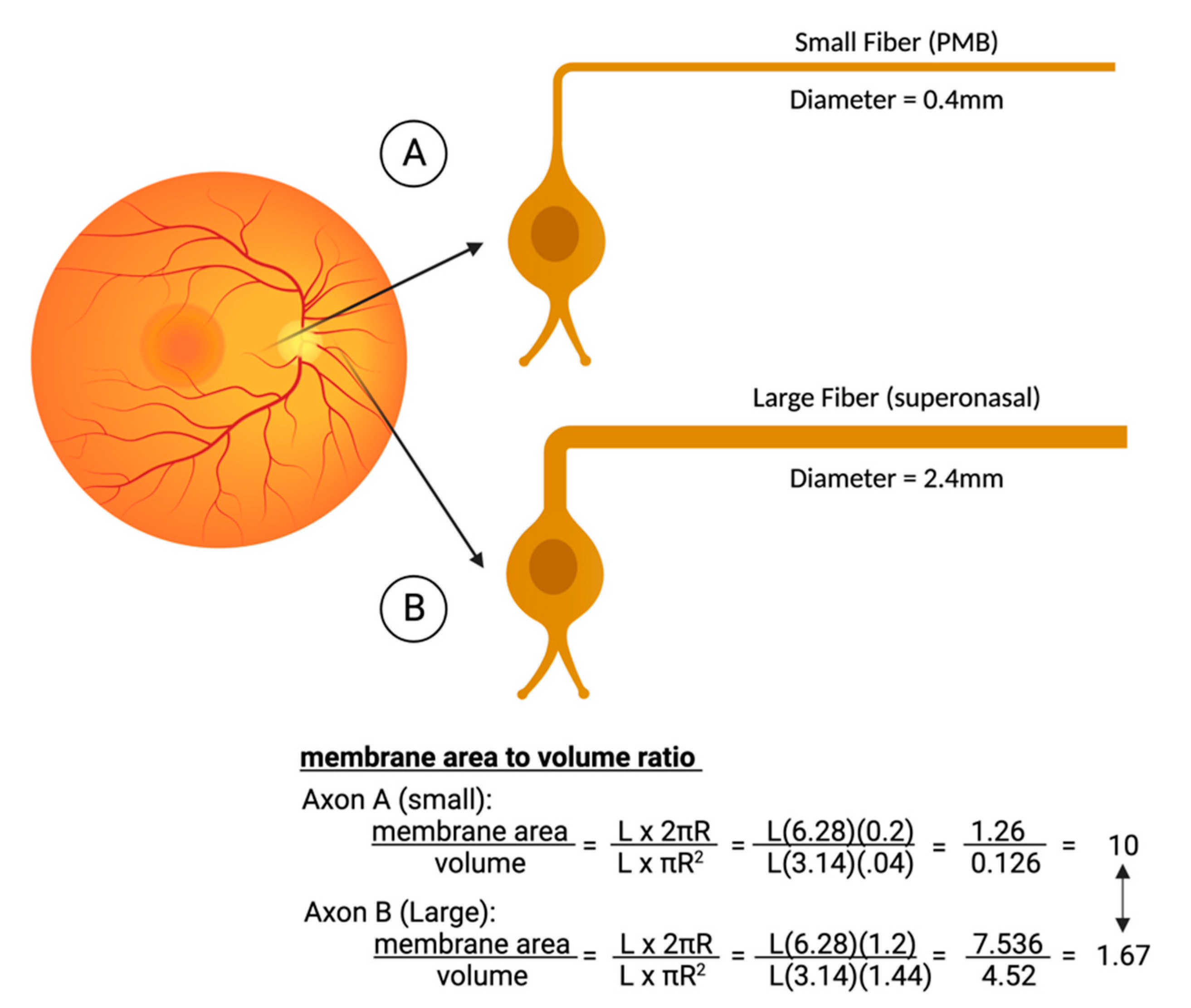
- Pan, B.X.; Ross-Cisneros, F.N.; Carelli, V.; Rue, K.S.; Salomao, S.R.; Moraes-Filho, M.N.; Moraes, M.N.; Berezovsky, A.; Belfort, R.; Sadun, A.A. Mathematically Modeling the Involvement of Axons in Leber’s Hereditary Optic Neuropathy. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7608–7617. [Google Scholar] [CrossRef] [PubMed]
- Sadun, A.A.; La Morgia, C.; Carelli, V. Mitochondrial Optic Neuropathies: Our Travels from Bench to Bedside and Back Again. Clin. Exp. Ophthalmol. 2013, 41, 702–712. [Google Scholar] [CrossRef]
- Sadun, A.A.; Win, P.H.; Ross-Cisneros, F.; Walker, S.; Carelli, V. Leber’s Hereditary Optic Neuropathy Differentially Affects Smaller Axons in the Optic Nerve. Trans. Am. Ophthalmol. Soc. 2000, 98, 223. [Google Scholar]
- Yu-Wai-Man, P.; Newman, N.J.; Carelli, V.; La Morgia, C.; Biousse, V.; Bandello, F.M.; Clermont, C.V.; Campillo, L.C.; Leruez, S.; Moster, M.L. Natural History of Patients with Leber Hereditary Optic Neuropathy—Results from the REALITY Study. Eye 2022, 36, 818–826. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Chinnery, P.F. Leber Hereditary Optic Neuropathy. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Spruijt, L.; Kolbach, D.N.; Rene, F.; Plomp, A.S.; Bauer, N.J.; Smeets, H.J.; de Die-Smulders, C.E. Influence of Mutation Type on Clinical Expression of Leber Hereditary Optic Neuropathy. Am. J. Ophthalmol. 2006, 141, 676. [Google Scholar] [CrossRef]
- Majander, A.; Bowman, R.; Poulton, J.; Antcliff, R.J.; Reddy, M.A.; Michaelides, M.; Webster, A.R.; Chinnery, P.F.; Votruba, M.; Moore, A.T. Childhood-Onset Leber Hereditary Optic Neuropathy. Br. J. Ophthalmol. 2017, 101, 1505–1509. [Google Scholar] [CrossRef]
- Caporali, L.; Maresca, A.; Capristo, M.; Del Dotto, V.; Tagliavini, F.; Valentino, M.L.; La Morgia, C.; Carelli, V. Incomplete Penetrance in Mitochondrial Optic Neuropathies. Mitochondrion 2017, 36, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.; Fearnley, I.M.; Shannon, R.J.; Hirst, J.; Walker, J.E. Analysis of the Subunit Composition of Complex I from Bovine Heart Mitochondria* S. Mol. Cell. Proteom. 2003, 2, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; Ross-Cisneros, F.N.; Sadun, A.A. Mitochondrial Dysfunction as a Cause of Optic Neuropathies. Prog. Retin. Eye Res. 2004, 23, 53–89. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.-Y.; Cringle, S.J. Oxygen Distribution and Consumption within the Retina in Vascularised and Avascular Retinas and in Animal Models of Retinal Disease. Prog. Retin. Eye Res. 2001, 20, 175–208. [Google Scholar] [CrossRef]
- Adapted from “Electron Transport Chain”. by BioRender.Com. 2022. Available online: https://app.biorender.com/biorender-templates (accessed on 1 September 2022).
- Kang, E.; Wu, J.; Gutierrez, N.M.; Koski, A.; Tippner-Hedges, R.; Agaronyan, K.; Platero-Luengo, A.; Martinez-Redondo, P.; Ma, H.; Lee, Y. Mitochondrial Replacement in Human Oocytes Carrying Pathogenic Mitochondrial DNA Mutations. Nature 2016, 540, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Silva-Pinheiro, P.; Minczuk, M. The Potential of Mitochondrial Genome Engineering. Nat. Rev. Genet. 2022, 23, 199–214. [Google Scholar] [CrossRef]
- Gómez-Durán, A.; Pacheu-Grau, D.; Martínez-Romero, Í.; López-Gallardo, E.; López-Pérez, M.J.; Montoya, J.; Ruiz-Pesini, E. Oxidative Phosphorylation Differences between Mitochondrial DNA Haplogroups Modify the Risk of Leber’s Hereditary Optic Neuropathy. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 1216–1222. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Hudson, G.; Klopstock, T.; Chinnery, P.F. Reply: Parsing the Differences in Affected with LHON: Genetic versus Environmental Triggers of Disease Conversion. Brain 2016, 139, e18. [Google Scholar] [CrossRef]
- Giordano, L.; Deceglie, S.; d’Adamo, P.; Valentino, M.; La Morgia, C.; Fracasso, F.; Roberti, M.; Cappellari, M.; Petrosillo, G.; Ciaravolo, S. Cigarette Toxicity Triggers Leber’s Hereditary Optic Neuropathy by Affecting MtDNA Copy Number, Oxidative Phosphorylation and ROS Detoxification Pathways. Cell Death Dis. 2015, 6, e2021. [Google Scholar] [CrossRef]
- Zaslavsky, K.; Margolin, E.A. Leber’s Hereditary Optic Neuropathy in Older Individuals Because of Increased Alcohol Consumption During the COVID-19 Pandemic. J. Neuro-Ophthalmol. 2021, 41, 316–320. [Google Scholar] [CrossRef]
- Giordano, C.; Iommarini, L.; Giordano, L.; Maresca, A.; Pisano, A.; Valentino, M.L.; Caporali, L.; Liguori, R.; Deceglie, S.; Roberti, M. Efficient Mitochondrial Biogenesis Drives Incomplete Penetrance in Leber’s Hereditary Optic Neuropathy. Brain 2014, 137, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Bahr, T.; Welburn, K.; Donnelly, J.; Bai, Y. Emerging Model Systems and Treatment Approaches for Leber’s Hereditary Optic Neuropathy: Challenges and Opportunities. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165743. [Google Scholar] [CrossRef] [PubMed]
- Uittenbogaard, M.; Chiaramello, A. Mitochondrial Biogenesis: A Therapeutic Target for Neurodevelopmental Disorders and Neurodegenerative Diseases. Curr. Pharm. Des. 2014, 20, 5574–5593. [Google Scholar] [CrossRef]
- Danese, A.; Patergnani, S.; Maresca, A.; Peron, C.; Raimondi, A.; Caporali, L.; Marchi, S.; La Morgia, C.; Del Dotto, V.; Zanna, C. Pathological Mitophagy Disrupts Mitochondrial Homeostasis in Leber’s Hereditary Optic Neuropathy. Cell Rep. 2022, 40, 111124. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, R.; Pirinen, E.; Lamperti, C.; Marchet, S.; Sauve, A.A.; Li, W.; Leoni, V.; Schon, E.A.; Dantzer, F.; Auwerx, J. NAD+-Dependent Activation of Sirt1 Corrects the Phenotype in a Mouse Model of Mitochondrial Disease. Cell. Metab. 2014, 19, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Garone, C.; Viscomi, C. Towards a Therapy for Mitochondrial Disease: An Update. Biochem. Soc. Trans. 2018, 46, 1247–1261. [Google Scholar] [CrossRef]
- Pirinen, E.; Auranen, M.; Khan, N.A.; Brilhante, V.; Urho, N.; Pessia, A.; Hakkarainen, A.; Kuula, J.; Heinonen, U.; Schmidt, M.S. Niacin Cures Systemic NAD+ Deficiency and Improves Muscle Performance in Adult-Onset Mitochondrial Myopathy. Cell Metab. 2020, 31, 1078–1090. [Google Scholar] [CrossRef]
- Ruiz-Pesini, E.; Emperador, S.; López-Gallardo, E.; Hernández-Ainsa, C.; Montoya, J. Increasing MtDNA Levels as Therapy for Mitochondrial Optic Neuropathies. Drug Discov. Today 2018, 23, 493–498. [Google Scholar] [CrossRef]
- Giordano, C.; Montopoli, M.; Perli, E.; Orlandi, M.; Fantin, M.; Ross-Cisneros, F.N.; Caparrotta, L.; Martinuzzi, A.; Ragazzi, E.; Ghelli, A. Oestrogens Ameliorate Mitochondrial Dysfunction in Leber’s Hereditary Optic Neuropathy. Brain 2011, 134, 220–234. [Google Scholar] [CrossRef]
- Pisano, A.; Preziuso, C.; Iommarini, L.; Perli, E.; Grazioli, P.; Campese, A.F.; Maresca, A.; Montopoli, M.; Masuelli, L.; Sadun, A.A. Targeting Estrogen Receptor β as Preventive Therapeutic Strategy for Leber’s Hereditary Optic Neuropathy. Hum. Mol. Genet. 2015, 24, 6921–6931. [Google Scholar] [CrossRef]
- Sun, J.; Zhu, H.; Wang, X.; Gao, Q.; Li, Z.; Huang, H. CoQ10 Ameliorates Mitochondrial Dysfunction in Diabetic Nephropathy through Mitophagy. J. Endocrinol. 2019, 240, 445–465. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L. Protective Effects of Trimetazidine and Coenzyme Q10 on Cisplatin-Induced Cardiotoxicity by Alleviating Oxidative Stress and Mitochondrial Dysfunction. Anatol. J. Cardiol. 2019, 22, 232. [Google Scholar] [CrossRef] [PubMed]
- Lyseng-Williamson, K.A. Idebenone: A Review in Leber’s Hereditary Optic Neuropathy. Drugs 2016, 76, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Jaber, S.M.; Shealinna, X.G.; Milstein, J.L.; VanRyzin, J.W.; Waddell, J.; Polster, B.M. Idebenone Has Distinct Effects on Mitochondrial Respiration in Cortical Astrocytes Compared to Cortical Neurons Due to Differential NQO1 Activity. J. Neurosci. 2020, 40, 4609–4619. [Google Scholar] [CrossRef] [PubMed]
- Amore, G.; Romagnoli, M.; Carbonelli, M.; Barboni, P.; Carelli, V.; La Morgia, C. Therapeutic Options in Hereditary Optic Neuropathies. Drugs 2021, 81, 57–86. [Google Scholar] [CrossRef]
- Varricchio, C.; Beirne, K.; Heard, C.; Newland, B.; Rozanowska, M.; Brancale, A.; Votruba, M. The Ying and Yang of Idebenone: Not Too Little, Not Too Much–Cell Death in NQO1 Deficient Cells and the Mouse Retina. Free Radic. Biol. Med. 2020, 152, 551–560. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Soiferman, D.; Moore, D.G.; Burté, F.; Saada, A. Evaluating the Therapeutic Potential of Idebenone and Related Quinone Analogues in Leber Hereditary Optic Neuropathy. Mitochondrion 2017, 36, 36–42. [Google Scholar] [CrossRef]
- Heitz, F.D.; Erb, M.; Anklin, C.; Robay, D.; Pernet, V.; Gueven, N. Idebenone Protects against Retinal Damage and Loss of Vision in a Mouse Model of Leber’s Hereditary Optic Neuropathy. PLoS ONE 2012, 7, e45182. [Google Scholar] [CrossRef]
- Klopstock, T.; Yu-Wai-Man, P.; Dimitriadis, K.; Rouleau, J.; Heck, S.; Bailie, M.; Atawan, A.; Chattopadhyay, S.; Schubert, M.; Garip, A. A Randomized Placebo-Controlled Trial of Idebenone in Leber’s Hereditary Optic Neuropathy. Brain 2011, 134, 2677–2686. [Google Scholar] [CrossRef]
- Carelli, V.; La Morgia, C.; Valentino, M.L.; Rizzo, G.; Carbonelli, M.; De Negri, A.M.; Sadun, F.; Carta, A.; Guerriero, S.; Simonelli, F. Idebenone Treatment in Leber’s Hereditary Optic Neuropathy. Brain 2011, 134, e188. [Google Scholar] [CrossRef]
- Van Everdingen, J.A.; Tjon-Fo-Sang, M.; van den Born, L.; Pott, J.W.R. New Treatment Option for Leber Hereditary Optic Neuropathy: Early Diagnosis Is Required. Ned. Tijdschr. Voor Geneeskd. 2021, 165, D5444. [Google Scholar]
- Catarino, C.B.; von Livonius, B.; Priglinger, C.; Banik, R.; Matloob, S.; Tamhankar, M.A.; Castillo, L.; Friedburg, C.; Halfpenny, C.A.; Lincoln, J.A. Real-World Clinical Experience with Idebenone in the Treatment of Leber Hereditary Optic Neuropathy. J. Neuro-Ophthalmol. 2020, 40, 558. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Masuda, Y.; Ishikawa, H.; Shikisima, K.; Goseki, T.; Kezuka, T.; Terao, M.; Miyazaki, A.; Matsumoto, K.; Nishikawa, H. Characteristics of Japanese Patients with Leber’s Hereditary Optic Neuropathy and Idebenone Trial: A Prospective, Interventional, Non-Comparative Study. Jpn. J. Ophthalmol. 2021, 65, 133–142. [Google Scholar] [CrossRef]
- Van Everdingen, J.A.; Pott, J.W.R.; Bauer, N.J.; Krijnen, A.M.; Lushchyk, T.; Wubbels, R.J. Clinical Outcomes of Treatment with Idebenone in Leber’s Hereditary Optic Neuropathy in the Netherlands: A National Cohort Study. Acta Ophthalmol. 2022, 100, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H. First-in-class Cardiolipin-protective Compound as a Therapeutic Agent to Restore Mitochondrial Bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef] [PubMed]
- Enns, G.M.; Cohen, B.H. Clinical Trials in Mitochondrial Disease: An Update on EPI-743 and RP103. J. Inborn Errors Metab. Screen. 2019, 5. [Google Scholar] [CrossRef]
- Sadun, A.A.; Chicani, C.F.; Ross-Cisneros, F.N.; Barboni, P.; Thoolen, M.; Shrader, W.D.; Kubis, K.; Carelli, V.; Miller, G. Effect of EPI-743 on the Clinical Course of the Mitochondrial Disease Leber Hereditary Optic Neuropathy. Arch. Neurol. 2012, 69, 331–338. [Google Scholar] [CrossRef]
- Leruez, S.; Verny, C.; Bonneau, D.; Procaccio, V.; Lenaers, G.; Amati-Bonneau, P.; Reynier, P.; Scherer, C.; Prundean, A.; Orssaud, C. Cyclosporine A Does Not Prevent Second-Eye Involvement in Leber’s Hereditary Optic Neuropathy. Orphanet J. Rare Dis. 2018, 13, 1–9. [Google Scholar] [CrossRef]
- Eells, J.T.; Wong-Riley, M.T.; VerHoeve, J.; Henry, M.; Buchman, E.V.; Kane, M.P.; Gould, L.J.; Das, R.; Jett, M.; Hodgson, B.D. Mitochondrial Signal Transduction in Accelerated Wound and Retinal Healing by Near-Infrared Light Therapy. Mitochondrion 2004, 4, 559–567. [Google Scholar] [CrossRef]
- Rojas, J.C.; Lee, J.; John, J.M.; Gonzalez-Lima, F. Neuroprotective Effects of Near-Infrared Light in an in Vivo Model of Mitochondrial Optic Neuropathy. J. Neurosci. 2008, 28, 13511–13521. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Xiao, S.; Hua, Z.; Yang, D.; Hu, M.; Zhu, Y.-T.; Zhong, H. Near Infrared (NIR) Light Therapy of Eye Diseases: A Review. Int. J. Med. Sci. 2021, 18, 109. [Google Scholar] [CrossRef] [PubMed]
- Sehic, A.; Guo, S.; Cho, K.-S.; Corraya, R.M.; Chen, D.F.; Utheim, T.P. Electrical Stimulation as a Means for Improving Vision. Am. J. Pathol. 2016, 186, 2783–2797. [Google Scholar] [CrossRef] [PubMed]
- Perin, C.; Viganò, B.; Piscitelli, D.; Matteo, B.M.; Meroni, R.; Cerri, C.G. Non-Invasive Current Stimulation in Vision Recovery: A Review of the Literature. Restor. Neurol. Neurosci. 2020, 38, 239–250. [Google Scholar] [CrossRef]
- Kurimoto, T.; Ueda, K.; Mori, S.; Kamada, S.; Sakamoto, M.; Yamada-Nakanishi, Y.; Matsumiya, W.; Nakamura, M. A Single-Arm, Prospective, Exploratory Study to Preliminarily Test Effectiveness and Safety of Skin Electrical Stimulation for Leber Hereditary Optic Neuropathy. J. Clin. Med. 2020, 9, 1359. [Google Scholar] [CrossRef]
- Bykov, Y.S.; Rapaport, D.; Herrmann, J.M.; Schuldiner, M. Cytosolic Events in the Biogenesis of Mitochondrial Proteins. Trends Biochem. Sci. 2020, 45, 650–667. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.; Augustin, S.; Ellouze, S.; Bénit, P.; Bouaita, A.; Rustin, P.; Sahel, J.-A.; Corral-Debrinski, M. The Optimized Allotopic Expression of ND1 or ND4 Genes Restores Respiratory Chain Complex I Activity in Fibroblasts Harboring Mutations in These Genes. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 1707–1717. [Google Scholar] [CrossRef] [PubMed]
- Moster, M.; Sadun, A.; Klopstock, T.; Newman, N.; Vignal-Clermont, C.; Carelli, V.; Yu-Wai-Man, P.; Biousse, V.; Sergott, R.; Katz, B. rAAV2/2-ND4 for the Treatment of Leber Hereditary Optic Neuropathy (LHON): Final Results from the RESCUE and REVERSE Phase III Clinical Trials and Experimental Data in Nonhuman Primates to Support a Bilateral Effect. Neurology 2020, 94, 2339. [Google Scholar]
- Yu-Wai-Man, P.; Newman, N.J.; Carelli, V.; Moster, M.L.; Biousse, V.; Sadun, A.A.; Klopstock, T.; Vignal-Clermont, C.; Sergott, R.C.; Rudolph, G. Bilateral Visual Improvement with Unilateral Gene Therapy Injection for Leber Hereditary Optic Neuropathy. Sci. Transl. Med. 2020, 12, eaaz7423. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.; Yu-Wai-Man, P.; Carelli, V.; Subramanian, P.; Moster, M.; Wang, A.-G.; Donahue, S.; Leroy, B.; Biousse, V.; Vignal-Clermont, C. The Phase III REFLECT Trial: Efficacy and Safety of Bilateral Gene Therapy for Leber Hereditary Optic Neuropathy (LHON)(P17-12.002). Neurology 2022, 98, 928. [Google Scholar]
- van Nispen, R.M.; Virgili, G.; Hoeben, M.; Langelaan, M.; Klevering, J.; Keunen, J.E.; van Rens, G.H. Low Vision Rehabilitation for Better Quality of Life in Visually Impaired Adults. Cochrane Database Syst. Rev. 2020. [Google Scholar] [CrossRef]
- Chen, B.S.; Holzinger, E.; Taiel, M.; Yu-Wai-Man, P. The Impact of Leber Hereditary Optic Neuropathy on the Quality of Life of Patients and Their Relatives: A Qualitative Study. J. Neuro-Ophthalmol. 2022, 42, 316–322. [Google Scholar] [CrossRef]
- Gale, J.; Khoshnevis, M.; Frousiakis, S.E.; Karanjia, R.; Poincenot, L.; Sadun, A.A.; Baron, D.A. An International Study of Emotional Response to Bilateral Vision Loss Using a Novel Graphical Online Assessment Tool. Psychosomatics 2017, 58, 38–45. [Google Scholar] [CrossRef]
- Garcia, G.A.; Khoshnevis, M.; Gale, J.; Frousiakis, S.E.; Hwang, T.J.; Poincenot, L.; Karanjia, R.; Baron, D.; Sadun, A.A. Profound Vision Loss Impairs Psychological Well-Being in Young and Middle-Aged Individuals. Clin. Ophthalmol. 2017, 11, 417. [Google Scholar] [CrossRef]
- Kurup, M.; Ching, J.; Yu-Wai-Man, P. Vision-Related Quality of Life and Mental Health in Patients with Leber Hereditary Optic Neuropathy. Investig. Ophthalmol. Vis. Sci. 2021, 62, 2393. [Google Scholar]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Cowan, C.S.; Renner, M.; De Gennaro, M.; Gross-Scherf, B.; Goldblum, D.; Hou, Y.; Munz, M.; Rodrigues, T.M.; Krol, J.; Szikra, T. Cell Types of the Human Retina and Its Organoids at Single-Cell Resolution. Cell 2020, 182, 1623–1640. [Google Scholar] [CrossRef]
- Kruczek, K.; Swaroop, A. Pluripotent Stem Cell-Derived Retinal Organoids for Disease Modeling and Development of Therapies. Stem Cells 2020, 38, 1206–1215. [Google Scholar] [CrossRef]
- Laha, B.; Stafford, B.K.; Huberman, A.D. Regenerating Optic Pathways from the Eye to the Brain. Science 2017, 356, 1031–1034. [Google Scholar] [CrossRef]
- Fligor, C.M.; Huang, K.-C.; Lavekar, S.S.; VanderWall, K.B.; Meyer, J.S. Differentiation of Retinal Organoids from Human Pluripotent Stem Cells. In Methods in Cell Biology; Elsevier: Amsterdam, The Netherlands, 2020; Volume 159, pp. 279–302. ISBN 0091-679X. [Google Scholar]
- De Lima, S.; Koriyama, Y.; Kurimoto, T.; Oliveira, J.T.; Yin, Y.; Li, Y.; Gilbert, H.-Y.; Fagiolini, M.; Martinez, A.M.B.; Benowitz, L. Full-Length Axon Regeneration in the Adult Mouse Optic Nerve and Partial Recovery of Simple Visual Behaviors. Proc. Natl. Acad. Sci. USA 2012, 109, 9149–9154. [Google Scholar] [CrossRef]
- Peng, M.; Lam, P.; Machnoor, M.; Paknahad, J.; Iseri, E.; Shao, X.; Shahidi, M.; Thomas, B.; Lazzi, G.; Gokoffski, K. Electric Fields Direct Full-Length Optic Nerve Regeneration and Partial Restoration of Visual Function. Investig. Ophthalmol. Vis. Sci. 2022, 63, 1139. [Google Scholar]
- Morgan, M.A.; Lange, L.; Schambach, A. Prime Time for Base Editing in the Mitochondria. Signal Transduct. Target. Ther. 2022, 7, 1–2. [Google Scholar] [CrossRef]
- Cho, S.-I.; Lee, S.; Mok, Y.G.; Lim, K.; Lee, J.; Lee, J.M.; Chung, E.; Kim, J.-S. Targeted A-to-G Base Editing in Human Mitochondrial DNA with Programmable Deaminases. Cell 2022, 185, 1764–1776. [Google Scholar] [CrossRef]
- Barrera-Paez, J.D.; Moraes, C.T. Mitochondrial Genome Engineering Coming-of-Age. Trends Genet. 2022, 38, 869–880. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Disease Classification |
|---|
| Asymptomatic (mutation carriers) |
| Preclinical |
| Subacute (<6 months) |
| Dynamic (6–12 months) |
| Chronic (>12 months) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spiegel, S.J.; Sadun, A.A. Solutions to a Radical Problem: Overview of Current and Future Treatment Strategies in Leber’s Hereditary Opic Neuropathy. Int. J. Mol. Sci. 2022, 23, 13205. https://doi.org/10.3390/ijms232113205
Spiegel SJ, Sadun AA. Solutions to a Radical Problem: Overview of Current and Future Treatment Strategies in Leber’s Hereditary Opic Neuropathy. International Journal of Molecular Sciences. 2022; 23(21):13205. https://doi.org/10.3390/ijms232113205
Chicago/Turabian StyleSpiegel, Samuel J., and Alfredo A. Sadun. 2022. "Solutions to a Radical Problem: Overview of Current and Future Treatment Strategies in Leber’s Hereditary Opic Neuropathy" International Journal of Molecular Sciences 23, no. 21: 13205. https://doi.org/10.3390/ijms232113205
APA StyleSpiegel, S. J., & Sadun, A. A. (2022). Solutions to a Radical Problem: Overview of Current and Future Treatment Strategies in Leber’s Hereditary Opic Neuropathy. International Journal of Molecular Sciences, 23(21), 13205. https://doi.org/10.3390/ijms232113205