
Enhanced Expression of ARK5 in Hepatic Stellate Cell and Hepatocyte Synergistically Promote Liver Fibrosis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
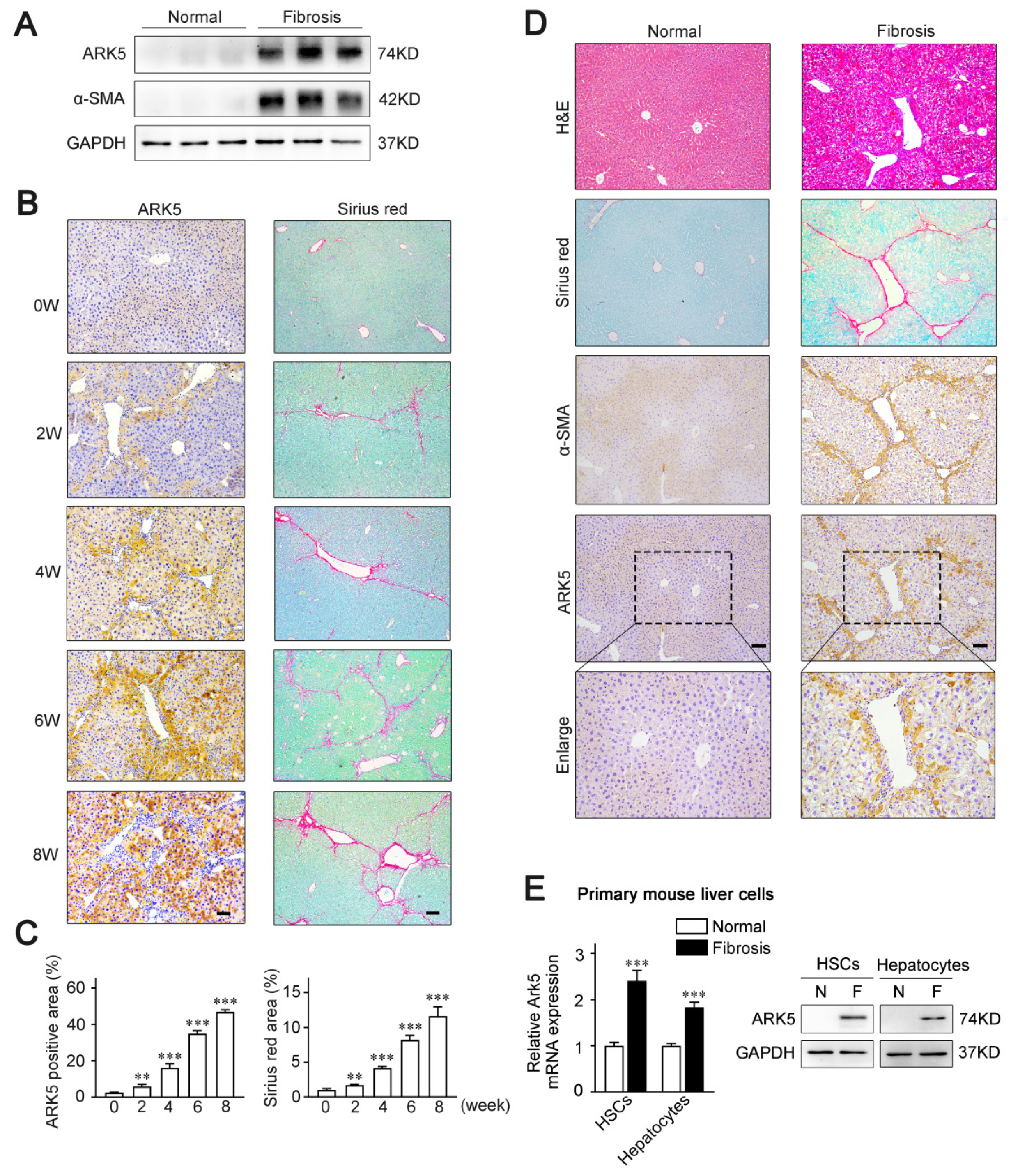
2.1. ARK5 Expression Level Correlates with the Progression of Liver Fibrosis, and Mainly Expresses in HSCs and Hepatocytes
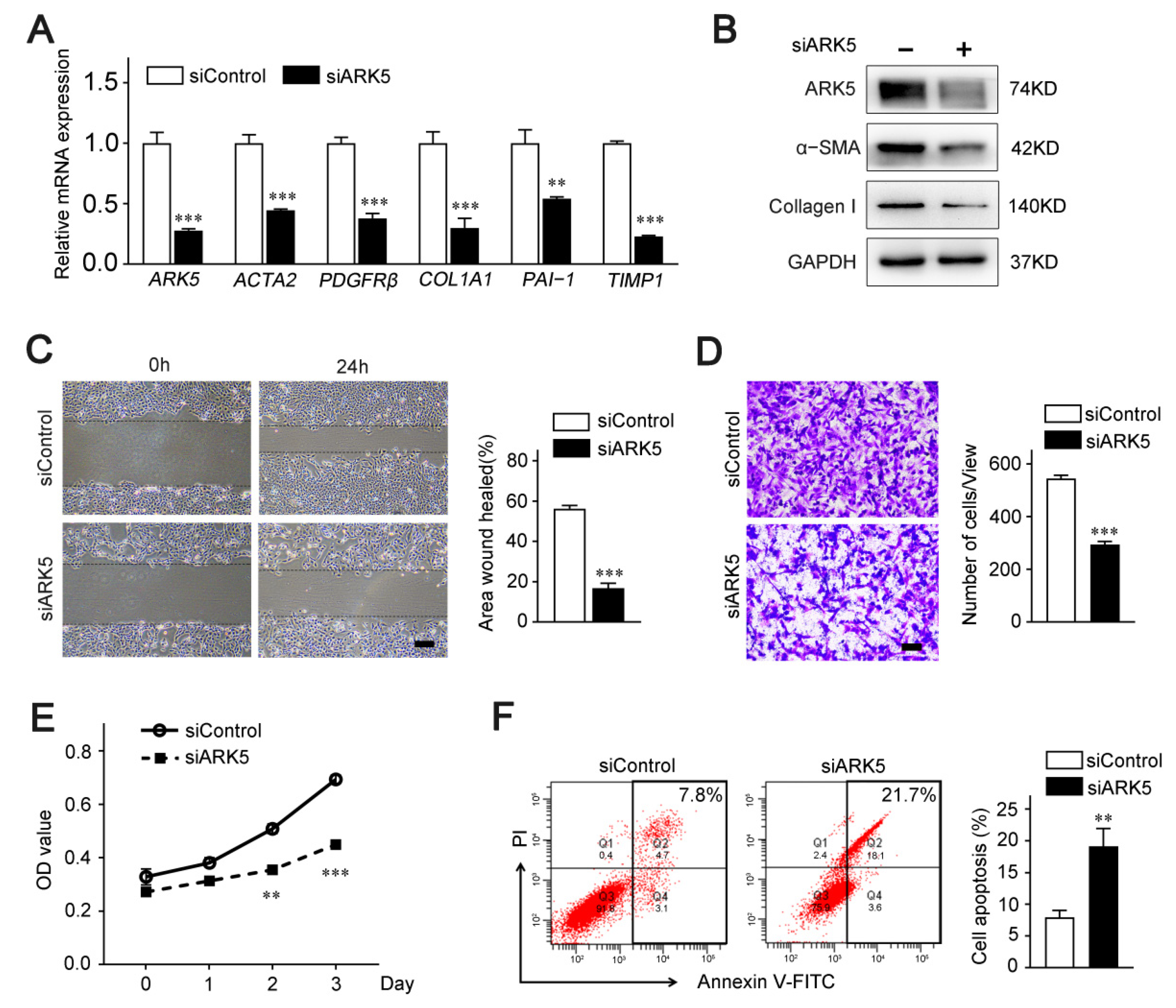
2.2. ARK5 Plays a Crucial Role in the Activation, Proliferation and Survival of HSCs
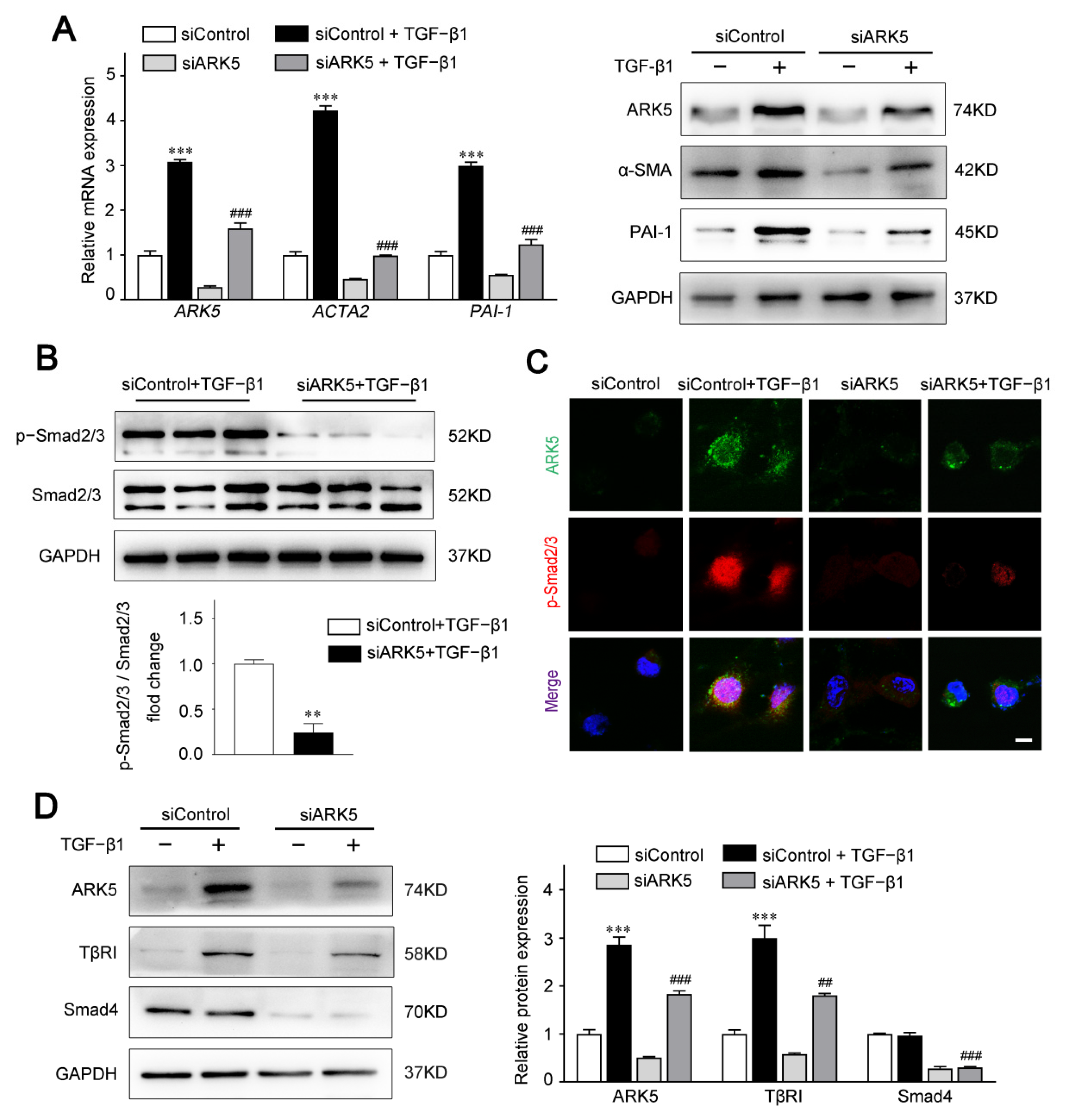
2.3. ARK5 Promotes HSCs Activation via TGFβ-Smad2/3 Activity
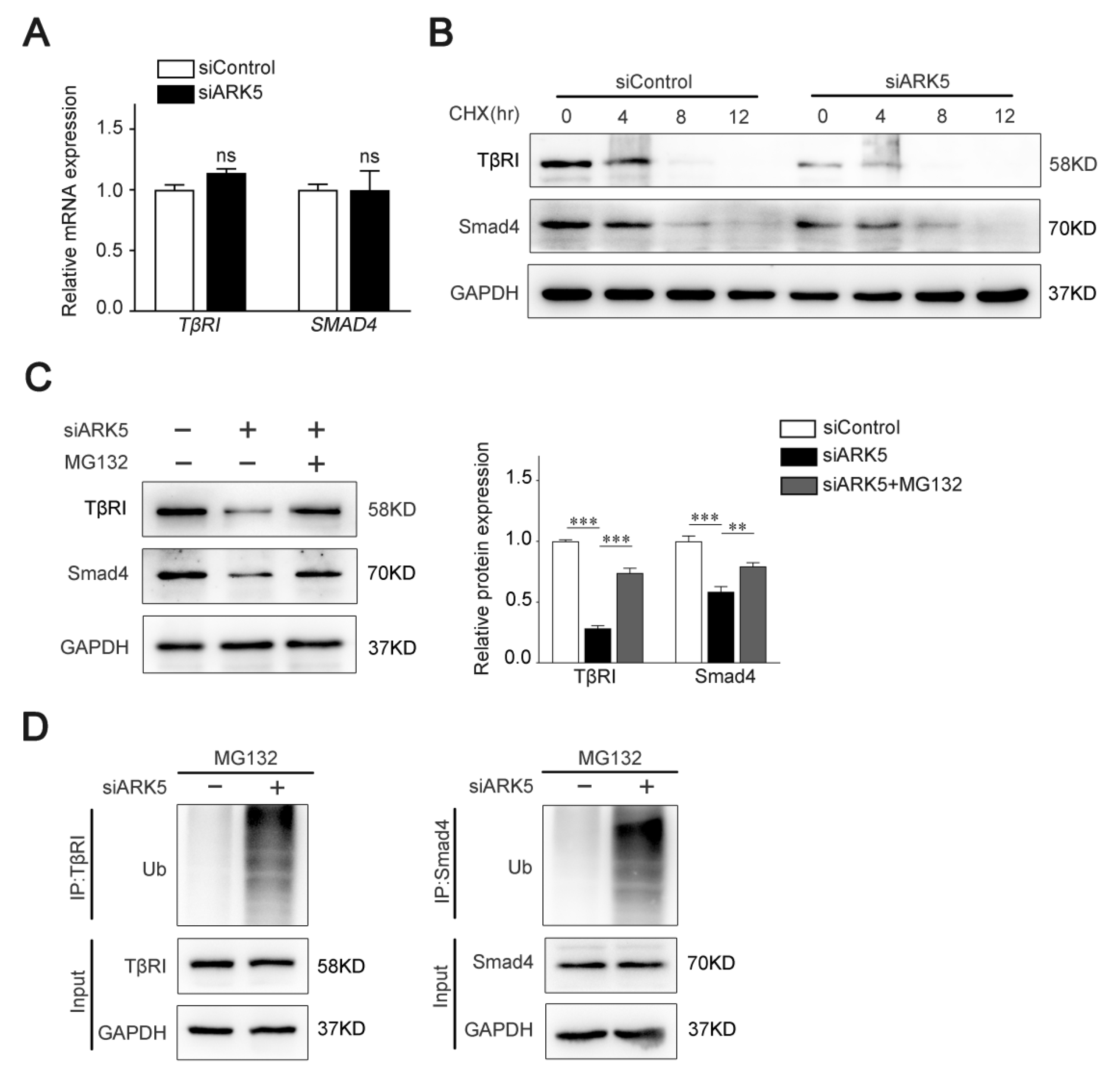
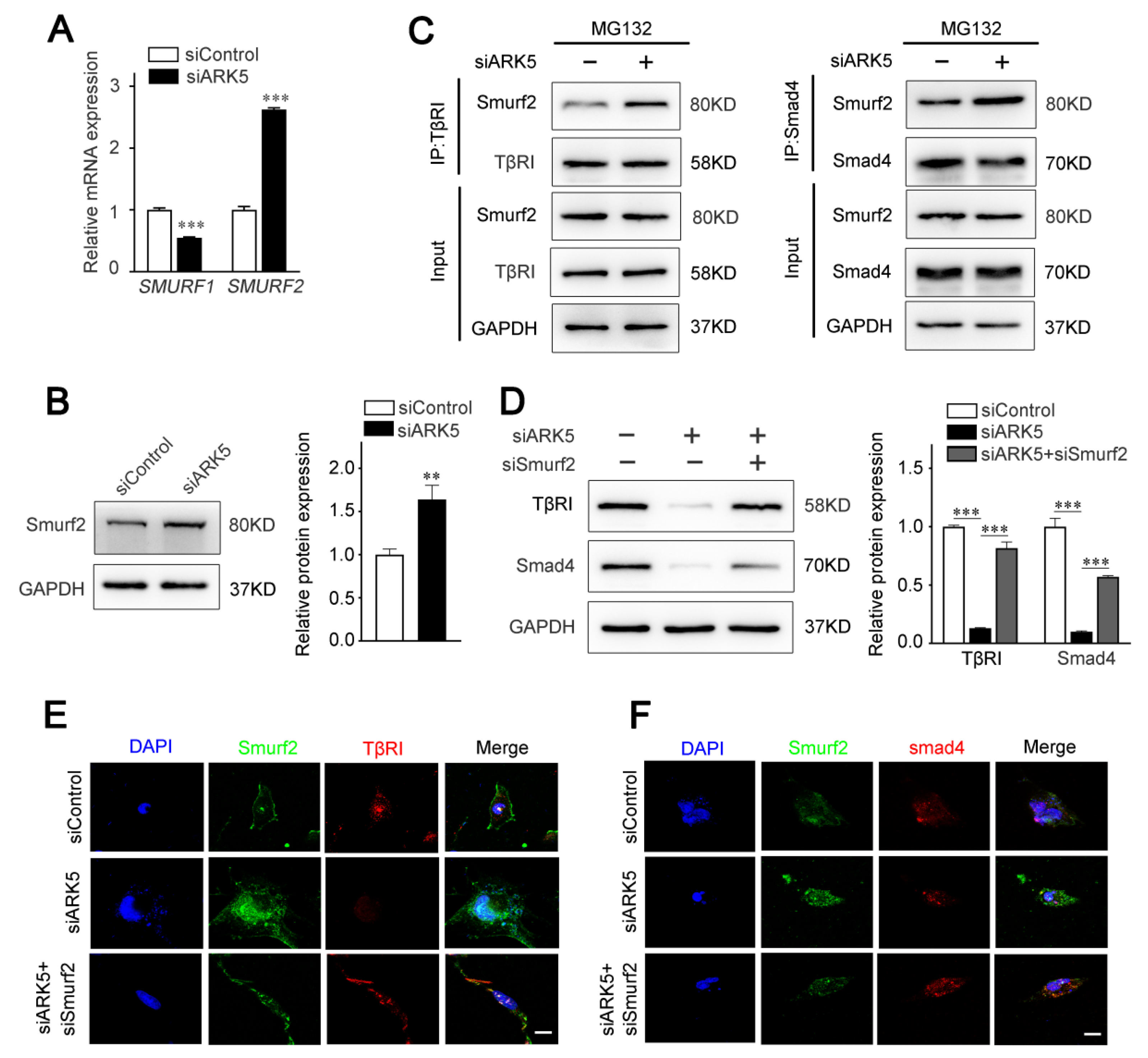
2.4. ARK5 Enhances TβRI and Smad4 Protein Stability by Suppressing Its Ubiquitination
2.5. ARK5 Reduces the Degradation of TβRI and Smad4 by Inhibiting the Expression of Smurf2
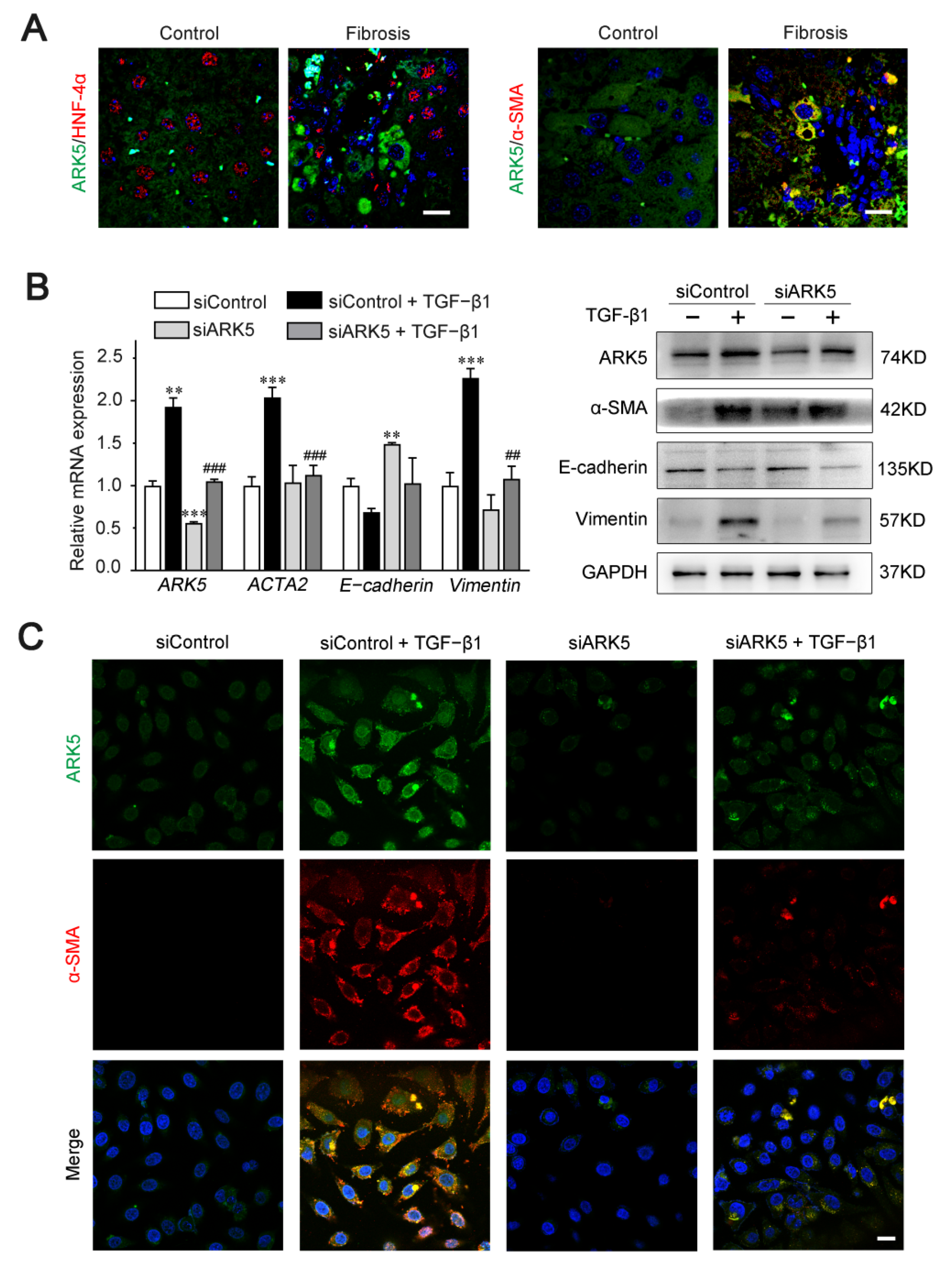
2.6. Enhanced Expression of ARK5 Promotes the Hepatocyte EMT during Liver Fibrosis
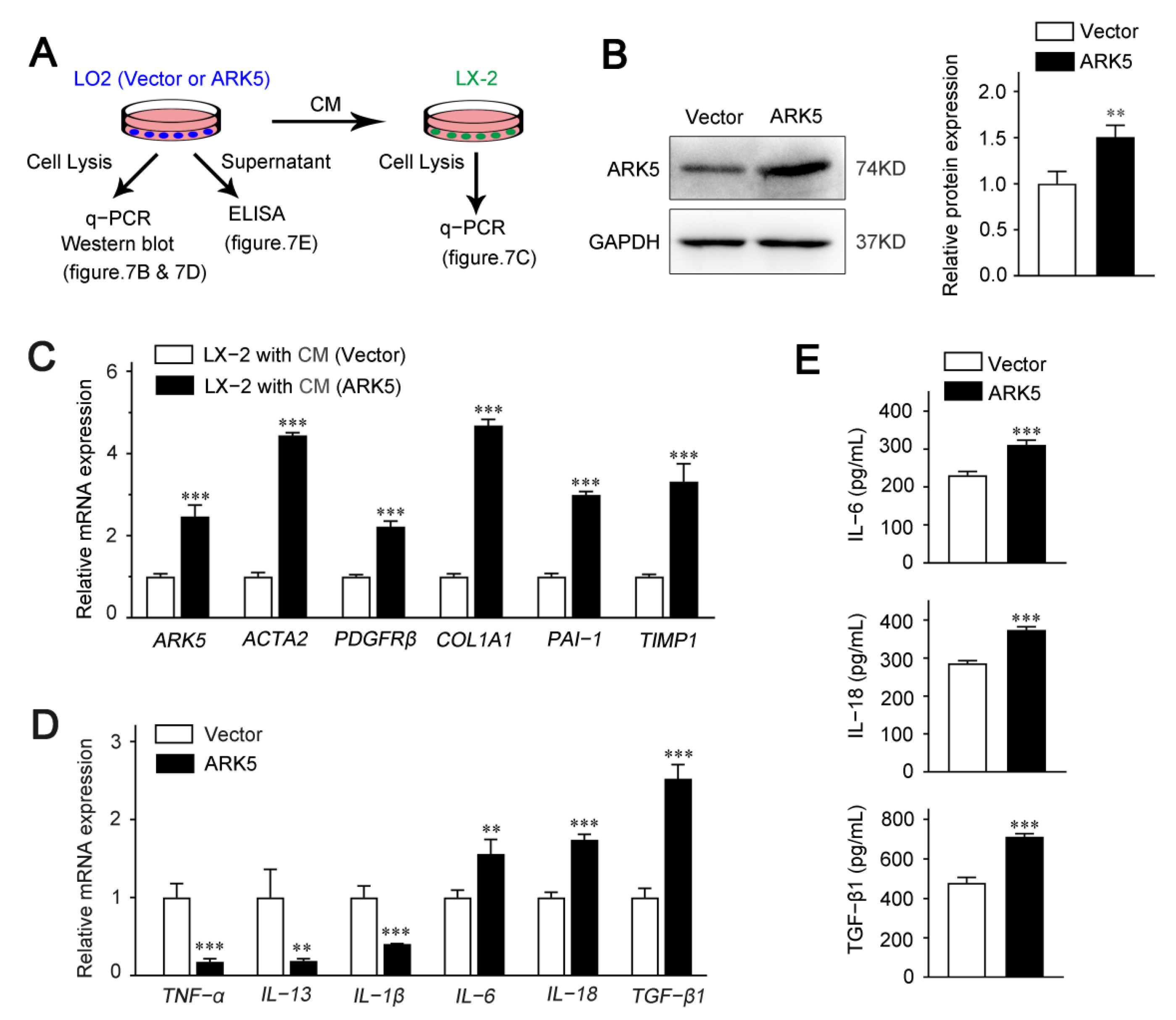
2.7. Hepatocyte ARK5 Mediates the Activation of HSCs by Regulating the Release of Inflammatory Factors
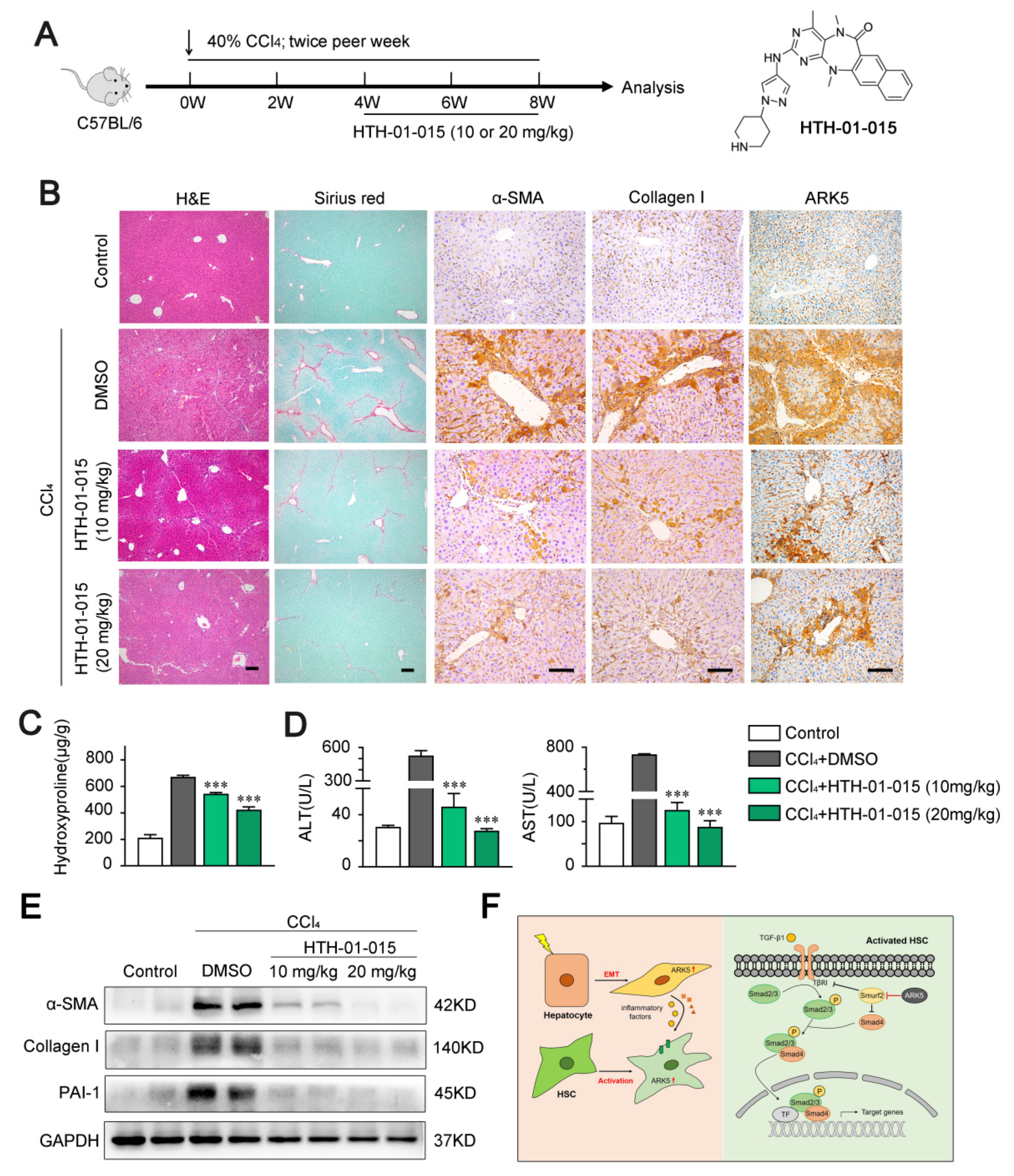
2.8. ARK5 Inhibitor Treatment Attenuates Liver Fibrosis in Mice
3. Discussion
4. Materials and Methods
4.1. Mouse Liver Fibrosis Models
4.2. Cell Culture and Treatments
4.3. Isolation of Primary HSCs and Hepatocytes from Mouse Liver
4.4. Histology and Immunostaining
4.5. Quantitative PCR Analysis
4.6. Western Blotting and Immunoprecipitation
4.7. Cell Apoptosis Analysis
4.8. Wound-Healing Assay
4.9. Transwell Assay
4.10. ELISA
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, L.; Guo, P.; Li, W.; Fang, F.; Zhu, W.; Fan, J.; Wang, F.; Gao, Y.; Zhao, Q.; Wang, Q.; et al. Perturbation of Specific Signaling Pathways Is Involved in Initiation of Mouse Liver Fibrosis. Hepatology 2021, 73, 1551–1569. [Google Scholar] [CrossRef]
- Wang, J.N.; Li, L.; Li, L.Y.; Yan, Q.; Li, J.; Xu, T. Emerging role and therapeutic implication of Wnt signaling pathways in liver fibrosis. Gene 2018, 674, 57–69. [Google Scholar] [CrossRef]
- Martí-Rodrigo, A.; Alegre, F.; Moragrega, Á.B.; García-García, F.; Martí-Rodrigo, P.; Fernández-Iglesias, A.; Gracia-Sancho, J.; Apostolova, N.; Esplugues, J.V.; Blas-García, A. Rilpivirine attenuates liver fibrosis through selective STAT1-mediated apoptosis in hepatic stellate cells. Gut 2020, 69, 920–932. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Qu, C.; Zheng, D.; Li, S.; Liu, Y.; Lidofsky, A.; Holmes, J.A.; Chen, J.; He, L.; Wei, L.; Liao, Y.; et al. Tyrosine kinase SYK is a potential therapeutic target for liver fibrosis. Hepatology 2018, 68, 1125–1139. [Google Scholar] [CrossRef]
- Huang, M.; Kim, H.G.; Zhong, X.; Dong, C.; Zhang, B.; Fang, Z.; Zhang, Y.; Lu, X.; Saxena, R.; Liu, Y.; et al. Sestrin 3 Protects Against Diet-Induced Nonalcoholic Steatohepatitis in Mice Through Suppression of Transforming Growth Factor β Signal Transduction. Hepatology 2020, 71, 76–92. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Cai, J.; Wang, J.; Qiu, Y.; Jiang, C.; Wang, Y.; Wang, Y.; Yi, C.; Guo, L.; Pan, L.; et al. Targeting Nestin(+) hepatic stellate cells ameliorates liver fibrosis by facilitating TβRI degradation. J. Hepatol. 2021, 74, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Gaul, S.; Leszczynska, A.; Alegre, F.; Kaufmann, B.; Johnson, C.D.; Adams, L.A.; Wree, A.; Damm, G.; Seehofer, D.; Calvente, C.J.; et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J. Hepatol. 2021, 74, 156–167. [Google Scholar] [CrossRef]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783.e4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Y.; Chang, X.; Zhang, X. Imbalance in mitochondrial dynamics induced by low PGC-1α expression contributes to hepatocyte EMT and liver fibrosis. Cell Death Dis. 2020, 11, 226. [Google Scholar] [CrossRef]
- Tsay, H.C.; Yuan, Q.; Balakrishnan, A.; Kaiser, M.; Möbus, S.; Kozdrowska, E.; Farid, M.; Tegtmeyer, P.K.; Borst, K.; Vondran, F.W.R.; et al. Hepatocyte-specific suppression of microRNA-221-3p mitigates liver fibrosis. J. Hepatol. 2019, 70, 722–734. [Google Scholar] [CrossRef]
- Zhao, Y.L.; Zhu, R.T.; Sun, Y.L. Epithelial-mesenchymal transition in liver fibrosis. Biomed. Rep. 2016, 4, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 23337–23347. [Google Scholar] [CrossRef] [PubMed]
- Mo, G.; Zhang, B.; Jiang, Q. Role of ARK5 in cancer and other diseases (Review). Exp. Ther. Med. 2021, 22, 697. [Google Scholar] [CrossRef]
- Faisal, M.; Kim, J.H.; Yoo, K.H.; Roh, E.J.; Hong, S.S.; Lee, S.H. Development and Therapeutic Potential of NUAKs Inhibitors. J. Med. Chem. 2021, 64, 2–25. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, X.; Wang, C.; Yin, F.; Qu, L.; Shi, C.; Zhao, J.; Li, S.; Ji, L.; Peng, W.; et al. Optimization of WZ4003 as NUAK inhibitors against human colorectal cancer. Eur. J. Med. Chem. 2021, 210, 113080. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yang, B.; Zhang, Y.; Feng, X.; He, B.; Xie, H.; Zhou, L.; Wu, J.; Zheng, S. miR-424-5p represses the metastasis and invasion of intrahepatic cholangiocarcinoma by targeting ARK5. Int. J. Biol. Sci. 2019, 15, 1591–1599. [Google Scholar] [CrossRef]
- Xu, T.; Zhang, J.; Chen, W.; Pan, S.; Zhi, X.; Wen, L.; Zhou, Y.; Chen, B.W.; Qiu, J.; Zhang, Y.; et al. ARK5 promotes doxorubicin resistance in hepatocellular carcinoma via epithelial-mesenchymal transition. Cancer Lett. 2016, 377, 140–148. [Google Scholar] [CrossRef]
- van de Vis, R.A.J.; Moustakas, A.; van der Heide, L.P. NUAK1 and NUAK2 Fine-Tune TGF-β Signaling. Cancers 2021, 13, 3377. [Google Scholar] [CrossRef]
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef]
- Min, Y.; Hao, L.; Liu, X.; Tan, S.; Song, H.; Ni, H.; Sheng, Z.; Jooss, N.; Liu, X.; Malmström, R.E.; et al. Platelets fine-tune effector responses of naïve CD4(+) T cells via platelet factor 4-regulated transforming growth factor β signaling. Cell. Mol. Life Sci. CMLS 2022, 79, 247. [Google Scholar] [CrossRef]
- Niu, B.; Liu, J.; Lv, B.; Lin, J.; Li, X.; Wu, C.; Jiang, X.; Zeng, Z.; Zhang, X.K.; Zhou, H. Interplay between transforming growth factor-β and Nur77 in dual regulations of inhibitor of differentiation 1 for colonic tumorigenesis. Nat. Commun. 2021, 12, 2809. [Google Scholar] [CrossRef]
- Sinha, A.; Iyengar, P.V.; Ten Dijke, P. E3 Ubiquitin Ligases: Key Regulators of TGFβ Signaling in Cancer Progression. Int. J. Mol. Sci. 2021, 22, 476. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, Z.; Ho, C.; Zhang, G.; Li, Q. Ubiquitin-Specific Protease 15 Maintains Transforming Growth Factor-β Pathway Activity by Deubiquitinating Transforming Growth Factor-β Receptor I during Wound Healing. Am. J. Pathol. 2019, 189, 1351–1362. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Xie, F.; Zhang, Z.; van Dam, H.; Zhang, L.; Zhou, F. The regulation of TGF-β/SMAD signaling by protein deubiquitination. Protein Cell 2014, 5, 503–517. [Google Scholar] [CrossRef]
- Yang, L.; Zhou, W.; Lin, H. Posttranslational Modifications of Smurfs: Emerging Regulation in Cancer. Front. Oncol. 2020, 10, 610663. [Google Scholar] [CrossRef]
- Su, J.; Morgani, S.M.; David, C.J.; Wang, Q.; Er, E.E.; Huang, Y.H.; Basnet, H.; Zou, Y.; Shu, W.; Soni, R.K.; et al. TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature 2020, 577, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, T.; Morishita, A.; Masaki, T. Diagnosis and Therapeutic Management of Liver Fibrosis by MicroRNA. Int. J. Mol. Sci. 2021, 22, 8139. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, H.; Yin, M.; Pei, X.; Hausser, A.; Ishikawa, E.; Yamasaki, S.; Jin, Z.G. Deletion of Protein Kinase D3 Promotes Liver Fibrosis in Mice. Hepatology 2020, 72, 1717–1734. [Google Scholar] [CrossRef]
- Cui, J.; Yu, Y.; Lu, G.F.; Liu, C.; Liu, X.; Xu, Y.X.; Zheng, P.Y. Overexpression of ARK5 is associated with poor prognosis in hepatocellular carcinoma. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2013, 34, 1913–1918. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; He, Q.; Wang, Q.; Lin, Y.; Cen, K.; Chen, X. LINC00922 promotes the proliferation, migration, invasion and EMT process of liver cancer cells by regulating miR-424-5p/ARK5. Mol. Cell. Biochem. 2021, 476, 3757–3769. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T. The origin of fibrogenic myofibroblasts in fibrotic liver. Hepatology 2017, 65, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Serna-Salas, S.; Damba, T.; Borghesan, M.; Demaria, M.; Moshage, H. Hepatic stellate cell senescence in liver fibrosis: Characteristics, mechanisms and perspectives. Mech. Ageing Dev. 2021, 199, 111572. [Google Scholar] [CrossRef] [PubMed]
- Zisser, A.; Ipsen, D.H.; Tveden-Nyborg, P. Hepatic Stellate Cell Activation and Inactivation in NASH-Fibrosis-Roles as Putative Treatment Targets? Biomedicines 2021, 9, 365. [Google Scholar] [CrossRef]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef]
- Chen, Y.; Di, C.; Zhang, X.; Wang, J.; Wang, F.; Yan, J.F.; Xu, C.; Zhang, J.; Zhang, Q.; Li, H.; et al. Transforming growth factor β signaling pathway: A promising therapeutic target for cancer. J. Cell. Physiol. 2020, 235, 1903–1914. [Google Scholar] [CrossRef]
- Yang, T.; Poenisch, M.; Khanal, R.; Hu, Q.; Dai, Z.; Li, R.; Song, G.; Yuan, Q.; Yao, Q.; Shen, X.; et al. Therapeutic HNF4A mRNA attenuates liver fibrosis in a preclinical model. J. Hepatol. 2021, 75, 1420–1433. [Google Scholar] [CrossRef]
- Ramundo, V.; Zanirato, G.; Aldieri, E. The Epithelial-to-Mesenchymal Transition (EMT) in the Development and Metastasis of Malignant Pleural Mesothelioma. Int. J. Mol. Sci. 2021, 22, 12216. [Google Scholar] [CrossRef]
- Ahmadi, A.; Najafi, M.; Farhood, B.; Mortezaee, K. Transforming growth factor-β signaling: Tumorigenesis and targeting for cancer therapy. J. Cell. Physiol. 2019, 234, 12173–12187. [Google Scholar] [CrossRef]
- Yang, F.; Li, H.; Li, Y.; Hao, Y.; Wang, C.; Jia, P.; Chen, X.; Ma, S.; Xiao, Z. Crosstalk between hepatic stellate cells and surrounding cells in hepatic fibrosis. Int. Immunopharmacol. 2021, 99, 108051. [Google Scholar] [CrossRef] [PubMed]
- Xiang, D.M.; Sun, W.; Ning, B.F.; Zhou, T.F.; Li, X.F.; Zhong, W.; Cheng, Z.; Xia, M.Y.; Wang, X.; Deng, X.; et al. The HLF/IL-6/STAT3 feedforward circuit drives hepatic stellate cell activation to promote liver fibrosis. Gut 2018, 67, 1704–1715. [Google Scholar] [CrossRef] [PubMed]
- Knorr, J.; Kaufmann, B.; Inzaugarat, M.E.; Holtmann, T.M.; Geisler, L.; Hundertmark, J.; Kohlhepp, M.S.; Boosheri, L.M.; Chilin-Fuentes, D.R.; Birmingham, A.; et al. Interleukin-18 signaling promotes activation of hepatic stellate cells in murine liver fibrosis. Hepatology 2022. [Google Scholar]
- Banerjee, S.; Buhrlage, S.J.; Huang, H.T.; Deng, X.; Zhou, W.; Wang, J.; Traynor, R.; Prescott, A.R.; Alessi, D.R.; Gray, N.S. Characterization of WZ4003 and HTH-01-015 as selective inhibitors of the LKB1-tumour-suppressor-activated NUAK kinases. Biochem. J. 2014, 457, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Mederacke, I.; Dapito, D.H.; Affò, S.; Uchinami, H.; Schwabe, R.F. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat. Protoc. 2015, 10, 305–315. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, Y.; Gao, C.; Wu, J.; Qu, H.; Xiao, Y.; Kang, Z.; Li, J.; Hong, J. Enhanced Expression of ARK5 in Hepatic Stellate Cell and Hepatocyte Synergistically Promote Liver Fibrosis. Int. J. Mol. Sci. 2022, 23, 13084. https://doi.org/10.3390/ijms232113084
You Y, Gao C, Wu J, Qu H, Xiao Y, Kang Z, Li J, Hong J. Enhanced Expression of ARK5 in Hepatic Stellate Cell and Hepatocyte Synergistically Promote Liver Fibrosis. International Journal of Molecular Sciences. 2022; 23(21):13084. https://doi.org/10.3390/ijms232113084
Chicago/Turabian StyleYou, Yang, Chongqing Gao, Junru Wu, Hengdong Qu, Yang Xiao, Ziwei Kang, Jinying Li, and Jian Hong. 2022. "Enhanced Expression of ARK5 in Hepatic Stellate Cell and Hepatocyte Synergistically Promote Liver Fibrosis" International Journal of Molecular Sciences 23, no. 21: 13084. https://doi.org/10.3390/ijms232113084
APA StyleYou, Y., Gao, C., Wu, J., Qu, H., Xiao, Y., Kang, Z., Li, J., & Hong, J. (2022). Enhanced Expression of ARK5 in Hepatic Stellate Cell and Hepatocyte Synergistically Promote Liver Fibrosis. International Journal of Molecular Sciences, 23(21), 13084. https://doi.org/10.3390/ijms232113084