

Necroptosis: A Pathogenic Negotiator in Human Diseases

 ,
,
Abstract
1. Introduction
2. General Mechanism of Necroptosis
2.1. Necroptosis Signaling: Necrosome Formation
2.2. Non-Canonical Necroptosis Signaling
3. Role of a Necrosome Complex in Inflammation
4. Necroptosis: A Hallmark of Human Pathological Diseases
4.1. Necroptosis in Neurodegenerative Diseases (ND)
4.2. In Renal Diseases
4.3. In Hepatic Diseases
4.4. In Pulmonary Diseases
4.5. In Cardiovascular Diseases
4.6. Autoimmune Diseases
4.7. In Cancer Diseases
4.7.1. Necroptosis Defense against Cancer
4.7.2. Necroptosis in Cancer Progression
5. Therapeutic Applications of Necroptosis Regulation
5.1. RIPK1 Antagonists
5.2. RIPK3 Antagonists
5.3. RIPK1/-3 Antagonists
5.4. MLKL Antagonists
5.5. Hsps 90/70s Inhibitors
5.6. Cancer Antagonists
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 3-BrPA | 3-Bromopyruvate |
| 5-AC | 5-azacytidine |
| 6-OHDA | 6-hydroxydopamine |
| Aβ | Amyloid-β |
| AC | Adenocarcinomas |
| AD | Alzheimer’s diseases |
| ADKFM | Adenine diet kidney fibrosis model |
| ALD | Alcoholic liver disease |
| ALS | Amyotrophic lateral sclerosis |
| AKI | Acute kidney injury |
| AML | Acute myeloid leukemia |
| APOE | Apolipoprotein E |
| ARDS | Acute respiratory distress syndrome |
| BMDMs | Bone marrow-derived macrophages |
| CaMKII | Ca2+/calmodulin-dependent protein kinase II |
| Ch25h | Cholesterol 25-hydroxylase |
| cIAPs | Cellular inhibitor of apoptosis proteins |
| CKD | Chronic kidney disease |
| CLL | Chronic lymphocytic leukemia |
| CNLS | Ceramide nano-liposomes |
| COPD | Chronic obstructive pulmonary disease |
| CYLD | Cylindromatosis |
| CXCL1 | C-X-C Motif Chemokine Ligand 1 |
| DAC | 5-aza-2’-deoxycytidine |
| DAI | DNA-dependent activator of IRFs |
| DAMPs | Damage-associated molecular patterns |
| DAPE | 1,2-Diarachidonoyl-sn-glycero-3-phosphoethanolamine |
| DMA | Disease-associated microglia |
| DMF | Dimethyl fumarate |
| DRs | Death receptors |
| Drp1 | Dynamin-related protein 1 |
| EAE | Experimental allergic encephalomyelitis |
| FADD | Fas-associated protein with death domain |
| FA-AKI | Folic acid-induced AKI model |
| FAO | Fatty acid β-oxidation |
| FLIPL | FLICE-like inhibitory protein long form |
| Fn-14 | Fibroblast growth factor-inducible 14 |
| FTD | Frontotemporal dementia |
| Fer-1 | Ferrostatin-1 |
| GAT | Ganoderic acid T |
| GLUD1 | Glutamate dehydrogenase 1 |
| GSK | GlaxoSmithKline |
| HD | Huntington disease |
| HFFC | High in fat, fructose, and cholesterol |
| HFD | High-fat diet |
| HMGB1 | High-mobility group box 1 |
| HPβCD | 2-hydroxypropyl-β-cyclodextrin |
| ICBs | Immune-checkpoint blockades |
| IFN-γ | Interferon-gamma |
| IPF | Idiopathic pulmonary fibrosis |
| I/R | Ischemia/reperfusion |
| MAPKs | Mitogen-activated protein kinases |
| MCAO | Middle cerebral artery occlusion |
| MCD | Methionine–choline-deficient |
| MLKL | Mixed-lineage kinase domain-like |
| MMS | Methyl methanesulfonate |
| MS | Multiple sclerosis |
| LAD | Left anterior descending coronary artery |
| LDL | Low-density lipoprotein |
| LOAD | Late-onset AD |
| LPS | Lipopolysaccharides |
| LRRK2 | Leucine-rich repeat kinase 2 |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NBC1 | Necroptosis-blocking compound 1 |
| Nec-1 | Necrostatin-1 |
| NF-κβ | Nuclear factor kappa B |
| NEMO | NF-κB essential modulator |
| NOX4 | NADPH Oxidase 4 |
| NSA | Necrosulfonamide |
| NSCLC | Non-small cell lung cancer tissue |
| PAMPs | Pathogen-associated molecular patterns |
| PBPs | Protein-bound polysaccharides |
| PCD | Programmed cell death |
| PD | Parkinson’s disease |
| PDA | Pancreatic ductal adenocarcinomas |
| PGAM5 | Phosphorylate phosphoglycerate mutase family member 5 |
| PI3K/AKT | Phosphoinositide-3-kinase–protein kinase B/Akt |
| Poly I:C | Polyionisic:polycytidylic acid |
| PYCARD | PYD And CARD Domain Containing |
| RA | Rheumatoid arthritis |
| RGMB | Repulsive guidance molecule B |
| RHIM | RIP homotypic interaction motif |
| RIPK | Receptor-interacting protein kinase |
| RNI | Reactive-nitrogen intermediates |
| SHIP | Src homology 2 domain-containing inositolphosphate 5’-phosphatase |
| SIRS | Sepsis/systemic inflammatory associated disorder |
| SCC | Squamous cell carcinoma |
| SCI | Spinal cord neuronal injury |
| TAB | TAK1-binding protein |
| tGCI | Transient global cerebral ischemia |
| TNF | Tumor necrosis factor |
| TLRs | Toll-like receptors |
| TRADD | TNFR-associated death domain |
| TRAF | TNFR-associated factor |
| TWEAK | Tumor necrosis factor-related weak inducer of apoptosis |
| UUO | Unilateral ureteral obstruction |
| ZBP1 | Z-DNA binding protein 1 |
| Z-VAD-FMK | Carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]- fluoromethylketone |
References
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef]
- Ge Yan, M.E. Thomas Efferth, Multiple cell death modalities and their key features. World Acad. Sci. 2020, 2, 39–48. [Google Scholar]
- Christofferson, D.E.; Yuan, J. Necroptosis as an alternative form of programmed cell death. Curr. Opin. Cell Biol. 2010, 22, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflamm. 2018, 15, 199. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat Rev Mol Cell Biol 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Chen, X.; Li, W.; Ren, J.; Huang, D.; He, W.T.; Song, Y.; Yang, C.; Li, W.; Zheng, X.; Chen, P.; et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014, 24, 105–121. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Chen, D.; Yu, J.; Zhang, L. Necroptosis: An alternative cell death program defending against cancer. Biochim. Biophys. Acta 2016, 1865, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Molnár, T.; Mázló, A.; Tslaf, V.; Szöllősi, A.G.; Emri, G.; Koncz, G. Current translational potential and underlying molecular mechanisms of necroptosis. Cell Death Dis. 2019, 10, 860. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Deveraux, Q.L.; Roy, N.; Stennicke, H.R.; Van Arsdale, T.; Zhou, Q.; Srinivasula, S.M.; Alnemri, E.S.; Salvesen, G.S.; Reed, J.C. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. Embo J. 1998, 17, 2215–2223. [Google Scholar] [CrossRef] [PubMed]
- Park, H.H.; Lo, Y.C.; Lin, S.C.; Wang, L.; Yang, J.K.; Wu, H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu. Rev. Immunol. 2007, 25, 561–586. [Google Scholar] [CrossRef]
- Moulin, M.; Anderton, H.; Voss, A.K.; Thomas, T.; Wong, W.W.; Bankovacki, A.; Feltham, R.; Chau, D.; Cook, W.D.; Silke, J.; et al. IAPs limit activation of RIP kinases by TNF receptor 1 during development. Embo J. 2012, 31, 1679–1691. [Google Scholar] [CrossRef]
- Feng, S.; Yang, Y.; Mei, Y.; Ma, L.; Zhu, D.E.; Hoti, N.; Castanares, M.; Wu, M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell. Signal. 2007, 19, 2056–2067. [Google Scholar] [CrossRef]
- Newton, K.; Dugger, D.L.; Wickliffe, K.E.; Kapoor, N.; de Almagro, M.C.; Vucic, D.; Komuves, L.; Ferrando, R.E.; French, D.M.; Webster, J.; et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 2014, 343, 1357–1360. [Google Scholar] [CrossRef]
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Orlowski, G.M.; Chan, F.K. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis. 2015, 6, e1636. [Google Scholar] [CrossRef] [PubMed]
- Duprez, L.; Takahashi, N.; Van Hauwermeiren, F.; Vandendriessche, B.; Goossens, V.; Vanden Berghe, T.; Declercq, W.; Libert, C.; Cauwels, A.; Vandenabeele, P. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 2011, 35, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. The mechanism of necroptosis in normal and cancer cells. Cancer Biol. Ther. 2013, 14, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Lee, J.; Navas, T.; Baldwin, D.T.; Stewart, T.A.; Dixit, V.M. RIP3, a novel apoptosis-inducing kinase. J. Biol. Chem. 1999, 274, 16871–16875. [Google Scholar] [CrossRef]
- Lu, W.; Sun, J.; Yoon, J.S.; Zhang, Y.; Zheng, L.; Murphy, E.; Mattson, M.P.; Lenardo, M.J. Mitochondrial Protein PGAM5 Regulates Mitophagic Protection against Cell Necroptosis. PLoS ONE 2016, 11, e0147792. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jiang, H.; Chen, S.; Du, F.; Wang, X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 2012, 148, 228–243. [Google Scholar] [CrossRef]
- Johnston, A.N.; Ma, Y.; Liu, H.; Liu, S.; Hanna-Addams, S.; Chen, S.; Chen, C.; Wang, Z. Necroptosis-blocking compound NBC1 targets heat shock protein 70 to inhibit MLKL polymerization and necroptosis. Proc. Natl. Acad. Sci. USA 2020, 117, 6521–6530. [Google Scholar] [CrossRef]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef]
- Hill, N.R.; Cook, H.T.; Pusey, C.D.; Tarzi, R.M. RIPK3-deficient mice were not protected from nephrotoxic nephritis. BMC Nephrol 2018, 19, 61. [Google Scholar] [CrossRef]
- Kalliolias, G.D.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef]
- Shan, B.; Pan, H.; Najafov, A.; Yuan, J. Necroptosis in development and diseases. Genes Dev. 2018, 32, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Song, Q.; Yu, A.; Tang, H.; Peng, Z.; Wang, X. Receptor-interacting protein kinase 3 is a predictor of survival and plays a tumor suppressive role in colorectal cancer. Neoplasma 2015, 62, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Höckendorf, U.; Yabal, M.; Herold, T.; Munkhbaatar, E.; Rott, S.; Jilg, S.; Kauschinger, J.; Magnani, G.; Reisinger, F.; Heuser, M.; et al. RIPK3 Restricts Myeloid Leukemogenesis by Promoting Cell Death and Differentiation of Leukemia Initiating Cells. Cancer Cell 2016, 30, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Koo, G.B.; Morgan, M.J.; Lee, D.G.; Kim, W.J.; Yoon, J.H.; Koo, J.S.; Kim, S.I.; Kim, S.J.; Son, M.K.; Hong, S.S.; et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015, 25, 707–725. [Google Scholar] [CrossRef]
- Han, J.; Zhong, C.Q.; Zhang, D.W. Programmed necrosis: Backup to and competitor with apoptosis in the immune system. Nat. Immunol. 2011, 12, 1143–1149. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Chen, X.; Zhuang, C.; Ren, Y.; Zhang, H.; Qin, X.; Hu, L.; Fu, J.; Miao, Z.; Chai, Y.; Liu, Z.G.; et al. Identification of the Raf kinase inhibitor TAK-632 and its analogues as potent inhibitors of necroptosis by targeting RIPK1 and RIPK3. Br. J. Pharmacol. 2019, 176, 2095–2108. [Google Scholar] [CrossRef]
- Ea, C.K.; Deng, L.; Xia, Z.P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 2006, 22, 245–257. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Jouan-Lanhouet, S.; Divert, T.; Theatre, E.; Bertin, J.; Gough, P.J.; Giansanti, P.; Heck, A.J.; Dejardin, E.; Vandenabeele, P.; et al. NF-κB-Independent Role of IKKα/IKKβ in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol. Cell 2015, 60, 63–76. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Delanghe, T.; Priem, D.; Wynosky-Dolfi, M.A.; Sorobetea, D.; Rojas-Rivera, D.; Giansanti, P.; Roelandt, R.; Gropengiesser, J.; Ruckdeschel, K.; et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat. Commun. 2019, 10, 1729. [Google Scholar] [CrossRef]
- O’Donnell, M.A.; Perez-Jimenez, E.; Oberst, A.; Ng, A.; Massoumi, R.; Xavier, R.; Green, D.R.; Ting, A.T. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat. Cell Biol. 2011, 13, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Pop, C.; Oberst, A.; Drag, M.; Van Raam, B.J.; Riedl, S.J.; Green, D.R.; Salvesen, G.S. FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem. J. 2011, 433, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Moquin, D.M.; McQuade, T.; Chan, F.K. CYLD deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS ONE 2013, 8, e76841. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.Q.; Chen, X.; Cai, Q.; Yang, Z.H.; Huang, D.; Wu, R.; et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schröder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef]
- Walsh, C.M. Grand challenges in cell death and survival: Apoptosis vs. necroptosis. Front. Cell Dev. Biol. 2014, 2, 3. [Google Scholar] [CrossRef]
- Saïd-Sadier, N.; Ojcius, D.M. Alarmins, inflammasomes and immunity. Biomed. J. 2012, 35, 437–449. [Google Scholar]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Zhu, K.; Liang, W.; Ma, Z.; Xu, D.; Cao, S.; Lu, X.; Liu, N.; Shan, B.; Qian, L.; Yuan, J. Necroptosis promotes cell-autonomous activation of proinflammatory cytokine gene expression. Cell Death Dis. 2018, 9, 500. [Google Scholar] [CrossRef] [PubMed]
- Najjar, M.; Saleh, D.; Zelic, M.; Nogusa, S.; Shah, S.; Tai, A.; Finger, J.N.; Polykratis, A.; Gough, P.J.; Bertin, J.; et al. RIPK1 and RIPK3 Kinases Promote Cell-Death-Independent Inflammation by Toll-like Receptor 4. Immunity 2016, 45, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Biton, S.; Ashkenazi, A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-α feedforward signaling. Cell 2011, 145, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Christofferson, D.E.; Li, Y.; Hitomi, J.; Zhou, W.; Upperman, C.; Zhu, H.; Gerber, S.A.; Gygi, S.; Yuan, J. A novel role for RIP1 kinase in mediating TNFα production. Cell Death Dis. 2012, 3, e320. [Google Scholar] [CrossRef]
- McNamara, C.R.; Ahuja, R.; Osafo-Addo, A.D.; Barrows, D.; Kettenbach, A.; Skidan, I.; Teng, X.; Cuny, G.D.; Gerber, S.; Degterev, A. Akt Regulates TNFα synthesis downstream of RIP1 kinase activation during necroptosis. PLoS ONE 2013, 8, e56576. [Google Scholar] [CrossRef]
- Saleh, D.; Najjar, M.; Zelic, M.; Shah, S.; Nogusa, S.; Polykratis, A.; Paczosa, M.K.; Gough, P.J.; Bertin, J.; Whalen, M.; et al. Kinase Activities of RIPK1 and RIPK3 Can Direct IFN-β Synthesis Induced by Lipopolysaccharide. J. Immunol. 2017, 198, 4435–4447. [Google Scholar] [CrossRef]
- Bury, J.J.; Highley, J.R.; Cooper-Knock, J.; Goodall, E.F.; Higginbottom, A.; McDermott, C.J.; Ince, P.G.; Shaw, P.J.; Kirby, J. Oligogenic inheritance of optineurin (OPTN) and C9ORF72 mutations in ALS highlights localisation of OPTN in the TDP-43-negative inclusions of C9ORF72-ALS. Neuropathology 2016, 36, 125–134. [Google Scholar] [CrossRef]
- Lukens, J.R.; Vogel, P.; Johnson, G.R.; Kelliher, M.A.; Iwakura, Y.; Lamkanfi, M.; Kanneganti, T.D. RIP1-driven autoinflammation targets IL-1α independently of inflammasomes and RIP3. Nature 2013, 498, 224–227. [Google Scholar] [CrossRef]
- Kang, T.B.; Jeong, J.S.; Yang, S.H.; Kovalenko, A.; Wallach, D. Caspase-8 deficiency in mouse embryos triggers chronic RIPK1-dependent activation of inflammatory genes, independently of RIPK3. Cell Death Differ. 2018, 25, 1107–1117. [Google Scholar] [CrossRef]
- Lawlor, K.E.; Khan, N.; Mildenhall, A.; Gerlic, M.; Croker, B.A.; D'Cruz, A.A.; Hall, C.; Kaur Spall, S.; Anderton, H.; Masters, S.L.; et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 2015, 6, 6282. [Google Scholar] [CrossRef]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Vince, J.E.; Wong, W.W.; Gentle, I.; Lawlor, K.E.; Allam, R.; O'Reilly, L.; Mason, K.; Gross, O.; Ma, S.; Guarda, G.; et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 2012, 36, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Yabal, M.; Müller, N.; Adler, H.; Knies, N.; Groß, C.J.; Damgaard, R.B.; Kanegane, H.; Ringelhan, M.; Kaufmann, T.; Heikenwälder, M.; et al. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep. 2014, 7, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Weng, D.; Marty-Roix, R.; Ganesan, S.; Proulx, M.K.; Vladimer, G.I.; Kaiser, W.J.; Mocarski, E.S.; Pouliot, K.; Chan, F.K.; Kelliher, M.A.; et al. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc. Natl. Acad. Sci. USA 2014, 111, 7391–7396. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.W.; Vince, J.E.; Lalaoui, N.; Lawlor, K.E.; Chau, D.; Bankovacki, A.; Anderton, H.; Metcalf, D.; O'Reilly, L.; Jost, P.J.; et al. cIAPs and XIAP regulate myelopoiesis through cytokine production in an RIPK1- and RIPK3-dependent manner. Blood 2014, 123, 2562–2572. [Google Scholar] [CrossRef]
- Antonopoulos, C.; El Sanadi, C.; Kaiser, W.J.; Mocarski, E.S.; Dubyak, G.R. Proapoptotic chemotherapeutic drugs induce noncanonical processing and release of IL-1β via caspase-8 in dendritic cells. J. Immunol. 2013, 191, 4789–4803. [Google Scholar] [CrossRef]
- Kang, T.B.; Yang, S.H.; Toth, B.; Kovalenko, A.; Wallach, D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 2013, 38, 27–40. [Google Scholar] [CrossRef]
- Kang, S.; Fernandes-Alnemri, T.; Rogers, C.; Mayes, L.; Wang, Y.; Dillon, C.; Roback, L.; Kaiser, W.; Oberst, A.; Sagara, J.; et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun. 2015, 6, 7515. [Google Scholar] [CrossRef]
- Ofengeim, D.; Ito, Y.; Najafov, A.; Zhang, Y.; Shan, B.; DeWitt, J.P.; Ye, J.; Zhang, X.; Chang, A.; Vakifahmetoglu-Norberg, H.; et al. Activation of necroptosis in multiple sclerosis. Cell Rep 2015, 10, 1836–1849. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O'Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Lens, S.; Gaide, O.; Alevizopoulos, K.; Tschopp, J. NF-kappaB signals induce the expression of c-FLIP. Mol. Cell Biol. 2001, 21, 5299–5305. [Google Scholar] [CrossRef] [PubMed]
- Picon, C.; Jayaraman, A.; James, R.; Beck, C.; Gallego, P.; Witte, M.E.; van Horssen, J.; Mazarakis, N.D.; Reynolds, R. Neuron-specific activation of necroptosis signaling in multiple sclerosis cortical grey matter. Acta Neuropathol. 2021, 141, 585–604. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L.; et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Li, Y.; Fukaya, M.; Lorenzini, I.; Cleveland, D.W.; Ostrow, L.W.; Rothstein, J.D.; Bergles, D.E. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. Neurosci. 2013, 16, 571–579. [Google Scholar] [CrossRef]
- Wang, T.; Perera, N.D.; Chiam, M.D.F.; Cuic, B.; Wanniarachchillage, N.; Tomas, D.; Samson, A.L.; Cawthorne, W.; Valor, E.N.; Murphy, J.M.; et al. Necroptosis is dispensable for motor neuron degeneration in a mouse model of ALS. Cell Death Differ. 2020, 27, 1728–1739. [Google Scholar] [CrossRef]
- Xu, D.; Jin, T.; Zhu, H.; Chen, H.; Ofengeim, D.; Zou, C.; Mifflin, L.; Pan, L.; Amin, P.; Li, W.; et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell 2018, 174, 1477–1491.e19. [Google Scholar] [CrossRef]
- Lafont, E.; Draber, P.; Rieser, E.; Reichert, M.; Kupka, S.; de Miguel, D.; Draberova, H.; von Mässenhausen, A.; Bhamra, A.; Henderson, S.; et al. TBK1 and IKKε prevent TNF-induced cell death by RIPK1 phosphorylation. Nat. Cell Biol. 2018, 20, 1389–1399. [Google Scholar] [CrossRef]
- Caccamo, A.; Branca, C.; Piras, I.S.; Ferreira, E.; Huentelman, M.J.; Liang, W.S.; Readhead, B.; Dudley, J.T.; Spangenberg, E.E.; Green, K.N.; et al. Necroptosis activation in Alzheimer's disease. Nat. Neurosci. 2017, 20, 1236–1246. [Google Scholar] [CrossRef]
- Ofengeim, D.; Mazzitelli, S.; Ito, Y.; DeWitt, J.P.; Mifflin, L.; Zou, C.; Das, S.; Adiconis, X.; Chen, H.; Zhu, H.; et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E8788-e8797. [Google Scholar] [CrossRef]
- Koper, M.J.; Van Schoor, E.; Ospitalieri, S.; Vandenberghe, R.; Vandenbulcke, M.; von Arnim, C.A.F.; Tousseyn, T.; Balusu, S.; De Strooper, B.; Thal, D.R. Necrosome complex detected in granulovacuolar degeneration is associated with neuronal loss in Alzheimer's disease. Acta Neuropathol. 2020, 139, 463–484. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer's Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.B.; Kasparcova, V.; Hoffman, S.; Swift, B.; Dare, L.; Schaeffer, M.; Capriotti, C.; Cook, M.; Finger, J.; Hughes-Earle, A.; et al. Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J. Immunol. 2014, 192, 5476–5480. [Google Scholar] [CrossRef] [PubMed]
- Iannielli, A.; Bido, S.; Folladori, L.; Segnali, A.; Cancellieri, C.; Maresca, A.; Massimino, L.; Rubio, A.; Morabito, G.; Caporali, L.; et al. Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson’s Disease Models. Cell Rep. 2018, 22, 2066–2079. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.R.; Wang, J.; Zhou, S.K.; Yang, L.; Yin, J.L.; Cao, J.P.; Cheng, Y.B. Necrostatin-1 protection of dopaminergic neurons. Neural Regen. Res. 2015, 10, 1120–1124. [Google Scholar]
- Liu, J.; Hu, H.; Wu, B. RIPK1 inhibitor ameliorates the MPP(+)/MPTP-induced Parkinson's disease through the ASK1/JNK signalling pathway. Brain Res. 2021, 1757, 147310. [Google Scholar] [CrossRef]
- Devalaraja-Narashimha, K.; Diener, A.M.; Padanilam, B.J. Cyclophilin D gene ablation protects mice from ischemic renal injury. Am. J. Physiol. Renal Physiol. 2009, 297, F749–F759. [Google Scholar] [CrossRef]
- Park, J.S.; Pasupulati, R.; Feldkamp, T.; Roeser, N.F.; Weinberg, J.M. Cyclophilin D and the mitochondrial permeability transition in kidney proximal tubules after hypoxic and ischemic injury. Am. J. Physiol. Renal Physiol. 2011, 301, F134–F150. [Google Scholar] [CrossRef]
- Linkermann, A.; Hackl, M.J.; Kunzendorf, U.; Walczak, H.; Krautwald, S.; Jevnikar, A.M. Necroptosis in immunity and ischemia-reperfusion injury. Am J Transplant 2013, 13, 2797–2804. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Carrasco, S.; Cannata-Ortiz, P.; Sanchez-Niño, M.D.; Ruiz Ortega, M.; Egido, J.; Linkermann, A.; Ortiz, A.; et al. Ferroptosis, but Not Necroptosis, Is Important in Nephrotoxic Folic Acid-Induced AKI. J. Am. Soc. Nephrol. 2017, 28, 218–229. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Fontecha-Barriuso, M.; Carrasco, S.; Sanchez-Niño, M.D.; Mässenhausen, A.V.; Linkermann, A.; Cannata-Ortiz, P.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; et al. TWEAK and RIPK1 mediate a second wave of cell death during AKI. Proc. Natl. Acad. Sci. USA 2018, 115, 4182–4187. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Bräsen, J.H.; Darding, M.; Jin, M.K.; Sanz, A.B.; Heller, J.O.; De Zen, F.; Weinlich, R.; Ortiz, A.; Walczak, H.; et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2013, 110, 12024–12029. [Google Scholar] [CrossRef] [PubMed]
- Mulay, S.R.; Desai, J.; Kumar, S.V.; Eberhard, J.N.; Thomasova, D.; Romoli, S.; Grigorescu, M.; Kulkarni, O.P.; Popper, B.; Vielhauer, V.; et al. Cytotoxicity of crystals involves RIPK3-MLKL-mediated necroptosis. Nat. Commun. 2016, 7, 10274. [Google Scholar] [CrossRef]
- Xu, Y.; Ma, H.; Shao, J.; Wu, J.; Zhou, L.; Zhang, Z.; Wang, Y.; Huang, Z.; Ren, J.; Liu, S.; et al. A Role for Tubular Necroptosis in Cisplatin-Induced AKI. J. Am. Soc. Nephrol. 2015, 26, 2647–2658. [Google Scholar] [CrossRef] [PubMed]
- Imamura, M.; Moon, J.S.; Chung, K.P.; Nakahira, K.; Muthukumar, T.; Shingarev, R.; Ryter, S.W.; Choi, A.M.; Choi, M.E. RIPK3 promotes kidney fibrosis via AKT-dependent ATP citrate lyase. JCI Insight 2018, 3, e94979. [Google Scholar] [CrossRef]
- Sureshbabu, A.; Patino, E.; Ma, K.C.; Laursen, K.; Finkelsztein, E.J.; Akchurin, O.; Muthukumar, T.; Ryter, S.W.; Gudas, L.; Choi, A.M.K.; et al. RIPK3 promotes sepsis-induced acute kidney injury via mitochondrial dysfunction. JCI Insight 2018, 3, e98411. [Google Scholar] [CrossRef]
- Chen, H.; Fang, Y.; Wu, J.; Chen, H.; Zou, Z.; Zhang, X.; Shao, J.; Xu, Y. RIPK3-MLKL-mediated necroinflammation contributes to AKI progression to CKD. Cell Death Dis. 2018, 9, 878. [Google Scholar] [CrossRef]
- Roychowdhury, S.; McMullen, M.R.; Pisano, S.G.; Liu, X.; Nagy, L.E. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology 2013, 57, 1773–1783. [Google Scholar] [CrossRef]
- Wang, S.; Ni, H.M.; Dorko, K.; Kumer, S.C.; Schmitt, T.M.; Nawabi, A.; Komatsu, M.; Huang, H.; Ding, W.X. Increased hepatic receptor interacting protein kinase 3 expression due to impaired proteasomal functions contributes to alcohol-induced steatosis and liver injury. Oncotarget 2016, 7, 17681–17698. [Google Scholar] [CrossRef]
- Bertola, A.; Mathews, S.; Ki, S.H.; Wang, H.; Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 2013, 8, 627–637. [Google Scholar] [CrossRef]
- Afonso, M.B.; Rodrigues, P.M.; Carvalho, T.; Caridade, M.; Borralho, P.; Cortez-Pinto, H.; Castro, R.E.; Rodrigues, C.M. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin. Sci. (Lond) 2015, 129, 721–739. [Google Scholar] [CrossRef] [PubMed]
- Gautheron, J.; Vucur, M.; Reisinger, F.; Cardenas, D.V.; Roderburg, C.; Koppe, C.; Kreggenwinkel, K.; Schneider, A.T.; Bartneck, M.; Neumann, U.P.; et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol. Med. 2014, 6, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Chao, X.; Kaseff, J.; Deng, F.; Wang, S.; Shi, Y.H.; Li, T.; Ding, W.X.; Jaeschke, H. Receptor-Interacting Serine/Threonine-Protein Kinase 3 (RIPK3)-Mixed Lineage Kinase Domain-Like Protein (MLKL)-Mediated Necroptosis Contributes to Ischemia-Reperfusion Injury of Steatotic Livers. Am. J. Pathol. 2019, 189, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Liss, K.H.H.; McCommis, K.S.; Chambers, K.T.; Pietka, T.A.; Schweitzer, G.G.; Park, S.L.; Nalbantoglu, I.; Weinheimer, C.J.; Hall, A.M.; Finck, B.N. The impact of diet-induced hepatic steatosis in a murine model of hepatic ischemia/reperfusion injury. Liver Transpl. 2018, 24, 908–921. [Google Scholar] [CrossRef]
- Wu, X.; Poulsen, K.L.; Sanz-Garcia, C.; Huang, E.; McMullen, M.R.; Roychowdhury, S.; Dasarathy, S.; Nagy, L.E. Erratum to: MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcoholic-associated fatty liver and steatohepatitis (J. Hepatol. 2020, 73, 616–627). J. Hepatol. 2021, 74, 1002. [Google Scholar] [CrossRef]
- Siempos, I.I.; Ma, K.C.; Imamura, M.; Baron, R.M.; Fredenburgh, L.E.; Huh, J.W.; Moon, J.S.; Finkelsztein, E.J.; Jones, D.S.; Lizardi, M.T.; et al. RIPK3 mediates pathogenesis of experimental ventilator-induced lung injury. JCI Insight 2018, 3, e97102. [Google Scholar] [CrossRef]
- Pan, L.; Yao, D.C.; Yu, Y.Z.; Chen, B.J.; Li, S.J.; Hu, G.H.; Xi, C.; Wang, Z.H.; Li, J.H.; Long, J.; et al. Activation of necroptosis in a rat model of acute respiratory distress syndrome induced by oleic acid. Sheng Li Xue Bao 2016, 68, 661–668. [Google Scholar]
- Lin, B.; Jin, Z.; Chen, X.; Zhao, L.; Weng, C.; Chen, B.; Tang, Y.; Lin, L. Necrostatin-1 protects mice from acute lung injury by suppressing necroptosis and reactive oxygen species. Mol. Med. Rep. 2020, 21, 2171–2181. [Google Scholar] [CrossRef]
- Wang, L.; Wang, T.; Li, H.; Liu, Q.; Zhang, Z.; Xie, W.; Feng, Y.; Socorburam, T.; Wu, G.; Xia, Z.; et al. Receptor Interacting Protein 3-Mediated Necroptosis Promotes Lipopolysaccharide-Induced Inflammation and Acute Respiratory Distress Syndrome in Mice. PLoS ONE 2016, 11, e0155723. [Google Scholar] [CrossRef]
- Bolognese, A.C.; Yang, W.L.; Hansen, L.W.; Denning, N.L.; Nicastro, J.M.; Coppa, G.F.; Wang, P. Inhibition of necroptosis attenuates lung injury and improves survival in neonatal sepsis. Surgery 2018, 164, 110–116. [Google Scholar] [CrossRef]
- Hansen, L.W.; Jacob, A.; Yang, W.L.; Bolognese, A.C.; Prince, J.; Nicastro, J.M.; Coppa, G.F.; Wang, P. Deficiency of receptor-interacting protein kinase 3 (RIPK3) attenuates inflammation and organ injury in neonatal sepsis. J. Pediatr. Surg. 2018, 53, 1699–1705. [Google Scholar] [CrossRef]
- Mizumura, K.; Cloonan, S.M.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Glass, K.; Owen, C.A.; Mahmood, A.; Washko, G.R.; et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Investig. 2014, 124, 3987–4003. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.H.; Kim, H.P.; Sciurba, F.C.; Lee, S.J.; Feghali-Bostwick, C.; Stolz, D.B.; Dhir, R.; Landreneau, R.J.; Schuchert, M.J.; Yousem, S.A.; et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS ONE 2008, 3, e3316. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.H.; Lam, H.C.; Jin, Y.; Kim, H.P.; Cao, J.; Lee, S.J.; Ifedigbo, E.; Parameswaran, H.; Ryter, S.W.; Choi, A.M. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc. Natl. Acad. Sci. USA 2010, 107, 18880–18885. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Van Eeckhoutte, H.P.; Liu, G.; Nair, P.M.; Jones, B.; Gillis, C.M.; Nalkurthi, B.C.; Verhamme, F.; Buyle-Huybrecht, T.; Vandenabeele, P.; et al. Necroptosis Signaling Promotes Inflammation, Airway Remodeling, and Emphysema in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021, 204, 667–681. [Google Scholar] [CrossRef]
- Chen, D.; Gregory, A.D.; Li, X.; Wei, J.; Burton, C.L.; Gibson, G.; Scott, S.J.; St Croix, C.M.; Zhang, Y.; Shapiro, S.D. RIP3-dependent necroptosis contributes to the pathogenesis of chronic obstructive pulmonary disease. JCI Insight 2021, 6, e144689. [Google Scholar] [CrossRef]
- Lee, J.M.; Yoshida, M.; Kim, M.S.; Lee, J.H.; Baek, A.R.; Jang, A.S.; Kim, D.J.; Minagawa, S.; Chin, S.S.; Park, C.S.; et al. Involvement of Alveolar Epithelial Cell Necroptosis in Idiopathic Pulmonary Fibrosis Pathogenesis. Am. J. Respir. Cell Mol. Biol. 2018, 59, 215–224. [Google Scholar] [CrossRef]
- Luedde, M.; Lutz, M.; Carter, N.; Sosna, J.; Jacoby, C.; Vucur, M.; Gautheron, J.; Roderburg, C.; Borg, N.; Reisinger, F.; et al. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc. Res. 2014, 103, 206–216. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, Y.; Cui, M.; Jin, L.; Wang, Y.; Lv, F.; Liu, Y.; Zheng, W.; Shang, H.; Zhang, J.; et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 2016, 22, 175–182. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Lim, S.Y.; Davidson, S.M.; Mocanu, M.M.; Yellon, D.M.; Smith, C.C. The cardioprotective effect of necrostatin requires the cyclophilin-D component of the mitochondrial permeability transition pore. Cardiovasc. Drugs Ther. 2007, 21, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Oerlemans, M.I.; Liu, J.; Arslan, F.; den Ouden, K.; van Middelaar, B.J.; Doevendans, P.A.; Sluijter, J.P. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic. Res. Cardiol. 2012, 107, 270. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Li, H.; Yang, M.; Ren, J.; Huang, Z.; Han, F.; Huang, J.; Ma, J.; Zhang, D.; Zhang, Z.; et al. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. 2013, 3, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Jin, W.; Wang, X. RIP3-mediated necrotic cell death accelerates systematic inflammation and mortality. Proc. Natl. Acad. Sci. USA 2015, 112, 11007–11012. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, Z.; Ren, J.; Morgan, S.; Assa, C.; Liu, B. Receptor-interacting protein kinase 3 contributes to abdominal aortic aneurysms via smooth muscle cell necrosis and inflammation. Circ. Res. 2015, 116, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Pavlosky, A.; Lau, A.; Su, Y.; Lian, D.; Huang, X.; Yin, Z.; Haig, A.; Jevnikar, A.M.; Zhang, Z.X. RIPK3-mediated necroptosis regulates cardiac allograft rejection. Am. J. Transplant. 2014, 14, 1778–1790. [Google Scholar] [CrossRef]
- Zhang, H.; Yin, Y.; Liu, Y.; Zou, G.; Huang, H.; Qian, P.; Zhang, G.; Zhang, J. Necroptosis mediated by impaired autophagy flux contributes to adverse ventricular remodeling after myocardial infarction. Biochem. Pharmacol. 2020, 175, 113915. [Google Scholar] [CrossRef]
- Honda, T.; Yamamoto, O.; Sawada, Y.; Egawa, G.; Kitoh, A.; Otsuka, A.; Dainichi, T.; Nakajima, S.; Miyachi, Y.; Kabashima, K. Receptor-interacting protein kinase 3 controls keratinocyte activation in a necroptosis-independent manner and promotes psoriatic dermatitis in mice. J. Allergy Clin. Immunol. 2017, 140, 619–622.e6. [Google Scholar] [CrossRef]
- Saito, N.; Honma, M.; Shibuya, T.; Iinuma, S.; Igawa, S.; Kishibe, M.; Ishida-Yamamoto, A. RIPK1 downregulation in keratinocyte enhances TRAIL signaling in psoriasis. J. Dermatol. Sci. 2018, 91, 79–86. [Google Scholar] [CrossRef]
- Duan, X.; Liu, X.; Liu, N.; Huang, Y.; Jin, Z.; Zhang, S.; Ming, Z.; Chen, H. Inhibition of keratinocyte necroptosis mediated by RIPK1/RIPK3/MLKL provides a protective effect against psoriatic inflammation. Cell Death Dis. 2020, 11, 134. [Google Scholar] [CrossRef]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Núñez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961-e969. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kwon, J.Y.; Kim, S.Y.; Jung, K.; Cho, M.L. Interferon-gamma regulates inflammatory cell death by targeting necroptosis in experimental autoimmune arthritis. Sci. Rep. 2017, 7, 10133. [Google Scholar] [CrossRef] [PubMed]
- Nugues, A.L.; El Bouazzati, H.; Hétuin, D.; Berthon, C.; Loyens, A.; Bertrand, E.; Jouy, N.; Idziorek, T.; Quesnel, B. RIP3 is downregulated in human myeloid leukemia cells and modulates apoptosis and caspase-mediated p65/RelA cleavage. Cell Death Dis. 2014, 5, e1384. [Google Scholar] [CrossRef]
- Liu, P.; Xu, B.; Shen, W.; Zhu, H.; Wu, W.; Fu, Y.; Chen, H.; Dong, H.; Zhu, Y.; Miao, K.; et al. Dysregulation of TNFα-induced necroptotic signaling in chronic lymphocytic leukemia: Suppression of CYLD gene by LEF1. Leukemia 2012, 26, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- McCormick, K.D.; Ghosh, A.; Trivedi, S.; Wang, L.; Coyne, C.B.; Ferris, R.L.; Sarkar, S.N. Innate immune signaling through differential RIPK1 expression promote tumor progression in head and neck squamous cell carcinoma. Carcinogenesis 2016, 37, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Lee, J.H.; Lee, S.Y.; Hong, M.J.; Choi, J.E.; Park, S.; Jeong, J.Y.; Lee, E.B.; Choi, S.H.; Lee, Y.H.; et al. Expression of key regulatory genes in necroptosis and its effect on the prognosis in non-small cell lung cancer. J. Cancer 2020, 11, 5503–5510. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Peng, K.; Liu, Y.; Xiong, J.; Zhu, F.F. Low expression of mixed lineage kinase domain-like protein is associated with poor prognosis in ovarian cancer patients. Onco Targets Ther. 2013, 6, 1539–1543. [Google Scholar]
- Takemura, R.; Takaki, H.; Okada, S.; Shime, H.; Akazawa, T.; Oshiumi, H.; Matsumoto, M.; Teshima, T.; Seya, T. PolyI:C-Induced, TLR3/RIP3-Dependent Necroptosis Backs Up Immune Effector-Mediated Tumor Elimination In Vivo. Cancer Immunol. Res. 2015, 3, 902–914. [Google Scholar] [CrossRef]
- Schmidt, S.V.; Seibert, S.; Walch-Rückheim, B.; Vicinus, B.; Kamionka, E.M.; Pahne-Zeppenfeld, J.; Solomayer, E.F.; Kim, Y.J.; Bohle, R.M.; Smola, S. RIPK3 expression in cervical cancer cells is required for PolyIC-induced necroptosis, IL-1α release, and efficient paracrine dendritic cell activation. Oncotarget 2015, 6, 8635–8647. [Google Scholar] [CrossRef]
- Werthmöller, N.; Frey, B.; Wunderlich, R.; Fietkau, R.; Gaipl, U.S. Modulation of radiochemoimmunotherapy-induced B16 melanoma cell death by the pan-caspase inhibitor zVAD-fmk induces anti-tumor immunity in a HMGB1-, nucleotide- and T-cell-dependent manner. Cell Death Dis. 2015, 6, e1761. [Google Scholar] [CrossRef]
- Kang, Y.J.; Bang, B.R.; Han, K.H.; Hong, L.; Shim, E.J.; Ma, J.; Lerner, R.A.; Otsuka, M. Regulation of NKT cell-mediated immune responses to tumours and liver inflammation by mitochondrial PGAM5-Drp1 signalling. Nat. Commun. 2015, 6, 8371. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, M.; Mei, L.; Ruan, J.; Hu, Q.; Peng, J.; Su, H.; Liao, H.; Liu, S.; Liu, W.; et al. Key roles of necroptotic factors in promoting tumor growth. Oncotarget 2016, 7, 22219–22233. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Hatanpaa, K.J.; Xie, Y.; Mickey, B.E.; Madden, C.J.; Raisanen, J.M.; Ramnarain, D.B.; Xiao, G.; Saha, D.; Boothman, D.A.; et al. The receptor interacting protein 1 inhibits p53 induction through NF-kappaB activation and confers a worse prognosis in glioblastoma. Cancer Res. 2009, 69, 2809–2816. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, W.; Xu, X.; Li, B.; He, W.; Padilla, M.T.; Jang, J.H.; Nyunoya, T.; Amin, S.; Wang, X.; et al. RIP1 potentiates BPDE-induced transformation in human bronchial epithelial cells through catalase-mediated suppression of excessive reactive oxygen species. Carcinogenesis 2013, 34, 2119–2128. [Google Scholar] [CrossRef] [PubMed]
- Seifert, L.; Werba, G.; Tiwari, S.; Giao Ly, N.N.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Barilla, R.; Daley, D.; Greco, S.H.; et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 2016, 532, 245–249. [Google Scholar] [CrossRef]
- Kim, S.J.; Li, J. Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. 2013, 4, e716. [Google Scholar] [CrossRef]
- Re, D.B.; Le Verche, V.; Yu, C.; Amoroso, M.W.; Politi, K.A.; Phani, S.; Ikiz, B.; Hoffmann, L.; Koolen, M.; Nagata, T.; et al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 2014, 81, 1001–1008. [Google Scholar] [CrossRef]
- Yuan, J.; Amin, P.; Ofengeim, D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 2019, 20, 19–33. [Google Scholar] [CrossRef]
- Papassotiropoulos, A.; Lambert, J.C.; Wavrant-De Vrièze, F.; Wollmer, M.A.; von der Kammer, H.; Streffer, J.R.; Maddalena, A.; Huynh, K.D.; Wolleb, S.; Lutjohann, D.; et al. Cholesterol 25-hydroxylase on chromosome 10q is a susceptibility gene for sporadic Alzheimer’s disease. Neurodegener. Dis. 2005, 2, 233–241. [Google Scholar] [CrossRef]
- Asanomi, Y.; Shigemizu, D.; Miyashita, A.; Mitsumori, R.; Mori, T.; Hara, N.; Ito, K.; Niida, S.; Ikeuchi, T.; Ozaki, K. A rare functional variant of SHARPIN attenuates the inflammatory response and associates with increased risk of late-onset Alzheimer’s disease. Mol. Med. 2019, 25, 20. [Google Scholar] [CrossRef]
- Linkermann, A.; Bräsen, J.H.; Himmerkus, N.; Liu, S.; Huber, T.B.; Kunzendorf, U.; Krautwald, S. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 2012, 81, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Heller, J.O.; Prókai, A.; Weinberg, J.M.; De Zen, F.; Himmerkus, N.; Szabó, A.J.; Bräsen, J.H.; Kunzendorf, U.; Krautwald, S. The RIP1-kinase inhibitor necrostatin-1 prevents osmotic nephrosis and contrast-induced AKI in mice. J. Am. Soc. Nephrol. 2013, 24, 1545–1557. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Wang, S.; Jiang, J.; Haig, A.; Pavlosky, A.; Linkermann, A.; Zhang, Z.X.; Jevnikar, A.M. RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am. J. Transplant. 2013, 13, 2805–2818. [Google Scholar] [CrossRef]
- Liu, S.S.; Chen, Y.Y.; Wang, S.X.; Yu, Q. Protective effect of dabrafenib on renal ischemia-reperfusion injury in vivo and in vitro. Biochem. Biophys. Res. Commun. 2020, 522, 395–401. [Google Scholar] [CrossRef]
- Shen, B.; Mei, M.; Pu, Y.; Zhang, H.; Liu, H.; Tang, M.; Pan, Q.; He, Y.; Wu, X.; Zhao, H. Necrostatin-1 Attenuates Renal Ischemia and Reperfusion Injury via Meditation of HIF-1α/mir-26a/TRPC6/PARP1 Signaling. Mol. Ther. Nucleic Acids 2019, 17, 701–713. [Google Scholar] [CrossRef]
- Newton, K.; Dugger, D.L.; Maltzman, A.; Greve, J.M.; Hedehus, M.; Martin-McNulty, B.; Carano, R.A.; Cao, T.C.; van Bruggen, N.; Bernstein, L.; et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016, 23, 1565–1576. [Google Scholar] [CrossRef]
- Wu, H.; Ma, J.; Wang, P.; Corpuz, T.M.; Panchapakesan, U.; Wyburn, K.R.; Chadban, S.J. HMGB1 contributes to kidney ischemia reperfusion injury. J. Am. Soc. Nephrol. 2010, 21, 1878–1890. [Google Scholar] [CrossRef]
- Ning, Y.; Shi, Y.; Chen, J.; Song, N.; Cai, J.; Fang, Y.; Yu, X.; Ji, J.; Ding, X. Necrostatin-1 Attenuates Cisplatin-Induced Nephrotoxicity Through Suppression of Apoptosis and Oxidative Stress and Retains Klotho Expression. Front. Pharmacol. 2018, 9, 384. [Google Scholar] [CrossRef]
- Tristão, V.R.; Gonçalves, P.F.; Dalboni, M.A.; Batista, M.C.; Durão Mde, S., Jr.; Monte, J.C. Nec-1 protects against nonapoptotic cell death in cisplatin-induced kidney injury. Ren. Fail. 2012, 34, 373–377. [Google Scholar] [CrossRef]
- Tristão, V.R.; Pessoa, E.A.; Nakamichi, R.; Reis, L.A.; Batista, M.C.; Durão Junior Mde, S.; Monte, J.C. Synergistic effect of apoptosis and necroptosis inhibitors in cisplatin-induced nephrotoxicity. Apoptosis 2016, 21, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chen, B.; Wang, Y.; Meng, C.; Huang, H.; Huang, X.R.; Qin, J.; Mulay, S.R.; Anders, H.J.; Qiu, A.; et al. RGMb protects against acute kidney injury by inhibiting tubular cell necroptosis via an MLKL-dependent mechanism. Proc. Natl. Acad. Sci. USA 2018, 115, E1475-e1484. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, M.; Graffeo, C.S.; Rokosh, R.; Pansari, M.; Ochi, A.; Levie, E.M.; Van Heerden, E.; Tippens, D.M.; Greco, S.; Barilla, R.; et al. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis. 2015, 6, e1759. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, K.; Hatano, E.; Iwaisako, K.; Takeiri, M.; Noma, N.; Ohmae, S.; Toriguchi, K.; Tanabe, K.; Tanaka, H.; Seo, S.; et al. Necrostatin-1 protects against reactive oxygen species (ROS)-induced hepatotoxicity in acetaminophen-induced acute liver failure. FEBS Open Bio 2014, 4, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; McGill, M.R.; Xie, Y.; Ni, H.M.; Ding, W.X.; Jaeschke, H. Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology 2013, 58, 2099–2108. [Google Scholar] [CrossRef]
- Li, J.X.; Feng, J.M.; Wang, Y.; Li, X.H.; Chen, X.X.; Su, Y.; Shen, Y.Y.; Chen, Y.; Xiong, B.; Yang, C.H.; et al. The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis. 2014, 5, e1278. [Google Scholar] [CrossRef]
- Dara, L.; Johnson, H.; Suda, J.; Win, S.; Gaarde, W.; Han, D.; Kaplowitz, N. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology 2015, 62, 1847–1857. [Google Scholar] [CrossRef]
- Saeed, W.K.; Jun, D.W.; Jang, K.; Chae, Y.J.; Lee, J.S.; Kang, H.T. Does necroptosis have a crucial role in hepatic ischemia-reperfusion injury? PLoS ONE 2017, 12, e0184752. [Google Scholar] [CrossRef]
- Farooq, M.; Filliol, A.; Simoes Eugénio, M.; Piquet-Pellorce, C.; Dion, S.; Raguenes-Nicol, C.; Jan, A.; Dimanche-Boitrel, M.T.; Le Seyec, J.; Samson, M. Depletion of RIPK1 in hepatocytes exacerbates liver damage in fulminant viral hepatitis. Cell Death Dis. 2019, 10, 12. [Google Scholar] [CrossRef]
- Mizumura, K.; Maruoka, S.; Gon, Y.; Choi, A.M.; Hashimoto, S. The role of necroptosis in pulmonary diseases. Respir. Investig. 2016, 54, 407–412. [Google Scholar] [CrossRef]
- Moreno-Gonzalez, G.; Vandenabeele, P.; Krysko, D.V. Necroptosis: A Novel Cell Death Modality and Its Potential Relevance for Critical Care Medicine. Am. J. Respir. Crit. Care Med. 2016, 194, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Sauler, M.; Bazan, I.S.; Lee, P.J. Cell Death in the Lung: The Apoptosis-Necroptosis Axis. Annu. Rev. Physiol. 2019, 81, 375–402. [Google Scholar] [CrossRef] [PubMed]
- Zelic, M.; Roderick, J.E.; O'Donnell, J.A.; Lehman, J.; Lim, S.E.; Janardhan, H.P.; Trivedi, C.M.; Pasparakis, M.; Kelliher, M.A. RIP kinase 1-dependent endothelial necroptosis underlies systemic inflammatory response syndrome. J. Clin. Investig. 2018, 128, 2064–2075. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.J.; Wang, K.Y.; Zhang, H.Z.; Meng, X.Y.; Chen, J.F.; Wang, P.; Jiang, J.H.; Ma, Q. Up-Regulation of RIP3 Alleviates Prostate Cancer Progression by Activation of RIP3/MLKL Signaling Pathway and Induction of Necroptosis. Front. Oncol. 2020, 10, 1720. [Google Scholar] [CrossRef]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Chan, F.K.; Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. 2017, 12, 103–130. [Google Scholar] [CrossRef]
- Gupta, K.; Phan, N.; Wang, Q.; Liu, B. Necroptosis in cardiovascular disease—A new therapeutic target. J. Mol. Cell Cardiol. 2018, 118, 26–35. [Google Scholar] [CrossRef]
- Jun-Long, H.; Yi, L.; Bao-Lian, Z.; Jia-Si, L.; Ning, Z.; Zhou-Heng, Y.; Xue-Jun, S.; Wen-Wu, L. Necroptosis Signaling Pathways in Stroke: From Mechanisms to Therapies. Curr. Neuropharmacol. 2018, 16, 1327–1339. [Google Scholar] [CrossRef]
- Koshinuma, S.; Miyamae, M.; Kaneda, K.; Kotani, J.; Figueredo, V.M. Combination of necroptosis and apoptosis inhibition enhances cardioprotection against myocardial ischemia-reperfusion injury. J. Anesth. 2014, 28, 235–241. [Google Scholar] [CrossRef]
- Guo, X.; Yin, H.; Li, L.; Chen, Y.; Li, J.; Doan, J.; Steinmetz, R.; Liu, Q. Cardioprotective Role of Tumor Necrosis Factor Receptor-Associated Factor 2 by Suppressing Apoptosis and Necroptosis. Circulation 2017, 136, 729–742. [Google Scholar] [CrossRef]
- Geserick, P.; Wang, J.; Schilling, R.; Horn, S.; Harris, P.A.; Bertin, J.; Gough, P.J.; Feoktistova, M.; Leverkus, M. Absence of RIPK3 predicts necroptosis resistance in malignant melanoma. Cell Death Dis. 2015, 6, e1884. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Augustine, C.K.; Gandham, V.D.; Jin, J.Y.; Tyler, D.S.; Akiyama, S.K.; Hall, R.P.; Zhang, J.Y. CYLD inhibits melanoma growth and progression through suppression of the JNK/AP-1 and β1-integrin signaling pathways. J. Investig. Dermatol. 2013, 133, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Kuo, W.T.; Huang, Y.C.; Lee, T.C.; Yu, L.C. Resistance to hypoxia-induced necroptosis is conferred by glycolytic pyruvate scavenging of mitochondrial superoxide in colorectal cancer cells. Cell Death Dis. 2013, 4, e622. [Google Scholar] [CrossRef] [PubMed]
- Colbert, L.E.; Fisher, S.B.; Hardy, C.W.; Hall, W.A.; Saka, B.; Shelton, J.W.; Petrova, A.V.; Warren, M.D.; Pantazides, B.G.; Gandhi, K.; et al. Pronecrotic mixed lineage kinase domain-like protein expression is a prognostic biomarker in patients with early-stage resected pancreatic adenocarcinoma. Cancer 2013, 119, 3148–3155. [Google Scholar] [CrossRef]
- Buchheit, C.L.; Rayavarapu, R.R.; Schafer, Z.T. The regulation of cancer cell death and metabolism by extracellular matrix attachment. Semin. Cell Dev. Biol. 2012, 23, 402–411. [Google Scholar] [CrossRef]
- Philipp, S.; Sosna, J.; Adam, D. Cancer and necroptosis: Friend or foe? Cell. Mol. Life Sci. 2016, 73, 2183–2193. [Google Scholar] [CrossRef]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O.; Barreira da Silva, R.; Reis e Sousa, C.; Green, D.R.; Oberst, A.; Albert, M.L. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8+ T cells. Science 2015, 350, 328–334. [Google Scholar] [CrossRef]
- Liu, X.Y.; Lai, F.; Yan, X.G.; Jiang, C.C.; Guo, S.T.; Wang, C.Y.; Croft, A.; Tseng, H.Y.; Wilmott, J.S.; Scolyer, R.A.; et al. RIP1 Kinase Is an Oncogenic Driver in Melanoma. Cancer Res. 2015, 75, 1736–1748. [Google Scholar] [CrossRef]
- Lim, J.H.; Oh, S.; Kim, L.; Suh, Y.J.; Ha, Y.J.; Kim, J.S.; Kim, H.J.; Park, M.H.; Kim, Y.S.; Cho, Y.; et al. Low-level expression of necroptosis factors indicates a poor prognosis of the squamous cell carcinoma subtype of non-small-cell lung cancer. Transl. Lung Cancer Res. 2021, 10, 1221–1230. [Google Scholar] [CrossRef]
- Roessner, A.; Kuester, D.; Malfertheiner, P.; Schneider-Stock, R. Oxidative stress in ulcerative colitis-associated carcinogenesis. Pathol. Res. Pract. 2008, 204, 511–524. [Google Scholar] [CrossRef]
- Strilic, B.; Yang, L.; Albarrán-Juárez, J.; Wachsmuth, L.; Han, K.; Müller, U.C.; Pasparakis, M.; Offermanns, S. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 2016, 536, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Ohuchida, K.; Otsubo, Y.; Kibe, S.; Takesue, S.; Abe, T.; Iwamoto, C.; Shindo, K.; Moriyama, T.; Nakata, K.; et al. Necroptosis in pancreatic cancer promotes cancer cell migration and invasion by release of CXCL5. PLoS ONE 2020, 15, e0228015. [Google Scholar] [CrossRef]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Mu, W. Necrostatin-1 and necroptosis inhibition: Pathophysiology and therapeutic implications. Pharmacol. Res. 2021, 163, 105297. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Su, Y.; Sun, L.; He, S.; Meng, L.; Liao, D.; Liu, X.; Ma, Y.; Liu, C.; Li, S.; et al. Discovery of a Highly Potent, Selective, and Metabolically Stable Inhibitor of Receptor-Interacting Protein 1 (RIP1) for the Treatment of Systemic Inflammatory Response Syndrome. J. Med. Chem. 2017, 60, 972–986. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.A.; Berger, S.B.; Jeong, J.U.; Nagilla, R.; Bandyopadhyay, D.; Campobasso, N.; Capriotti, C.A.; Cox, J.A.; Dare, L.; Dong, X.; et al. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J. Med. Chem. 2017, 60, 1247–1261. [Google Scholar] [CrossRef]
- Weisel, K.; Scott, N.E.; Tompson, D.J.; Votta, B.J.; Madhavan, S.; Povey, K.; Wolstenholme, A.; Simeoni, M.; Rudo, T.; Richards-Peterson, L.; et al. Randomized clinical study of safety, pharmacokinetics, and pharmacodynamics of RIPK1 inhibitor GSK2982772 in healthy volunteers. Pharmacol. Res. Perspect. 2017, 5, e00365. [Google Scholar] [CrossRef]
- Patel, S.; Webster, J.D.; Varfolomeev, E.; Kwon, Y.C.; Cheng, J.H.; Zhang, J.; Dugger, D.L.; Wickliffe, K.E.; Maltzman, A.; Sujatha-Bhaskar, S.; et al. RIP1 inhibition blocks inflammatory diseases but not tumor growth or metastases. Cell Death Differ. 2020, 27, 161–175. [Google Scholar] [CrossRef]
- von Schaper, E. Everything but amyloid: New thinking prompts FDA revamp. Nat. Biotechnol. 2018, 36, 483–484. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Saitoh, M.; Katoh, T.; Seki, T.; Bigi, S.V.; Shimizu, Y.; Ishii, T.; Okai, T.; Kuno, M.; Hattori, H.; et al. Discovery of 7-Oxo-2,4,5,7-tetrahydro-6 H-pyrazolo[3,4-c]pyridine Derivatives as Potent, Orally Available, and Brain-Penetrating Receptor Interacting Protein 1 (RIP1) Kinase Inhibitors: Analysis of Structure-Kinetic Relationships. J. Med. Chem. 2018, 61, 2384–2409. [Google Scholar] [CrossRef]
- Cougnoux, A.; Clifford, S.; Salman, A.; Ng, S.L.; Bertin, J.; Porter, F.D. Necroptosis inhibition as a therapy for Niemann-Pick disease, type C1: Inhibition of RIP kinases and combination therapy with 2-hydroxypropyl-β-cyclodextrin. Mol. Genet. Metab. 2018, 125, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Wang, J.; Zhang, H.; Cao, Y.; Qu, Y.; Huang, S.; Kong, X.; Song, C.; Li, J.; Li, Q.; et al. Inhibition of microglial receptor-interacting protein kinase 1 ameliorates neuroinflammation following cerebral ischaemic stroke. J. Cell. Mol. Med. 2020, 24, 12585–12598. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.; Berger, S.B.; Pillay, S.; Moriwaki, K.; Huang, C.; Guo, H.; Lich, J.D.; Finger, J.; Kasparcova, V.; Votta, B.; et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 2014, 56, 481–495. [Google Scholar] [CrossRef]
- Nogusa, S.; Thapa, R.J.; Dillon, C.P.; Liedmann, S.; Oguin, T.H., 3rd; Ingram, J.P.; Rodriguez, D.A.; Kosoff, R.; Sharma, S.; Sturm, O.; et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe 2016, 20, 13–24. [Google Scholar] [CrossRef]
- Cruz, S.A.; Qin, Z.; Stewart, A.F.R.; Chen, H.H. Dabrafenib, an inhibitor of RIP3 kinase-dependent necroptosis, reduces ischemic brain injury. Neural Regen. Res. 2018, 13, 252–256. [Google Scholar]
- Syed, M.A.; Shah, D.; Das, P.; Andersson, S.; Pryhuber, G.; Bhandari, V. Retraction: TREM-1 Attenuates RIPK3-mediated Necroptosis in Hyperoxia-induced Lung Injury in Neonatal Mice. Am. J. Respir. Cell Mol. Biol. 2021, 64, 518. [Google Scholar]
- Park, H.H.; Park, S.Y.; Mah, S.; Park, J.H.; Hong, S.S.; Hong, S.; Kim, Y.S. HS-1371, a novel kinase inhibitor of RIP3-mediated necroptosis. Exp. Mol. Med. 2018, 50, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Noh, H.J.; Lee, H.; Park, H.H.; Ha, Y.J.; Park, S.H.; Lee, H.; Kim, S.J.; Kang, H.C.; Eyun, S.I.; et al. TRIM24-RIP3 axis perturbation accelerates osteoarthritis pathogenesis. Ann. Rheum. Dis. 2020, 79, 1635–1643. [Google Scholar] [CrossRef]
- Martens, S.; Hofmans, S.; Declercq, W.; Augustyns, K.; Vandenabeele, P. Inhibitors Targeting RIPK1/RIPK3: Old and New Drugs. Trends Pharmacol Sci 2020, 41, 209–224. [Google Scholar] [CrossRef]
- Tu, H.; Zhou, Y.J.; Tang, L.J.; Xiong, X.M.; Zhang, X.J.; Ali Sheikh, M.S.; Zhang, J.J.; Luo, X.J.; Yuan, C.; Peng, J. Combination of ponatinib with deferoxamine synergistically mitigates ischemic heart injury via simultaneous prevention of necroptosis and ferroptosis. Eur. J. Pharmacol. 2021, 898, 173999. [Google Scholar] [CrossRef]
- Yue, L.J.; Zhu, X.Y.; Li, R.S.; Chang, H.J.; Gong, B.; Tian, C.C.; Liu, C.; Xue, Y.X.; Zhou, Q.; Xu, T.S.; et al. S-allyl-cysteine sulfoxide (alliin) alleviates myocardial infarction by modulating cardiomyocyte necroptosis and autophagy. Int. J. Mol. Med. 2019, 44, 1943–1951. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Liu, W.N.; Li, Y.Q.; Zhang, X.J.; Yang, J.; Luo, X.J.; Peng, J. Ligustroflavone reduces necroptosis in rat brain after ischemic stroke through targeting RIPK1/RIPK3/MLKL pathway. Naunyn Schmiedebergs Arch. Pharmacol. 2019, 392, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Nikseresht, S.; Khodagholi, F.; Ahmadiani, A. Protective effects of ex-527 on cerebral ischemia-reperfusion injury through necroptosis signaling pathway attenuation. J. Cell Physiol. 2019, 234, 1816–1826. [Google Scholar] [CrossRef] [PubMed]
- Ryan, F.; Khodagholi, F.; Dargahi, L.; Minai-Tehrani, D.; Ahmadiani, A. Temporal Pattern and Crosstalk of Necroptosis Markers with Autophagy and Apoptosis Associated Proteins in Ischemic Hippocampus. Neurotox. Res. 2018, 34, 79–92. [Google Scholar] [CrossRef]
- Zhan, L.; Lu, Z.; Zhu, X.; Xu, W.; Li, L.; Li, X.; Chen, S.; Sun, W.; Xu, E. Hypoxic preconditioning attenuates necroptotic neuronal death induced by global cerebral ischemia via Drp1-dependent signaling pathway mediated by CaMKIIα inactivation in adult rats. FASEB J. 2019, 33, 1313–1329. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, S.R.; Dionísio, P.A.; Gaspar, M.M.; Ferreira, M.B.T.; Rodrigues, C.A.B.; Pereira, R.G.; Estevão, M.S.; Perry, M.J.; Moreira, R.; Afonso, C.A.M.; et al. Discovery of a Necroptosis Inhibitor Improving Dopaminergic Neuronal Loss after MPTP Exposure in Mice. Int. J. Mol. Sci. 2021, 22, 5289. [Google Scholar] [CrossRef]
- Yan, B.; Liu, L.; Huang, S.; Ren, Y.; Wang, H.; Yao, Z.; Li, L.; Chen, S.; Wang, X.; Zhang, Z. Discovery of a new class of highly potent necroptosis inhibitors targeting the mixed lineage kinase domain-like protein. Chem. Commun. (Camb) 2017, 53, 3637–3640. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, D.; Zhang, M.; Zhang, Y. Programmed necrosis in cardiomyocytes: Mitochondria, death receptors and beyond. Br. J. Pharmacol. 2019, 176, 4319–4339. [Google Scholar] [CrossRef]
- Font-Belmonte, E.; Ugidos, I.F.; Santos-Galdiano, M.; González-Rodríguez, P.; Anuncibay-Soto, B.; Pérez-Rodríguez, D.; Gonzalo-Orden, J.M.; Fernández-López, A. Post-ischemic salubrinal administration reduces necroptosis in a rat model of global cerebral ischemia. J. Neurochem. 2019, 151, 777–794. [Google Scholar] [CrossRef]
- Han, F.; Guan, X.; Guo, W.; Lu, B. Therapeutic potential of a TrkB agonistic antibody for ischemic brain injury. Neurobiol. Dis. 2019, 127, 570–581. [Google Scholar] [CrossRef]
- Li, D.; Li, C.; Li, L.; Chen, S.; Wang, L.; Li, Q.; Wang, X.; Lei, X.; Shen, Z. Natural Product Kongensin A is a Non-Canonical HSP90 Inhibitor that Blocks RIP3-dependent Necroptosis. Cell Chem. Biol. 2016, 23, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, A.V.; Lowes, K.N.; Tanzer, M.C.; Lucet, I.S.; Hildebrand, J.M.; Petrie, E.J.; van Delft, M.F.; Liu, Z.; Conos, S.A.; Zhang, J.G.; et al. HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis. 2016, 7, e2051. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.M.; Chen, Z.; Zhao, J.B.; Zhang, P.P.; Pu, Y.F.; Jiang, S.H.; Hou, J.J.; Cui, Y.M.; Jia, X.L.; Zhang, S.Q. Hsp90 modulates the stability of MLKL and is required for TNF-induced necroptosis. Cell Death Dis. 2016, 7, e2089. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, H.M.; Zhou, C.; Li, Q.; Ma, L.; Zhang, Z.; Sun, Y.; Wang, L.; Zhang, X.; Zhu, B.; et al. Non-benzoquinone geldanamycin analogs trigger various forms of death in human breast cancer cells. J. Exp. Clin. Cancer Res. 2016, 35, 149. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Shim, J.H.; Chae, J.I.; Cho, Y.S. Heat shock protein 90 inhibitor regulates necroptotic cell death via down-regulation of receptor interacting proteins. Pharmazie 2015, 70, 193–198. [Google Scholar]
- Marunouchi, T.; Nishiumi, C.; Iinuma, S.; Yano, E.; Tanonaka, K. Effects of Hsp90 inhibitor on the RIP1-RIP3-MLKL pathway during the development of heart failure in mice. Eur. J. Pharmacol. 2021, 898, 173987. [Google Scholar] [CrossRef]
- Chen, J.; Kos, R.; Garssen, J.; Redegeld, F. Molecular Insights into the Mechanism of Necroptosis: The Necrosome As a Potential Therapeutic Target. Cells 2019, 8, 1486. [Google Scholar] [CrossRef]
- Wagner, A.J.; Chugh, R.; Rosen, L.S.; Morgan, J.A.; George, S.; Gordon, M.; Dunbar, J.; Normant, E.; Grayzel, D.; Demetri, G.D. A phase I study of the HSP90 inhibitor retaspimycin hydrochloride (IPI-504) in patients with gastrointestinal stromal tumors or soft-tissue sarcomas. Clin. Cancer Res. 2013, 19, 6020–6029. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, H.; Huang, J.; Yao, Z.; Yu, J.; Zhang, W.; Zhang, L.; Wang, Z.; Zhuang, C. Discovery of bardoxolone derivatives as novel orally active necroptosis inhibitors. Eur. J. Med. Chem. 2021, 212, 113030. [Google Scholar] [CrossRef]
- Srinivasan, S.R.; Cesa, L.C.; Li, X.; Julien, O.; Zhuang, M.; Shao, H.; Chung, J.; Maillard, I.; Wells, J.A.; Duckett, C.S.; et al. Heat Shock Protein 70 (Hsp70) Suppresses RIP1-Dependent Apoptotic and Necroptotic Cascades. Mol. Cancer Res. 2018, 16, 58–68. [Google Scholar] [CrossRef]
- Yang, M.; Lv, Y.; Tian, X.; Lou, J.; An, R.; Zhang, Q.; Li, M.; Xu, L.; Dong, Z. Neuroprotective Effect of β-Caryophyllene on Cerebral Ischemia-Reperfusion Injury via Regulation of Necroptotic Neuronal Death and Inflammation: In Vivo and in Vitro. Front. Neurosci. 2017, 11, 583. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Zhang, Y.; Liao, X.; Jiang, Y.; Lv, X.; Yuan, X.; Meng, J.; Xie, Y.; Peng, Z.; Yuan, Q.; et al. Fluorofenidone Alleviates Renal Fibrosis by Inhibiting Necroptosis Through RIPK3/MLKL Pathway. Front. Pharmacol. 2020, 11, 534775. [Google Scholar] [CrossRef]
- von Mässenhausen, A.; Tonnus, W.; Himmerkus, N.; Parmentier, S.; Saleh, D.; Rodriguez, D.; Ousingsawat, J.; Ang, R.L.; Weinberg, J.M.; Sanz, A.B.; et al. Phenytoin inhibits necroptosis. Cell Death Dis. 2018, 9, 359. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Li, H.D.; Wu, W.F.; Ming-Kuen Tang, P.; Ren, G.L.; Gao, L.; Li, X.F.; Yang, Y.; Xu, T.; Ma, T.T.; et al. Wogonin protects against cisplatin-induced acute kidney injury by targeting RIPK1-mediated necroptosis. Lab. Investig. 2018, 98, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tang, L.J.; Tu, H.; Zhou, Y.J.; Li, N.S.; Luo, X.J.; Peng, J. Arctiin protects rat heart against ischemia/reperfusion injury via a mechanism involving reduction of necroptosis. Eur. J. Pharmacol. 2020, 875, 173053. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, M.; Nazarinia, D.; Ramezani, F.; Azizi, Y.; Naderi, N.; Aboutaleb, N. Necroptosis and RhoA/ROCK pathways: Molecular targets of Nesfatin-1 in cardioprotection against myocardial ischemia/reperfusion injury in a rat model. Mol. Biol. Rep. 2021, 48, 2507–2518. [Google Scholar] [CrossRef]
- Hu, Y.; Pan, H.; Peng, J.; He, J.; Tang, M.; Yan, S.; Rong, J.; Li, J.; Zheng, Z.; Wang, H.; et al. Resveratrol inhibits necroptosis by mediating the TNF-α/RIP1/RIP3/MLKL pathway in myocardial hypoxia/reoxygenation injury. Acta Biochim. Biophys. Sin. (Shanghai) 2021, 53, 430–437. [Google Scholar] [CrossRef]
- Zhu, Y.; Yuan, R.; Chen, X.; He, J.; Chen, Y.; Zhang, C.; Sun, K.; Yang, S.; Liu, Z.; Gao, H. Tanshinone I Exerts Cardiovascular Protective Effects in Vivo and in Vitro Through Inhibiting Necroptosis via Akt/nrf2 Signaling Pathway. BMC Chin. Med. 2021, 16, 48. [Google Scholar]
- Chen, C.; Xiao, W.; Huang, L.; Yu, G.; Ni, J.; Yang, L.; Wan, R.; Hu, G. Shikonin induces apoptosis and necroptosis in pancreatic cancer via regulating the expression of RIP1/RIP3 and synergizes the activity of gemcitabine. Am. J. Transl. Res. 2017, 9, 5507–5517. [Google Scholar]
- Zhou, Z.; Lu, B.; Wang, C.; Wang, Z.; Luo, T.; Piao, M.; Meng, F.; Chi, G.; Luo, Y.; Ge, P. RIP1 and RIP3 contribute to shikonin-induced DNA double-strand breaks in glioma cells via increase of intracellular reactive oxygen species. Cancer Lett. 2017, 390, 77–90. [Google Scholar] [CrossRef]
- Wada, N.; Kawano, Y.; Fujiwara, S.; Kikukawa, Y.; Okuno, Y.; Tasaki, M.; Ueda, M.; Ando, Y.; Yoshinaga, K.; Ri, M.; et al. Shikonin, dually functions as a proteasome inhibitor and a necroptosis inducer in multiple myeloma cells. Int. J. Oncol. 2015, 46, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Deng, B.; Liao, Y.; Shan, L.; Yin, F.; Wang, Z.; Zeng, H.; Zuo, D.; Hua, Y.; Cai, Z. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer 2013, 13, 580. [Google Scholar] [CrossRef]
- Liu, L.; Fan, J.; Ai, G.; Liu, J.; Luo, N.; Li, C.; Cheng, Z. Berberine in combination with cisplatin induces necroptosis and apoptosis in ovarian cancer cells. Biol. Res. 2019, 52, 37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, Z.; Li, Q.; Jiao, H.; Chong, D.; Sun, X.; Zhang, P.; Huo, Q.; Liu, H. Shikonin induces necroptosis by reactive oxygen species activation in nasopharyngeal carcinoma cell line CNE-2Z. J. Bioenerg. Biomembr. 2017, 49, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Hwang, K.E.; Park, D.S.; Oh, S.H.; Jun, H.Y.; Yoon, K.H.; Jeong, E.T.; Kim, H.R.; Kim, Y.S. Shikonin-induced necroptosis is enhanced by the inhibition of autophagy in non-small cell lung cancer cells. J. Transl. Med. 2017, 15, 123. [Google Scholar] [CrossRef]
- Shahsavari, Z.; Karami-Tehrani, F.; Salami, S.; Ghasemzadeh, M. RIP1K and RIP3K provoked by shikonin induce cell cycle arrest in the triple negative breast cancer cell line, MDA-MB-468: Necroptosis as a desperate programmed suicide pathway. Tumour Biol. 2016, 37, 4479–4491. [Google Scholar] [CrossRef]
- Deng, Q.; Yu, X.; Xiao, L.; Hu, Z.; Luo, X.; Tao, Y.; Yang, L.; Liu, X.; Chen, H.; Ding, Z.; et al. Neoalbaconol induces energy depletion and multiple cell death in cancer cells by targeting PDK1-PI3-K/Akt signaling pathway. Cell Death Dis. 2013, 4, e804. [Google Scholar] [CrossRef]
- Yu, X.; Deng, Q.; Li, W.; Xiao, L.; Luo, X.; Liu, X.; Yang, L.; Peng, S.; Ding, Z.; Feng, T.; et al. Neoalbaconol induces cell death through necroptosis by regulating RIPK-dependent autocrine TNFα and ROS production. Oncotarget 2015, 6, 1995–2008. [Google Scholar] [CrossRef]
- Sun, W.; Bao, J.; Lin, W.; Gao, H.; Zhao, W.; Zhang, Q.; Leung, C.H.; Ma, D.L.; Lu, J.; Chen, X. 2-Methoxy-6-acetyl-7-methyljuglone (MAM), a natural naphthoquinone, induces NO-dependent apoptosis and necroptosis by H2O2-dependent JNK activation in cancer cells. Free Radic. Biol. Med. 2016, 92, 61–77. [Google Scholar] [CrossRef]
- Sun, W.; Wu, X.; Gao, H.; Yu, J.; Zhao, W.; Lu, J.J.; Wang, J.; Du, G.; Chen, X. Cytosolic calcium mediates RIP1/RIP3 complex-dependent necroptosis through JNK activation and mitochondrial ROS production in human colon cancer cells. Free Radic. Biol. Med. 2017, 108, 433–444. [Google Scholar] [CrossRef]
- Sun, W.; Yu, J.; Gao, H.; Wu, X.; Wang, S.; Hou, Y.; Lu, J.J.; Chen, X. Inhibition of Lung Cancer by 2-Methoxy-6-Acetyl-7-Methyljuglone Through Induction of Necroptosis by Targeting Receptor-Interacting Protein 1. Antioxid. Redox. Signal. 2019, 31, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; Tao, H.R.; Fang, L.; Kong, X.Q.; Han, L.N.; Li, N.; Xu, Y.X.; Li, L.Y.; Yu, M.; Zhang, H. 11-Methoxytabersonine Induces Necroptosis with Autophagy through AMPK/mTOR and JNK Pathways in Human Lung Cancer Cells. Chem. Pharm. Bull. (Tokyo) 2020, 68, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Hadis Alidadi, M.S. Azin Samimi, Anayatollah Salimi, Atefeh Ashtari, Layasadat Khorsandi, Allium jesdianum Extract Induces Oxidative Stress and Necroptosis in Human Colorectal Cancer (HT-29) Cell Line. Braz. Arch. Biol. Technol. 2021, 64. [Google Scholar]
- Lee, Y.J.; Nam, H.S.; Cho, M.K.; Lee, S.H. Arctigenin induces necroptosis through mitochondrial dysfunction with CCN1 upregulation in prostate cancer cells under lactic acidosis. Mol. Cell Biochem. 2020, 467, 45–56. [Google Scholar] [CrossRef]
- Kang, J.I.; Hong, J.Y.; Choi, J.S.; Lee, S.K. Columbianadin Inhibits Cell Proliferation by Inducing Apoptosis and Necroptosis in HCT116 Colon Cancer Cells. Biomol. Ther. (Seoul) 2016, 24, 320–327. [Google Scholar] [CrossRef]
- Xu, B.; Xu, M.; Tian, Y.; Yu, Q.; Zhao, Y.; Chen, X.; Mi, P.; Cao, H.; Zhang, B.; Song, G.; et al. Matrine induces RIP3-dependent necroptosis in cholangiocarcinoma cells. Cell Death Discov. 2017, 3, 16096. [Google Scholar] [CrossRef]
- Watanabe, S.; Suzuki, T.; Hara, F.; Yasui, T.; Uga, N.; Naoe, A. Polyphyllin D, a steroidal saponin in Paris polyphylla, induces apoptosis and necroptosis cell death of neuroblastoma cells. Pediatr. Surg. Int. 2017, 33, 713–719. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, C.; Lu, B.; Zhou, Z.; Jin, Y.; Wang, Z.; Zheng, L.; Liu, K.; Luo, T.; Zhu, D.; et al. Pristimerin triggers AIF-dependent programmed necrosis in glioma cells via activation of JNK. Cancer Lett. 2016, 374, 136–148. [Google Scholar] [CrossRef]
- Zhao, X.; Quan, J.; Tan, Y.; Liu, Y.; Liao, C.; Li, Z.; Liao, W.; Liu, J.; Cao, Y.; Luo, X. RIP3 mediates TCN-induced necroptosis through activating mitochondrial metabolism and ROS production in chemotherapy-resistant cancers. Am. J. Cancer Res. 2021, 11, 729–745. [Google Scholar]
- Liu, T.; Sun, X.; Cao, Z. Shikonin-induced necroptosis in nasopharyngeal carcinoma cells via ROS overproduction and upregulation of RIPK1/RIPK3/MLKL expression. Onco Targets Ther. 2019, 12, 2605–2614. [Google Scholar] [CrossRef]
- Xiong, J.; Wang, L.; Fei, X.C.; Jiang, X.F.; Zheng, Z.; Zhao, Y.; Wang, C.F.; Li, B.; Chen, S.J.; Janin, A.; et al. MYC is a positive regulator of choline metabolism and impedes mitophagy-dependent necroptosis in diffuse large B-cell lymphoma. Blood Cancer J. 2017, 7, e0. [Google Scholar] [CrossRef] [PubMed]
- LingHu, H.R.; Luo, H.; Gang, L. Bufalin Induces Glioma Cell Death by Apoptosis or Necroptosis. Onco Targets Ther. 2020, 13, 4767–4778. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, G.; Han, G.; Feng, S.; Liu, Y.; Chen, J.; Liu, C.; Zhao, L.; Jin, F. Emodin induced necroptosis in the glioma cell line U251 via the TNF-α/RIP1/RIP3 pathway. Investig. New Drugs 2020, 38, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Mbaveng, A.T.; Bitchagno, G.T.M.; Kuete, V.; Tane, P.; Efferth, T. Cytotoxicity of ungeremine towards multi-factorial drug resistant cancer cells and induction of apoptosis, ferroptosis, necroptosis and autophagy. Phytomedicine 2019, 60, 152832. [Google Scholar] [CrossRef]
- Han, Q.; Ma, Y.; Wang, H.; Dai, Y.; Chen, C.; Liu, Y.; Jing, L.; Sun, X. Resibufogenin suppresses colorectal cancer growth and metastasis through RIP3-mediated necroptosis. J. Transl. Med. 2018, 16, 201. [Google Scholar] [CrossRef]
- Lu, Z.; Wu, C.; Zhu, M.; Song, W.; Wang, H.; Wang, J.; Guo, J.; Li, N.; Liu, J.; Li, Y.; et al. Ophiopogonin D' induces RIPK1-dependent necroptosis in androgen-dependent LNCaP prostate cancer cells. Int. J. Oncol. 2020, 56, 439–447. [Google Scholar] [CrossRef]
- Shao, C.S.; Feng, N.; Zhou, S.; Zheng, X.X.; Wang, P.; Zhang, J.S.; Huang, Q. Ganoderic acid T improves the radiosensitivity of HeLa cells via converting apoptosis to necroptosis. Toxicol. Res. (Camb) 2021, 10, 531–541. [Google Scholar] [CrossRef]
- Delehouzé, C.; Comte, A.; Leon-Icaza, S.A.; Cougoule, C.; Hauteville, M.; Goekjian, P.; Bulinski, J.C.; Dimanche-Boitrel, M.T.; Meunier, E.; Rousselot, M.; et al. Nigratine as dual inhibitor of necroptosis and ferroptosis regulated cell death. Sci. Rep. 2022, 12, 5118. [Google Scholar] [CrossRef]
- Byun, H.S.; Ju, E.; Park, K.A.; Sohn, K.C.; Jung, C.S.; Hong, J.H.; Ro, H.; Lee, H.Y.; Quan, K.T.; Park, I.; et al. Rubiarbonol B induces RIPK1-dependent necroptosis via NOX1-derived ROS production. Cell Biol. Toxicol. 2022. [Google Scholar] [CrossRef]
- Bhosale, P.B.; Abusaliya, A.; Kim, H.H.; Ha, S.E.; Park, M.Y.; Jeong, S.H.; Vetrivel, P.; Heo, J.D.; Kim, J.A.; Won, C.K.; et al. Apigetrin Promotes TNFα-Induced Apoptosis, Necroptosis, G2/M Phase Cell Cycle Arrest, and ROS Generation through Inhibition of NF-κB Pathway in Hep3B Liver Cancer Cells. Cells 2022, 11, 2734. [Google Scholar] [CrossRef]
- Pawlikowska, M.; Jędrzejewski, T.; Brożyna, A.A.; Wrotek, S. Protein-Bound Polysaccharides from Coriolus Versicolor Induce RIPK1/RIPK3/MLKL-Mediated Necroptosis in ER-Positive Breast Cancer and Amelanotic Melanoma Cells. Cell Physiol. Biochem. 2020, 54, 591–604. [Google Scholar] [PubMed]
- Liu, X.; Zhang, Y.; Gao, H.; Hou, Y.; Lu, J.J.; Feng, Y.; Xu, Q.; Liu, B.; Chen, X. Induction of an MLKL mediated non-canonical necroptosis through reactive oxygen species by tanshinol A in lung cancer cells. Biochem. Pharmacol. 2020, 171, 113684. [Google Scholar] [CrossRef] [PubMed]
- Chromik, J.; Safferthal, C.; Serve, H.; Fulda, S. Smac mimetic primes apoptosis-resistant acute myeloid leukaemia cells for cytarabine-induced cell death by triggering necroptosis. Cancer Lett. 2014, 344, 101–109. [Google Scholar] [CrossRef]
- Hannes, S.; Abhari, B.A.; Fulda, S. Smac mimetic triggers necroptosis in pancreatic carcinoma cells when caspase activation is blocked. Cancer Lett. 2016, 380, 31–38. [Google Scholar] [CrossRef]
- Cabal-Hierro, L.; O’Dwyer, P.J. TNF Signaling through RIP1 Kinase Enhances SN38-Induced Death in Colon Adenocarcinoma. Mol. Cancer Res. 2017, 15, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Kaku, Y.; Tsuchiya, A.; Kanno, T.; Nakano, T.; Nishizaki, T. Diarachidonoylphosphoethanolamine induces necrosis/necroptosis of malignant pleural mesothelioma cells. Cell Signal. 2015, 27, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Konstantakou, E.G.; Voutsinas, G.E.; Velentzas, A.D.; Basogianni, A.S.; Paronis, E.; Balafas, E.; Kostomitsopoulos, N.; Syrigos, K.N.; Anastasiadou, E.; Stravopodis, D.J. 3-BrPA eliminates human bladder cancer cells with highly oncogenic signatures via engagement of specific death programs and perturbation of multiple signaling and metabolic determinants. Mol. Cancer 2015, 14, 135. [Google Scholar] [CrossRef]
- Guan, R.; Xie, L.; Wang, L.; Zhou, Y.; Chen, Y.; Ji, L.; Chao, H. Necroptosis-inducing iridium(iii) complexes as regulators of cyclin-dependent kinases. Inorg. Chem. Front. 2021, 8, 1788–1794. [Google Scholar] [CrossRef]
- Sulkshane, P.; Teni, T. BH3 mimetic Obatoclax (GX15-070) mediates mitochondrial stress predominantly via MCL-1 inhibition and induces autophagy-dependent necroptosis in human oral cancer cells. Oncotarget 2017, 8, 60060–60079. [Google Scholar] [CrossRef]
- English, C.; Aloi, J.J. New FDA-Approved Disease-Modifying Therapies for Multiple Sclerosis. Clin. Ther. 2015, 37, 691–715. [Google Scholar] [CrossRef]
- Saddoughi, S.A.; Gencer, S.; Peterson, Y.K.; Ward, K.E.; Mukhopadhyay, A.; Oaks, J.; Bielawski, J.; Szulc, Z.M.; Thomas, R.J.; Selvam, S.P.; et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol. Med. 2013, 5, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, H.; Ding, K.; Xu, J. FTY720 induces autophagy-related apoptosis and necroptosis in human glioblastoma cells. Toxicol. Lett. 2015, 236, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Zhao, Y.; Ma, C.Y.; Xu, X.M.; Zhang, Y.Q.; Wang, C.G.; Jin, J.; Shen, X.; Gao, J.L.; Li, N.; et al. Dimethyl fumarate induces necroptosis in colon cancer cells through GSH depletion/ROS increase/MAPKs activation pathway. Br. J. Pharmacol. 2015, 172, 3929–3943. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.; Yang, N.; Montpetit, R.; Kirschenman, R.; Lemieux, H.; Goping, I.S. BAD sensitizes breast cancer cells to docetaxel with increased mitotic arrest and necroptosis. Sci. Rep. 2020, 10, 355. [Google Scholar] [CrossRef]
- Jiang, Y.; Shan, S.; Chi, L.; Zhang, G.; Gao, X.; Li, H.; Zhu, X.; Yang, J. Methyl methanesulfonate induces necroptosis in human lung adenoma A549 cells through the PIG-3-reactive oxygen species pathway. Tumour Biol. 2016, 37, 3785–3795. [Google Scholar] [CrossRef]
- Santofimia-Castaño, P.; Xia, Y.; Lan, W.; Zhou, Z.; Huang, C.; Peng, L.; Soubeyran, P.; Velázquez-Campoy, A.; Abián, O.; Rizzuti, B.; et al. Ligand-based design identifies a potent NUPR1 inhibitor exerting anticancer activity via necroptosis. J. Clin. Investig. 2019, 129, 2500–2513. [Google Scholar] [CrossRef]
- Kong, Q.; Lv, J.; Yan, S.; Chang, K.J.; Wang, G. A Novel Naphthyridine Derivative, 3u, Induces Necroptosis at Low Concentrations and Apoptosis at High Concentrations in Human Melanoma A375 Cells. Int. J. Mol. Sci. 2018, 19, 2975. [Google Scholar] [CrossRef]
- Chengzhu, W.U.; Gao, M.; Shen, L.; Bohan, L.I.; Bai, X.; Gui, J.; Hongmei, L.I.; Huo, Q.; Tao, M.A. Miconazole triggers various forms of cell death in human breast cancer MDA-MB-231 cells. Pharmazie 2019, 74, 290–294. [Google Scholar]
- You, B.J.; Hour, M.J.; Chen, L.Y.; Luo, S.C.; Hsu, P.H.; Lee, H.Z. Fenofibrate induces human hepatoma Hep3B cells apoptosis and necroptosis through inhibition of thioesterase domain of fatty acid synthase. Sci. Rep. 2019, 9, 3306. [Google Scholar] [CrossRef]
- Ma, Y.M.; Peng, Y.M.; Zhu, Q.H.; Gao, A.H.; Chao, B.; He, Q.J.; Li, J.; Hu, Y.H.; Zhou, Y.B. Novel CHOP activator LGH00168 induces necroptosis in A549 human lung cancer cells via ROS-mediated ER stress and NF-κB inhibition. Acta Pharmacol. Sin. 2016, 37, 1381–1390. [Google Scholar] [CrossRef]
- Xie, Y.; Zhu, S.; Zhong, M.; Yang, M.; Sun, X.; Liu, J.; Kroemer, G.; Lotze, M.; Zeh, H.J., 3rd; Kang, R.; et al. Inhibition of Aurora Kinase A Induces Necroptosis in Pancreatic Carcinoma. Gastroenterology 2017, 153, 1429–1443.e5. [Google Scholar] [CrossRef] [PubMed]
- Chefetz, I.; Grimley, E.; Yang, K.; Hong, L.; Vinogradova, E.V.; Suciu, R.; Kovalenko, I.; Karnak, D.; Morgan, C.A.; Chtcherbinine, M.; et al. A Pan-ALDH1A Inhibitor Induces Necroptosis in Ovarian Cancer Stem-like Cells. Cell Rep. 2019, 26, 3061–3075.e6. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ni, H.; Wu, S.; Han, S.; Chen, C.; Li, L.; Li, Y.; Gui, F.; Han, J.; Deng, X. Targeting RIPK3 oligomerization blocks necroptosis without inducing apoptosis. FEBS Lett. 2020, 594, 2294–2302. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Lei, J.; Yin, M.M.; Chen, Y. Organoantimony (III) Derivative Induces Necroptosis in Human Breast Cancer MDA-MB-231 Cells. Anticancer Agents Med. Chem. 2022, 22, 2448–2457. [Google Scholar] [CrossRef] [PubMed]
- Sonkusre, P.; Cameotra, S.S. Biogenic selenium nanoparticles induce ROS-mediated necroptosis in PC-3 cancer cells through TNF activation. J. Nanobiotechnol. 2017, 15, 43. [Google Scholar] [CrossRef] [PubMed]
- Maryam Farasat, F.N. Layasadat Khorsandi, Zinc oxide nanoparticles induce necroptosis and inhibit autophagy in MCF-7 human breast cancer cells. Biologia 2020, 75, 161–174. [Google Scholar] [CrossRef]
- Palakorn Kaokaen, N.C. Phongsakorn Kunhorm, Kedkanya Mesil, Thunwa Binlateh, Parinya Noisa, Paiboon Jitprasertwong, Nanoencapsulation of Cordycepin Induces Switching from Necroptosis to Apoptosis in Human Oral Cancer Cells (HSC-4) Through Inhibition of Receptor-Interacting Serine/Threonine-Protein Kinase 3 (RIPK3) and Autophagy Modulation. Nat. Prod. Commun. SAGE J. 2022, 17, 1934578X221074838. [Google Scholar]
- Van Hoecke, L.; Van Lint, S.; Roose, K.; Van Parys, A.; Vandenabeele, P.; Grooten, J.; Tavernier, J.; De Koker, S.; Saelens, X. Treatment with mRNA coding for the necroptosis mediator MLKL induces antitumor immunity directed against neo-epitopes. Nat. Commun. 2018, 9, 3417. [Google Scholar] [CrossRef]
- Gulei, D.; Berindan-Neagoe, I. Activation of Necroptosis by Engineered Self Tumor-Derived Exosomes Loaded with CRISPR/Cas9. Mol. Ther. Nucleic Acids 2019, 17, 448–451. [Google Scholar] [CrossRef]
- Kang, T.; Huang, Y.; Zhu, Q.; Cheng, H.; Pei, Y.; Feng, J.; Xu, M.; Jiang, G.; Song, Q.; Jiang, T.; et al. Necroptotic cancer cells-mimicry nanovaccine boosts anti-tumor immunity with tailored immune-stimulatory modality. Biomaterials 2018, 164, 80–97. [Google Scholar] [CrossRef]
- Snyder, A.G.; Hubbard, N.W.; Messmer, M.N.; Kofman, S.B.; Hagan, C.E.; Orozco, S.L.; Chiang, K.; Daniels, B.P.; Baker, D.; Oberst, A. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci. Immunol. 2019, 4, eaaw2004. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kitatani, K.; Toyoshima, M.; Ishibashi, M.; Usui, T.; Minato, J.; Egiz, M.; Shigeta, S.; Fox, T.; Deering, T.; et al. Ceramide Nanoliposomes as a MLKL-Dependent, Necroptosis-Inducing, Chemotherapeutic Reagent in Ovarian Cancer. Mol. Cancer Ther. 2018, 17, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Akara-Amornthum, P.; Lomphithak, T.; Choksi, S.; Tohtong, R.; Jitkaew, S. Key necroptotic proteins are required for Smac mimetic-mediated sensitization of cholangiocarcinoma cells to TNF-α and chemotherapeutic gemcitabine-induced necroptosis. PLoS ONE 2020, 15, e0227454. [Google Scholar] [CrossRef] [PubMed]
- Gerges, S.; Rohde, K.; Fulda, S. Cotreatment with Smac mimetics and demethylating agents induces both apoptotic and necroptotic cell death pathways in acute lymphoblastic leukemia cells. Cancer Lett. 2016, 375, 127–132. [Google Scholar] [CrossRef]
- Rohde, K.; Kleinesudeik, L.; Roesler, S.; Löwe, O.; Heidler, J.; Schröder, K.; Wittig, I.; Dröse, S.; Fulda, S. A Bak-dependent mitochondrial amplification step contributes to Smac mimetic/glucocorticoid-induced necroptosis. Cell Death Differ. 2017, 24, 83–97. [Google Scholar] [CrossRef]
- Bolik, J.; Krause, F.; Stevanovic, M.; Gandraß, M.; Thomsen, I.; Schacht, S.S.; Rieser, E.; Müller, M.; Schumacher, N.; Fritsch, J.; et al. Inhibition of ADAM17 impairs endothelial cell necroptosis and blocks metastasis. J. Exp. Med. 2022, 219, e20201039. [Google Scholar] [CrossRef]






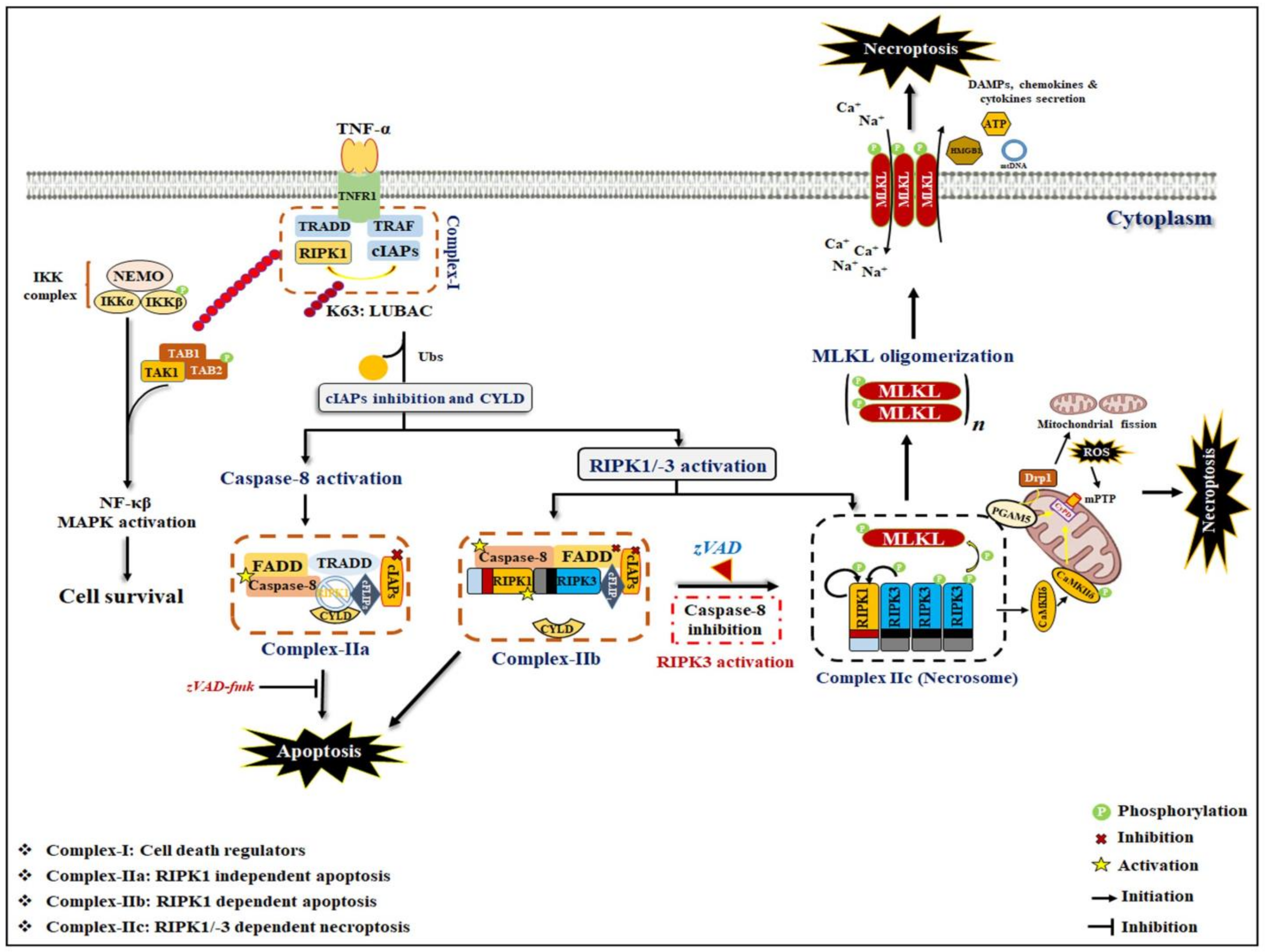{kind=link}
{kind=link}
{kind=link}
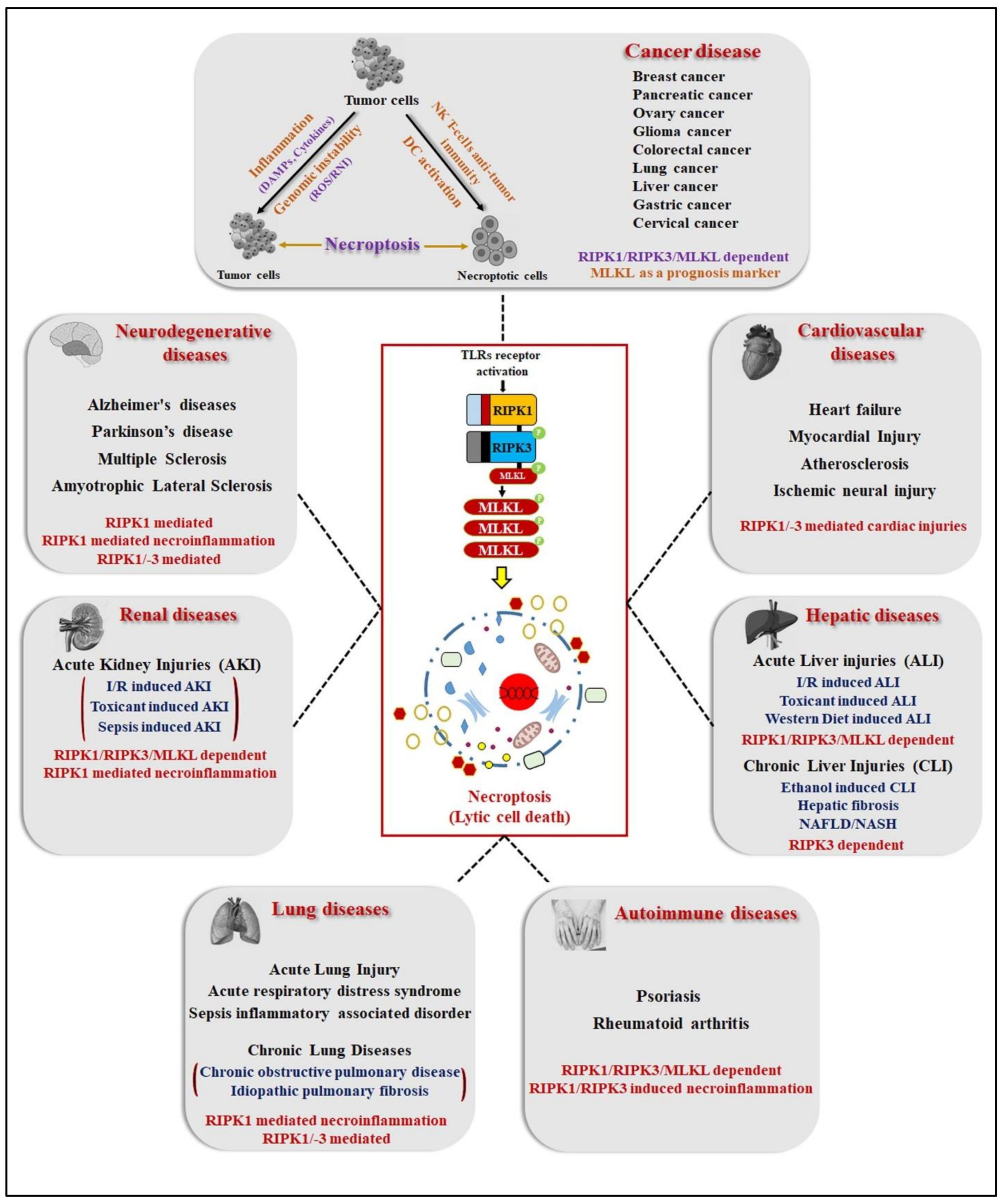{kind=link}
{kind=link}
| Apoptosis | Necroptosis | References | |
|---|---|---|---|
| Morphology | Cytoplasmic shrinkage, Chromatin condensation, DNA fragmentation, Cell membrane blebbing, Apoptotic body formation and shedding | Translucent cytoplasm, Swelling sub-cellular membrane organelles, Increased cell volume, Early loss of membrane integrity, Spillage of intracellular contents into extracellular space | [1,3,5,6,9] |
| Death signaling factors | BID, BAX, Bcl-2, Cytochrome c, APAF1, FADD, Caspase-8, Caspase-9 | RIPK1, RIPK3, FADD, Caspase-8, cFLIPs, cIAPs, CYLD | [1,3,9,14,23] |
| Death execution factors | Caspase-3, Caspase-7 | p-MLKL (core executioner), PGAM5, Drp1 | [24,25,26,27] |
| Death execution events | Caspase-3 activation after interacts with caspase-8 and -9 caused cell shrinkage, DNA fragmentation, shedding of apoptotic body formation | MLKL phosphorylation, oligomerization and plasma membrane translocation causes membrane permeabilization and necroptosis induction | [3,24,28] |
| Methods to examine death events | TUNEL assays, DNA laddering, Annexin-V/PI positive cells counting, Caspase-3/-7 activity assays | PI staining (staining with impermeant dyes), HMGB1 & LDH release assay, Detection of RIPK1, RIPK3 and MLKL expression and membrane localization, Detection of necroptosis cells morphology | [24,25,26,27,29] |
| No. | Pathological Diseases | Disease Models | Involvement of Necroptosis | References | |
|---|---|---|---|---|---|
| 1 | Neurological disease | Multiple sclerosis | Postmortem brain tissue samples of human patients; RIPK3−/− and Cuprizone-induced MS mice model | Increased RIPK1/-3 expression in microglia and macrophages in both human and mouse brain samples,Reduced cleaved caspase-8 expression | [69,70] |
| EAE mice model | NF-κβ signaling mediated inflammatory response promotes FLIPL expression leading to necroptosis without caspase-8 activation | [70,71,72] | |||
| Elevated expression of RIPK1, increased RIPK3 and MLKL phosphorylation in both microglia and oligodendrocytes primarily localized in the white matter of brain. | [70,73] | ||||
| Amyotrophic sclerosis | SOD1G93A-mutated mice | Increased levels of RIPK1 and RIPK3 in MNs and astrocytes primarily localized in the spinal cord | [74,75,76] | ||
| Optn−/− mice | Elevated levels of RIPK1, RIPK3 and pMLKL and necroptosis in the MNs of spinal cord | [76] | |||
| TBK1−/− mice; TBK1−/− cell line | Higher level of RIPK1 and TNF-α induced necroptosis | [77,78] | |||
| Alzheimer’s disease | Postmortem brain tissue samples and 23 AD, 24 p-preAD brain tissue samples from human patients; 5xFAD mice model | Elevated level of RIPK1/-3/MLKL at both mRNA and protein level | [79,80,81] | ||
| Tg2576 APP/PS1 mice model | Higher RIPK1 activation drives Cst7 expression in microglia leads to lysosomal dysfunction and blocked proteostasis of deposit Aβ-plaques in AD | [80,82] | |||
| cpdm mice model | Attenuation of M1 ubiquitination of RIPK1 to stimulate its activation | [83] | |||
| Parkinson’s disease | OPA1-mutant PD patients derived iPSC; OPA1-A495V and OPA1-G488R neuronal cell lines | Higher phosphorylation level of RIPK1/-3/MLKL and mitochondrial dysfunction-induced necroptosis | [84] | ||
| PD models (PC12 & SH-SY5Y cell line; 6-OHDA & MPTP mice model) | Promotes necrosome complex formation (RIPK1/-3/MLKL),ROS stimulated RIPK1 activation following with ASK1/JNK signaling activation | [84,85,86] | |||
| 2 | Renal disease | I/R-induced AKI | IRI C57BL/6 mouse model | RIPK3- and CypD and PPIF mediated mPT-dependent necroptosis | [87,88,89] |
| Toxicant-induced AKI | FA-AKI renal mouse model | RIPK3/MLKL- and TWEAK-Fn14 axis-induced necroinflammation mediated necroptosis | [90,91] | ||
| Cisplatin & Oxalate crystals nephrotoxicant mice model | RIPK3- and CypD-induced necroinflammation mediated necroptosis in the renal proximal tubular epithelial cells and whole kidney lysates,MLKL dependent necroptosis via higher MLKL membrane association in oxalate crystal-treated renal PTC cells | [92,93,94,95] | |||
| Sepsis-induced AKI | CLP C57BL/6N mice model | RIPK3 induced higher OS and mitochondrial dysfunction via upregulation of NOX4 and mitochondrial complex-I/III inhibition | [96] | ||
| Renal fibrosis CKD | RIPK3−/− UUO and AD fibrosis mice model | RIPK3 exacerbates kidney fibrogenesis via the PI3K-AKT dependent activation of ACL metabolic activity | [95] | ||
| IRI mice model; OGBD treated PTC cell line | RIPK3/MLKL-dependent necroptosis mediated necroinflammation promotes renal interstitial fibrogenesis | [97] | |||
| 3 | Hepatic disease | Alcoholic liver disease | Chronic alcoholic mice model | CYP2E1-mediated higher RIP3 expression induces hepatocyte necroptosis during chronic alcohol treatment | [98] |
| Gao-binge (chronic alcoholic) mice model | Impaired hepatic proteasome function via chronic alcohol exposure led to the higher accumulation of RIPK3 and induced necroptosis | [99,100] | |||
| NAFLD and NASH diseases | HFCD and MCD diet mice model | TNF-α mediated RIPK3-dependent necroptosis, -oxidative stress and -necroinflammation in mice liver lysates | [101,102,103,104] | ||
| FFC diet mice model | MLKL-dependent necroptotic signaling promotes FFC diet-induced liver injuries and necroinflammation via autophagy-flux inhibition | [105] | |||
| 4 | Pulmonary disease | Acute lung injuries (ALI) | VILI- induced ALI mice model | Impaired mitochondria-FAO metabolism pathway via MV exposure led to the RIPK3-dependent necroptosis mediates ALI pathogenesis | [106] |
| LPS/ATP-induced ALI mice model | RIPK3 mediated NLRP3 inflammasome activation promotes lung injury via markedly increase inflammatory cell influx,IL-1α/β, IL-6, IL-18 secretion and HMGB1 release in infiltrating macrophages/monocytes cells | 133 | |||
| Acute respiratory distress syndrome | Oleic acid-fed SD rat ARDS model | Elevated expression of TNF-α and RIPK1, RIPK3, MLKL in the BALF and lung tissue of OA-fed SD rat | [107] | ||
| LPS-induced ARDS mice model | RIPK3-dependent necroptosis enhanced necroinflammation, ROS overgeneration and lung tissue injuries via increasing neutrophil infiltration and cytokines secretion in lung parenchyma tissue | [108,109] | |||
| Sepsis/systemic inflammatory associated disorder (SIRS) | TNF-α induced SIRS mice model | RIPK1/-3 induced necroptosis with the DAMPs release and pro-inflammatory cytokines secretion associated with higher systemic inflammation and polymicrobial sepsis | [109] | ||
| Cecal slurry-induced neonatal sepsis C57BL/6 mouse model | Elevated expression of RIPK1 and RIPK3 associated with systemic and pulmonary inflammation and lung injuries in neonatal sepsis | [110,111] | |||
| Chronic obstructive pulmonary disease | Cigarette smoke (CS)-exposed Beas-2B cells and - C57BL/6 mice model | CS-induced mitoROS, mitochondrial dysfunction and mitophagy leads to necroptotic signaling activation with necroinflammation in pulmonary epithelial cells and alveolar macrophages | [112,113,114,115,116] | ||
| Idiopathic pulmonary fibrosis (IPF) | BLM-induced IPF mice model | RIPK3 mediated necroptosis and DAMPs release induced necroinflammation and lung injury via increasing neutrophil infiltration and cytokines secretion in the alveolar epithelial cells | [117] | ||
| 5 | Cardiovascular disease | Heart failure | I/R or cardiotoxicant (doxorubicin) induced cardiac injury murine and pig model | Elevated level of RIPK1, RIPK3 and MLKL (phosphorylation) in the rat myocardial tissues, Detection of RIPK1/RIPK3 necrosome complex formation,Higher RIPK3 level caused activation of CAMKII as a results MPT opening and myocardial necroptosis | [118,119,120,121,122] |
| Atherosclerosis | OxLDL- or APOE-deficient mice model | Higher RIPK3 activation and subsequent necroptosis induction atheroma macrophages leads to the local and systemic inflammation and atherosclerotic lesions | [123,124] | ||
| Aortic aneurysm | Elastase-induced AAA C57BL/6 mice model | Increased RIPK1 and RIPK3 level in aneurysmal tissues promotes AAA progression via caused SMC necroptosis and vascular inflammation (lymphocytes infiltration and DAMPs release) | [125,126] | ||
| Calcium phosphate-induced AAA C57BL/6 mice model | GSK'074 treatment diminished AAA progression by impeding SMC necroptosis, necroinflammation (macrophages infiltration and cytokines secretion) and aortic expansion in aneurysm-prone aortae | [127] | |||
| 6 | Autoimmune diseases | Psoriasis | Primary epidermal keratinocytes cultured cells; Aldara-/IMQ- induced psoriasis mice models | Enhanced RIPK3 expression facilitates psoriatic inflammation by neutrophil-mediated cytokines/chemokines secretion,Elevated RIPK1/-3/MLKL expression, detection of their localization in all layers of epidermal skin cells confirmed by confocal microscopy. | [128,129,130] |
| Rheumatoid arthritis (RA) | Collagen-induced arthritis mice model | Enhanced RIPK1/RIPK3/MLKL expression promotes NLRP3 inflammasome complex formation and cytokines secretion in the synovium joint tissue,IFN-γ diminished necroptosis-mediated RA progression by downregulation of MLKL expression | [131,132] | ||
| 7 | Cancer disease | Acute myeloid leukemia (AML) | DA1-3b p210BCR-ABL leukemia murine cell line; DA1-3b/C3HeOuJ mouse model | Reduced RIPK3 expression observed in clinical samples from human AML patients was independently associated with poor prognosis,RIPK3 downregulation suggest diminished necroptosis via p65/RelA cleavage and as results shift towards NF-κβ mediate cell survival in DA1-3b leukemia cells | [133] |
| Chronic lymphocytic leukemia (CLL) | Primary CLL cells from human CLL patients | Lower expression of RIPK3 and CYLD reduced TNF-α/zVAD mediated necroptosis in CML cells | [134] | ||
| Head and neck squamous cell carcinoma (HNSCC) | Primary and metastatic cell lines and tissues from human HNSCC patients | Enhanced genomic methylation of RIPK1 promoter caused reduced TLR3-mediated apoptosis/necroptosis with higher tumor cells migration in HNSCC metastatic cells,Loss of RIPK1 expression strongly linked with metastatic disease in a cohort of HNSCC patients | [135] | ||
| Non-small cell lung cancer (NSCLC) | Clinical samples from 253 and 394 NSCLC human patients | Lower RIPK1/-3/MLKL expression was linked with poor prognosis in a NSCLC tissues associated with reduced levels of necroptosis signaling, Lower RIPK1 and PELI with higher p53 expression found to be linked with a worse OS of stage-I NSCLC patients | [136] | ||
| Ovarian cancer | Clinical samples from 75 human ovarian cancer patients | Low MLKL level was significantly linked with both reduced DFS and OS of patients with ovarian cancer, MLKL serves as a potential prognosis marker in ovarian cancer patients | [137] | ||
| Colon cancer | HT29, Colo205, HCT116, SW480 colon cancer cell lines; Colon cancer tissues samples from human colon cancer patients | Reduce RIPK1 and RIPK3 expressions (due to hypoxia) resists cancer cells to chemotherapeutic agents-induced necroptotic cell death; Poly I:C induces TLR3/RIP3-dependent necroptosis and tumor growth inhibition via induction of DC cells mediated anti-tumor activity in colon cancer tissues | [138] | ||
| Cervical cancer | HPV18-positive HeLa and C4-I cervical carcinoma cell lines | RIPK3-upregulation stimulates IL1-α secretion from Poly I:C-induced necroptotic cervical cancer cells leads to activation DC cells, selective activation of DC cells shows anti-tumor effects via producing cytotoxic cytokine IL-12 or activating CD8+ T-cells | [139] | ||
| Melanoma | DM733, DM598, DM738 and DM833 melanoma cell lines; A2058 cells injected NCr-nu/nu miceB16 mouse melanoma cell lines and B16 cells injected C57Bl/6 mice melanoma metastasis model | Selective inhibition of CYLD expression and activation of JNK/AP-1 and β1-integrin signaling by Snail is critical to protects melanoma from necroptosis in melanocytes and nevus cells, zVAD-fmk in combination with RT, CT, HT increased necroptosis in B16-F10 melanoma cells concomitantly with HMGB1 secretion and increase cytotoxic DCs as well as CD8+ T-cells infiltration around tumor microenvironment. RIPK3 upregulation induces NK T-cells mediated anti-tumor responses via mitochondrial PGAMF-NFAT/Drp1 signaling activation in metastatic melanoma tumor cells;Increased RIPK1 expression positively linked with the higher metastatic rate and a poor prognosis in melanoma potentially due to NF-κβ-mediated pro-tumorigenic abilities | [140,141,142] | ||
| Glioblastoma | Tissue samples from human glioblastoma patients | Increased RIPK1 levels prevents p53 induction via augmenting mdm2 level and NF-κβ signaling activation to confers a poor prognosis in glioblastoma | [143] | ||
| Lung cancer | HBEC-2, HBEC-13 and BEAS-2B cell lines; Cigarette smoke/Benzo(a)pyrene-exposed mouse lung tumor model | Higher RIPK1 expression promotes BPDE-induced malignant transformation of HBEC cells by catalase-mediated suppression of ROS overproduction as well as MAPKs signaling deactivation | [144] | ||
| Breast cancer | Clinical samples of human breast cancer patients; MDA-MB231, 4T1, MCF10A and MCF12A breast cancer cell lines; MDA-MB-231/4T1 xenografted Balb/C mice model | Nearly 85% patients of breast cancer have reduced RIPK3 expression that was associated with a worse prognosis, Lower RIPK3 expression suggest by genomic methylation of transcription factor in its transcriptional sites in breast cancer cells; Lower RIPK1/-3/MLKL expression found to be linked with reduced breast cancer tumorigenicity due to impairment of NF-κβ-mediated pro-tumor growth cytokines secretion | [142] | ||
| Pancreatic adenocarcinoma (PAC) | Tissue samples from 80 human PAC patients; AsPC1, PANC1, and MIA PaCa-2 pancreatic cancer cell lines; KrasG12D PDEC/ FC1242 cells xenografted C57BL/6 mice | Decreased MLKL expression was strongly correlated with cancer progression with diminished necroptosis, MLKL serves as a therapeutic targets in PAC cells, RIPK1/-3 dependent necroptosis signaling promotes pancreatic tumorigenicity through enhancing CXCL1 and Mincle-induced adaptive immune suppression in tumor-infiltrating myeloid cells; Higher RIPK3 and MLKL levels promotes the pancreatic cancer cells migration and invasion via the activation of CXCL5-CXCR2 axis | [145] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaouhan, H.S.; Vinod, C.; Mahapatra, N.; Yu, S.-H.; Wang, I.-K.; Chen, K.-B.; Yu, T.-M.; Li, C.-Y. Necroptosis: A Pathogenic Negotiator in Human Diseases. Int. J. Mol. Sci. 2022, 23, 12714. https://doi.org/10.3390/ijms232112714
Chaouhan HS, Vinod C, Mahapatra N, Yu S-H, Wang I-K, Chen K-B, Yu T-M, Li C-Y. Necroptosis: A Pathogenic Negotiator in Human Diseases. International Journal of Molecular Sciences. 2022; 23(21):12714. https://doi.org/10.3390/ijms232112714
Chicago/Turabian StyleChaouhan, Hitesh Singh, Ch Vinod, Nikita Mahapatra, Shao-Hua Yu, I-Kuan Wang, Kuen-Bao Chen, Tung-Min Yu, and Chi-Yuan Li. 2022. "Necroptosis: A Pathogenic Negotiator in Human Diseases" International Journal of Molecular Sciences 23, no. 21: 12714. https://doi.org/10.3390/ijms232112714
APA StyleChaouhan, H. S., Vinod, C., Mahapatra, N., Yu, S.-H., Wang, I.-K., Chen, K.-B., Yu, T.-M., & Li, C.-Y. (2022). Necroptosis: A Pathogenic Negotiator in Human Diseases. International Journal of Molecular Sciences, 23(21), 12714. https://doi.org/10.3390/ijms232112714