


Internalized Amyloid-β (1-42) Peptide Inhibits the Store-Operated Calcium Entry in HT-22 Cells

 ,
,  ,
,  , and
, and 
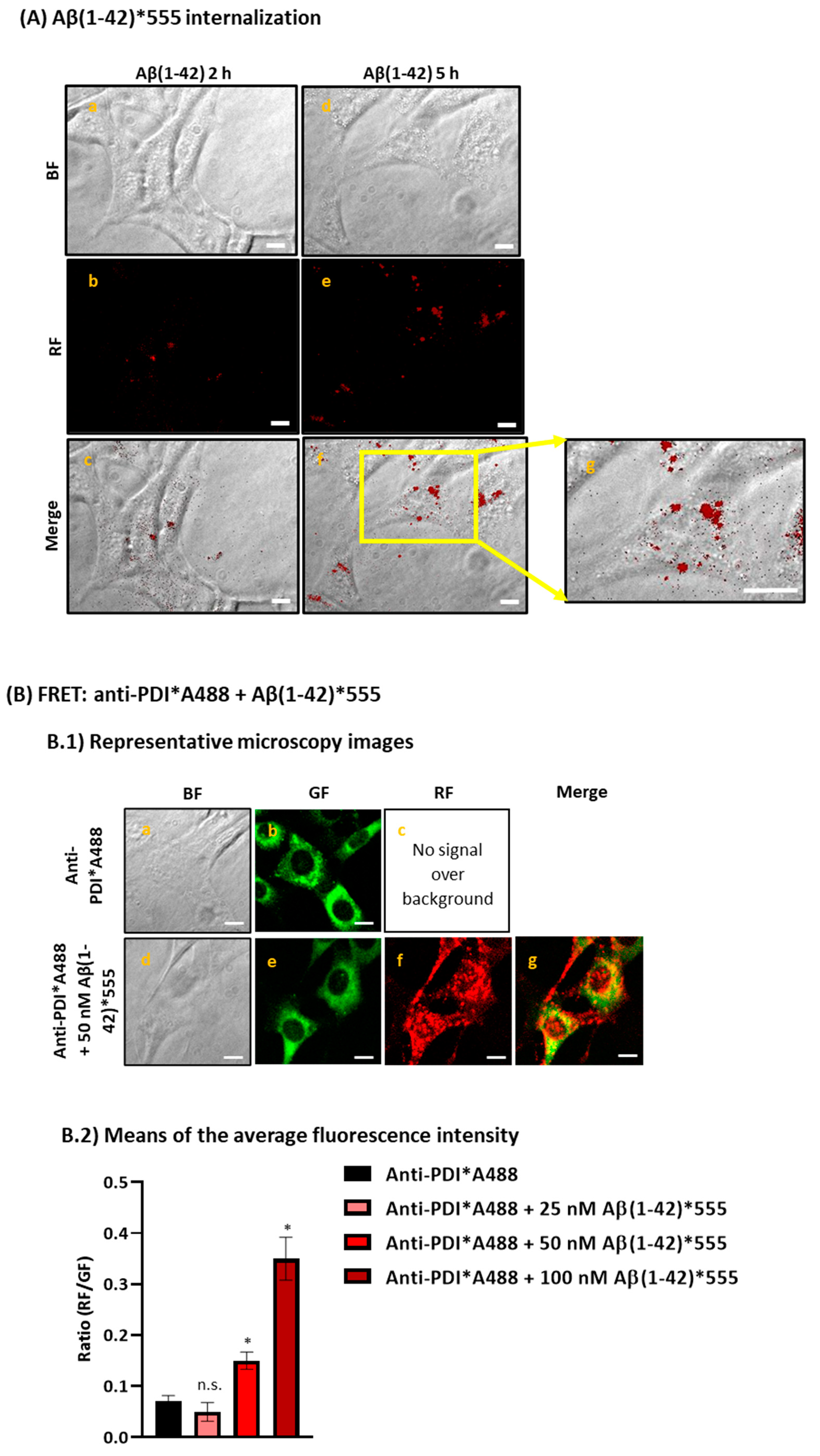{kind=link}
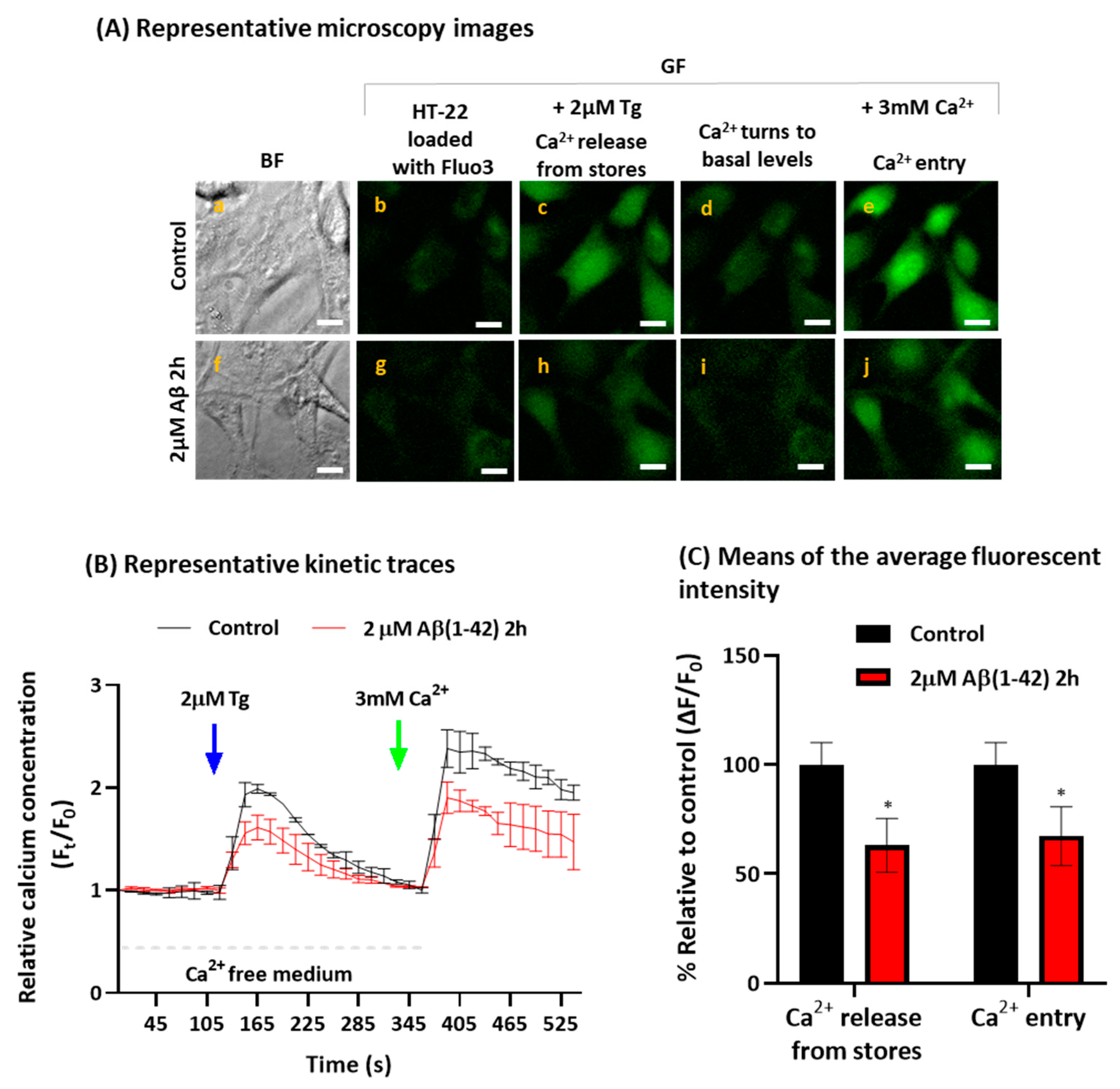{kind=link}
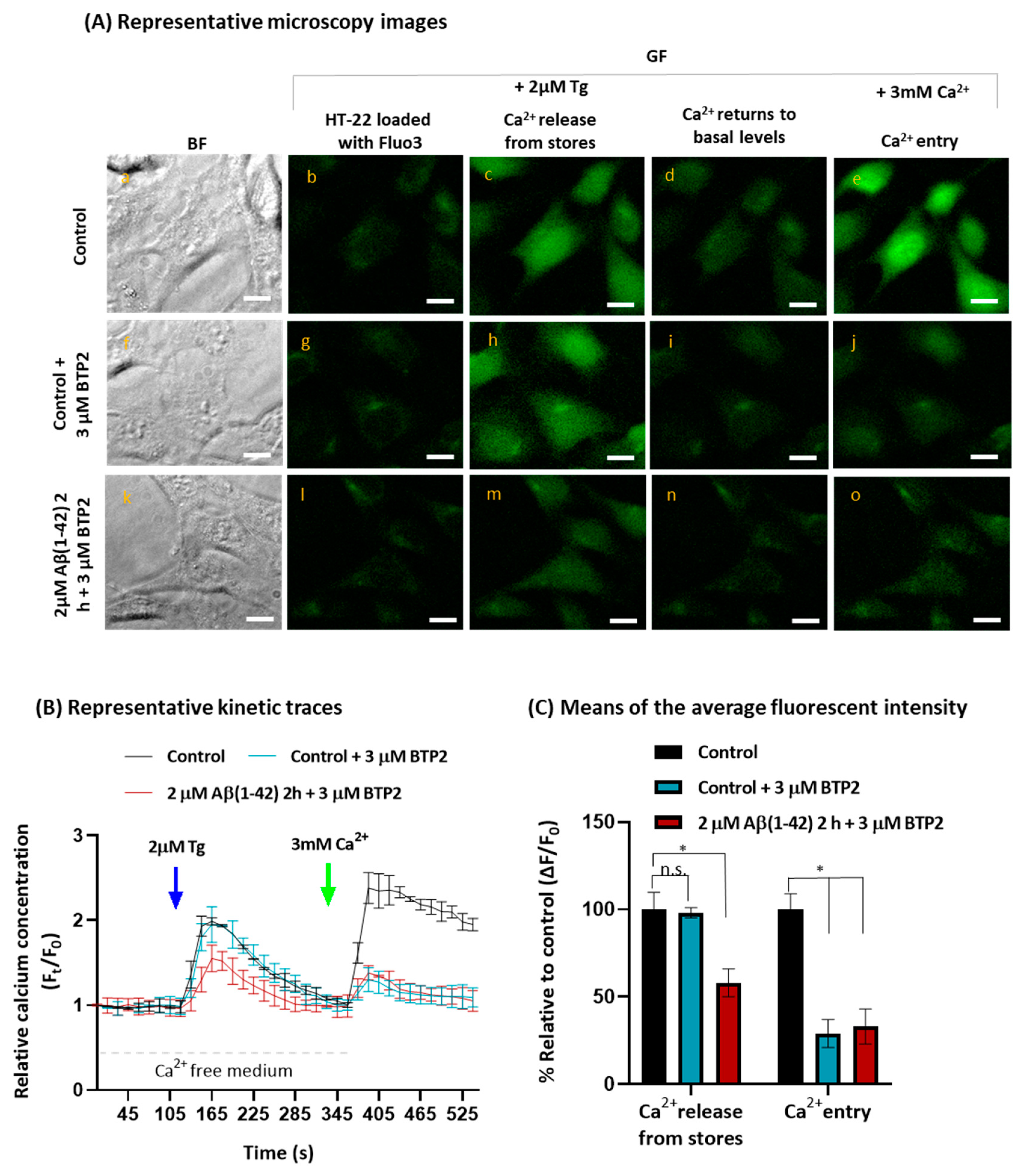{kind=link}
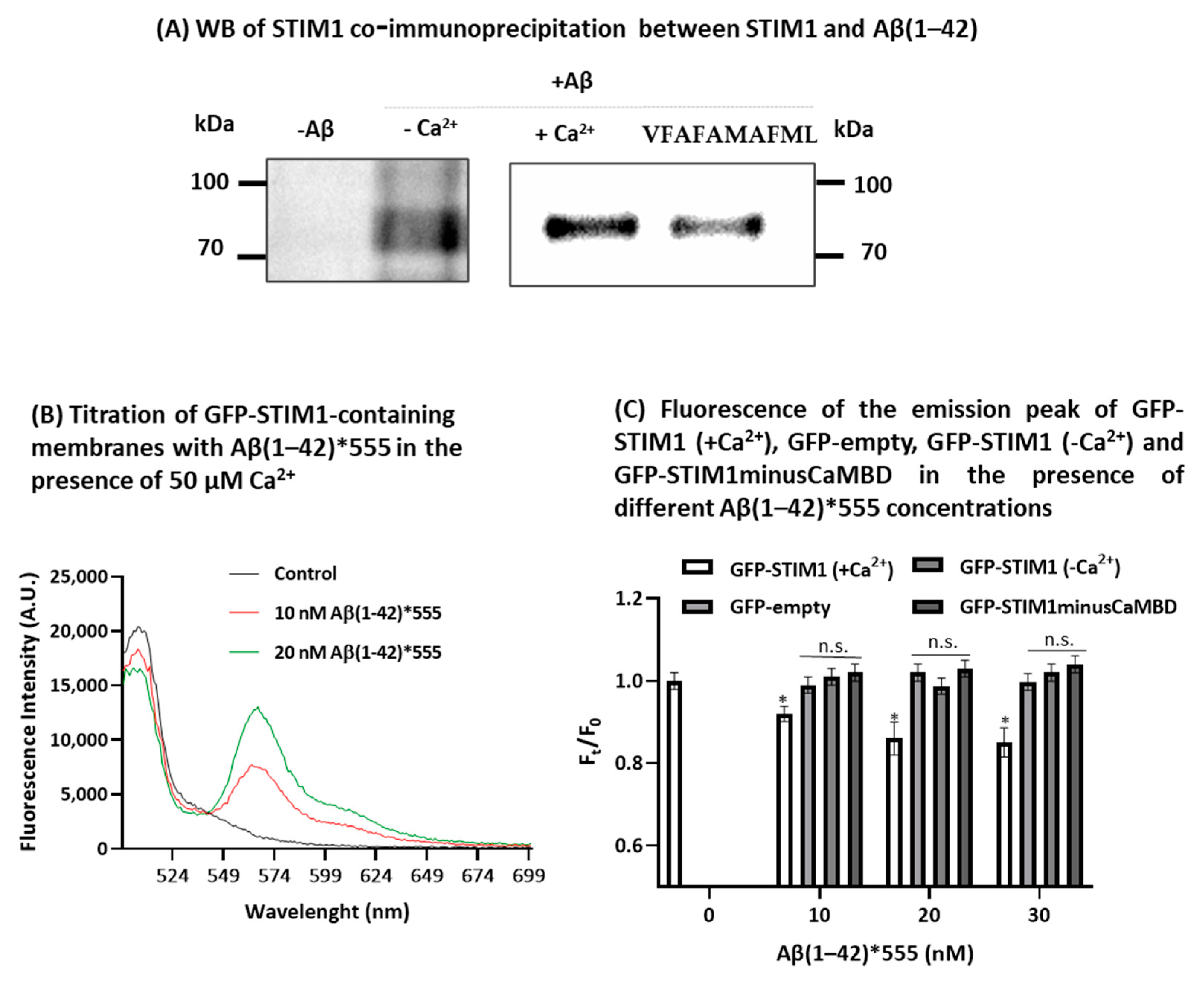{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
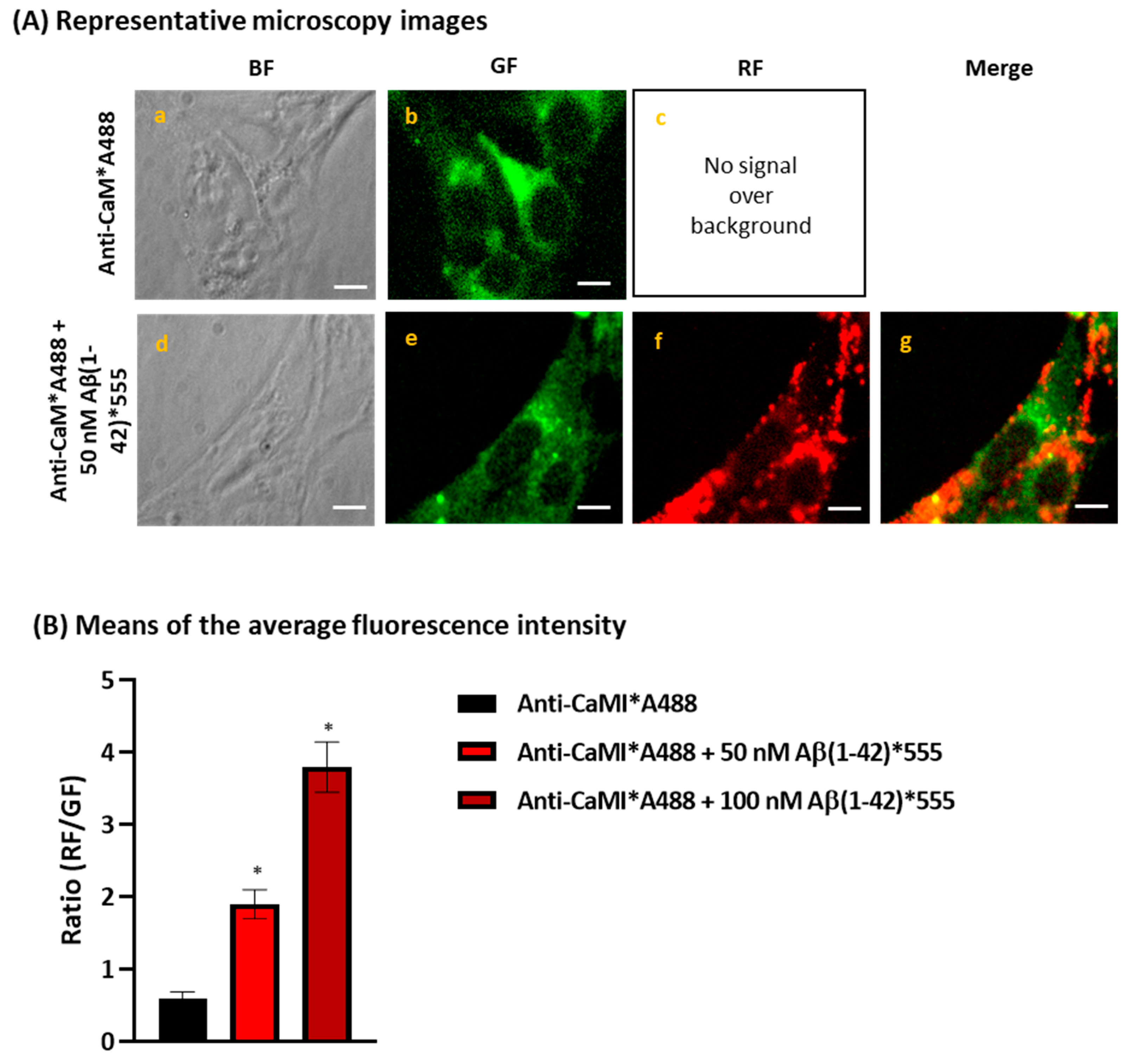
2.1. Aβ(1-42) Internalizes Inside HT-22 Cells after a Short Period of Incubation and Co-Localizes with ER
2.2. Aβ(1-42) Inhibits SOCE Activity after a Short Period of Incubation
2.3. STIM1 Is a Target for Aβ(1-42)
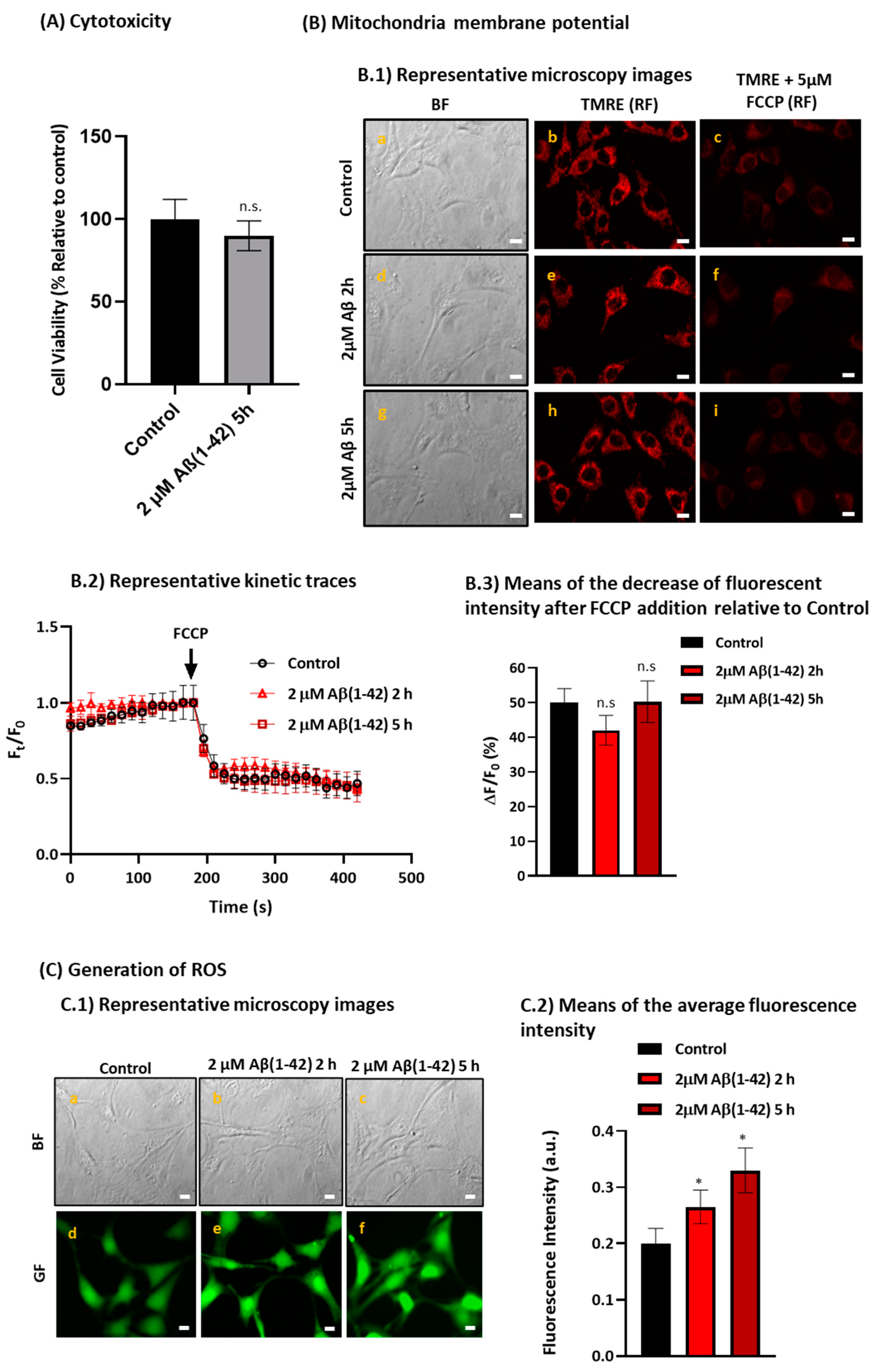
2.4. Aβ(1-42) Induces a Low to Moderate Increase in Reactive Oxygen Species (ROS) Production without Altering the Mitochondria Membrane Potential after a Short Period of Incubation in HT-22 Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. HT-22 Cell Culture
4.3. Cell Viability
4.4. Mitochondrial Membrane Potential
4.5. Measurement of Cellular Oxidative Stress
4.6. Measurement of Internalized Aβ(1-42)*555
4.7. FRET Imaging
4.8. Intracellular Cytosolic Calcium Measurements
4.9. Co-Immunoprecipitation
4.10. Western Blotting
4.11. STIM1-GFP Constructs and Expression
4.12. Fluorescence Measurements and Calculation of the FRET Parameters J and R0
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | amyloid β peptide |
| Aβ(1-42)*555 | Aβ(1-42)-HiLyte™-Fluor555 |
| AD | Alzheimer’s disease |
| Anti-CaM*A488 | complex anti-CaM + AlexaFluor488-labeled anti-IgG antibody |
| Anti-PDI*A488 | complex anti-PDI + AlexaFluor488-labeled anti-IgG antibody |
| a.u. | fluorescence intensity in arbitrary units |
| BTP2 | N-[4-[3,5-bis(trifluoromethyl)pyrazol-1-yl]phenyl]-4-methylthiadiazole-5-carboxamide |
| CaM | calmodulin |
| CaMBD | calmodulin binding domain |
| DMEM | Dulbecco’s modified Eagle’s medium |
| ER | endoplasmic reticulum |
| FCCP | trifluoromethoxy carbonylcyanide phenylhydrazone |
| Fluo3-AM | Fluo-3-pentaacetoxymethyl ester |
| FRET | fluorescence resonance energy transfer |
| Fura2-AM | Fura2 acetoxymethyl ester |
| GF | green fluorescence |
| GFP | green fluorescent protein |
| GSH | reduced glutathione |
| H2DCFDA | 2′,7′-dichlorodihydrofluorescein diacetate |
| HEPES | N-[2-hydroxyethyl] piperazine-N′-[2-ethanesulfonic acid] |
| IC50 | concentration producing 50% of the maximum effect |
| IgG | immunoglobulin G |
| IP3R | inositol 1,4,5-trisphosphate receptor |
| MCB | monocholorobimane |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PBS | phosphate-buffered saline |
| PBST | PBS supplemented with 0.2% 4-(1,1,3,3-tetramethyl butyl) phenyl-polyethyleneglycol |
| PDI | protein disulfide isomerase |
| RF | red fluorescence |
| RT | room temperature |
| ROS | reactive oxygen species |
| RyR | ryanodine receptor |
| SDS-PAGE | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| SEM | standard error of the mean |
| SERCA | Sarco-endoplasmic reticulum Ca2+-ATPase |
| SOCE | store-operated calcium entry |
| STIM1 | stromal interaction molecule 1 |
| TBST | Tris(hydroxymethyl)aminomethane-buffered saline supplemented with 0.05% polyoxyethylene sorbitan monolaurate |
| Tg | thapsigargin |
| TMRE | Tetramethylrhodamine ethyl ester |
| XeC | Xestonpongin C |
References
- Khachaturian, Z.S. Calcium Hypothesis of Alzheimer’s Disease and Brain Aging. Ann. N. Y. Acad. Sci. 1994, 747, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Calcium Signalling and Alzheimer’s Disease. Neurochem. Res. 2011, 36, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Kuchibhotla, K.V.; Goldman, S.T.; Lattarulo, C.R.; Wu, H.Y.; Hyman, B.T.; Bacskai, B.J. Aβ Plaques Lead to Aberrant Regulation of Calcium Homeostasis in Vivo Resulting in Structural and Functional Disruption of Neuronal Networks. Neuron 2008, 59, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Lopez, R.; Lyckman, A.; Oddo, S.; Laferla, F.M.; Querfurth, H.W.; Shtifman, A. Increased Intraneuronal Resting [Ca2+] in Adult Alzheimer’s Disease Mice. J. Neurochem. 2008, 105, 262–271. [Google Scholar] [CrossRef]
- Tong, B.C.; Wu, A.J.; Li, M.; Cheung, K.H. Calcium Signaling in Alzheimer’s Disease & Therapies. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1745–1760. [Google Scholar] [CrossRef]
- Poejo, J.; Salazar, J.; Mata, A.M.; Gutierrez-Merino, C. Binding of Amyloid β(1-42)-calmodulin Complexes to Plasma Membrane Lipid Rafts in Cerebellar Granule Neurons Alters Resting Cytosolic Calcium Homeostasis. Int. J. Mol. Sci. 2021, 22, 1984. [Google Scholar] [CrossRef]
- Poejo, J.; Salazar, J.; Mata, A.M.; Gutierrez-Merino, C. The Relevance of Amyloid β-Calmodulin Complexation in Neurons and Brain Degeneration in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4976. [Google Scholar] [CrossRef]
- Salazar, J.; Poejo, J.; Mata, A.M.; Samhan-Arias, A.K.; Gutierrez-Merino, C. Design and Experimental Evaluation of a Peptide Antagonist against Amyloid β(1-42) Interactions with Calmodulin and Calbindin-D28k. Int. J. Mol. Sci. 2022, 23, 2289. [Google Scholar] [CrossRef]
- Corbacho, I.; Berrocal, M.; Török, K.; Mata, A.M.; Gutierrez-Merino, C. High-Affinity Binding of Amyloid β-peptide to Calmodulin: Structural and Functional Implications. Biochem. Biophys. Res. Commun. 2017, 486, 992–997. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Podlisny, M.B.; Ostaszewski, B.L.; Squazzo, S.L.; Koo, E.H.; Rydell, R.E.; Teplow, D.B.; Selkoe, D.J. Aggregation of Secreted Amyloid β-Protein into Sodium Dodecyl Sulfate- Stable Oligomers in Cell Culture. J. Biol. Chem. 1995, 270, 9564–9570. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally Secreted Oligomers of Amyloid β Protein Potently Inhibit Hippocampal Long-Term Potentiation in Vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Leissring, M.A.; Adame, A.; Sun, X.; Spooner, E.; Masliah, E.; Selkoe, D.J.; Lemere, C.A.; Walsh, D.M. Biochemical and Immunohistochemical Analysis of an Alzheimer’s Disease Mouse Model Reveals the Presence of Multiple Cerebral Aβ Assembly Forms throughout Life. Neurobiol. Dis. 2009, 36, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Chang, L.; Viola, K.L.; Lacor, P.N.; Lambert, M.P.; Finch, C.E.; Krafft, G.A.; Klein, W.L. Alzheimer’s Disease-Affected Brain: Presence of Oligomeric Aβ Ligands (ADDLs) Suggests a Molecular Basis for Reversible Memory Loss. Proc. Natl. Acad. Sci. USA 2003, 100, 10417–10422. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid β-Protein Dimers Isolated Directly from Alzheimer Brains Impair Synaptic Plasticity and Memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of Amyloid β-Protein: Synaptic and Network Dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zheng, M.M.; Ma, Y.; Han, X.J.; Ma, X.Q.; Qu, C.Q.; Du, Y.F. Soluble Oligomers and Fibrillar Species of Amyloid β-Peptide Differentially Affect Cognitive Functions and Hippocampal Inflammatory Response. Biochem. Biophys. Res. Commun. 2012, 429, 125–130. [Google Scholar] [CrossRef]
- Forny-Germano, L.; Lyra e Silva, N.M.; Batista, A.F.; Brito-Moreira, J.; Gralle, M.; Boehnke, S.E.; Coe, B.C.; Lablans, A.; Marques, S.A.; Martinez, A.M.; et al. Alzheimer’s Disease-like Pathology Induced by Amyloid-β Oligomers in Nonhuman Primates. J. Neurosci. 2014, 34, 13629–13643. [Google Scholar] [CrossRef]
- Umeda, T.; Tomiyama, T.; Sakama, N.; Tanaka, S.; Lambert, M.P.; Klein, W.L.; Mori, H. Intraneuronal Amyloid β Oligomers Cause Cell Death Via Endoplasmic Reticulum Stress, Endosomal/Lysosomal Leakage, and Mitochondrial Dysfunction In Vivo. J. Neurosci. Res. 2011, 89, 1031–1042. [Google Scholar] [CrossRef]
- Cha, M.Y.; Han, S.H.; Son, S.M.; Hong, H.-S.; Choi, Y.-J.; Byun, J.; Mook-Jung, I. Mitochondria-Specific Accumulation of Amyloid β Induces Mitochondrial Dysfunction Leading to Apoptotic Cell Death. PLoS ONE 2012, 7, e34929. [Google Scholar] [CrossRef]
- Jiao, Y.; Zhang, Y.; Wei, Y.; Liu, Z.; An, W.; Guo, M. Direct Observation of Internalization and ROS Generation of Amyloid β-Peptide in Neuronal Cells at Subcellular Resolution. ChemBioChem 2012, 13, 2335–2338. [Google Scholar] [CrossRef] [PubMed]
- Lana, E.; Khanbolouki, M.; Degavre, C.; Samuelsson, E.B.; Åkesson, E.; Winblad, B.; Alici, E.; Lithner, C.U.; Behbahani, H. Perforin Promotes Amyloid Beta Internalisation in Neurons. Mol. Neurobiol. 2017, 54, 874–887. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.M.; Zhu, L.; Lan, X.; Zhao, D.W.; He, Y.J.; Sun, Z.Q.; Wu, D.; Li, H.Y. Endocytosis Is a Key Mode of Interaction between Extracellular β-Amyloid and the Cell Membrane. Biophys. J. 2020, 119, 1078–1090. [Google Scholar] [CrossRef] [PubMed]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Majewski, L.; Kuznicki, J. SOCE in Neurons: Signaling or Just Refilling? Biochim. Biophys. Acta Mol. Cell Res. 2014, 1853, 1940–1952. [Google Scholar] [CrossRef] [PubMed]
- Secondo, A.; Bagetta, G.; Amantea, D. On the Role of Store-Operated Calcium Entry in Acute and Chronic Neurodegenerative Diseases. Front. Mol. Neurosci. 2018, 11, 87. [Google Scholar] [CrossRef]
- Huang, A.S.; Tong, B.C.K.; Wu, A.J.; Chen, X.; Sreenivasmurthy, S.G.; Zhu, Z.; Liu, J.; Su, C.; Li, M.; Cheung, K.H. Rectifying Attenuated Store-Operated Calcium Entry as a Therapeutic Approach for Alzheimer’s Disease. Curr. Alzheimer Res. 2020, 17, 1072–1087. [Google Scholar] [CrossRef]
- Pascual-Caro, C.; Orantos-Aguilera, Y.; Sanchez-Lopez, I.; de Juan-Sanz, J.; Parys, J.B.; Area-Gomez, E.; Pozo-Guisado, E.; Martin-Romero, F.J. STIM1 Deficiency Leads to Specific Down-Regulation of ITPR3 in SH-SY5Y Cells. Int. J. Mol. Sci. 2020, 21, 6598. [Google Scholar] [CrossRef]
- Peterson, C.; Gibson, G.E.; Blass, J.P. Altered Calcium Uptake in Cultured Skin Fibroblasts from Patients with Alzheimer’s Disease. N. Engl. J. Med. 1985, 312, 1063–1065. [Google Scholar] [CrossRef]
- Ito, E.; Oka, K.; Etcheberrigaray, R.; Nelson, T.J.; McPhie, D.L.; Tofel-Grehl, B.; Gibson, G.E.; Alkon, D.L. Internal Ca2+ Mobilization Is Altered in Fibroblasts from Patients with Alzheimer`s Disease. Proc. Natl. Acad. Sci. USA 1994, 91, 534–538. [Google Scholar] [CrossRef]
- Tong, B.C.; Lee, C.S.; Cheng, W.H.; Lai, K.O.; Foskett, J.K.; Cheung, K.H. Familial Alzheimer’s Disease-Associated Presenilin 1 Mutants Promote γ-Secretase Cleavage of STIM1 to Impair Store-Operated Ca2+ Entry. Sci. Signal. 2016, 9, ra89. [Google Scholar] [CrossRef] [PubMed]
- Pannaccione, A.; Secondo, A.; Molinaro, P.; D’Avanzo, C.; Cantile, M.; Esposito, A.; Boscia, F.; Scorziello, A.; Sirabella, R.; Di Renzo, G.; et al. A New Concept: A/β 1-42 Generates a Hyper Functional Proteolytic NCX3 Fragment That Delays Caspase-12 Activation and Neuronal Death. J. Neurosci. 2012, 32, 10609–10617. [Google Scholar] [CrossRef] [PubMed]
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; LaFerla, F.M.; Parker, I. Enhanced Ryanodine Receptor Recruitment Contributes to Ca2+ Disruptions in Young, Adult, and Aged Alzheimer’s Disease Mice. J. Neurosci. 2006, 26, 5180–5189. [Google Scholar] [CrossRef] [PubMed]
- Chakroborty, S.; Goussakov, I.; Miller, M.B.; Stutzmann, G.E. Deviant Ryanodine Receptor-Mediated Calcium Release Resets Synaptic Homeostasis in Presymptomatic 3xTg-AD Mice. J. Neurosci. 2009, 29, 9458–9470. [Google Scholar] [CrossRef]
- Llano, I.; González, J.; Caputo, C.; Lai, F.A.; Blayney, L.M.; Tan, Y.P.; Marty, A. Presynaptic Calcium Stores Underlie Large-Amplitude Miniature IPSCs and Spontaneous Calcium Transients. Nat. Neurosci. 2000, 3, 1256–1265. [Google Scholar] [CrossRef]
- Chakroborty, S.; Briggs, C.; Miller, M.B.; Goussakov, I.; Schneider, C.; Kim, J.; Wicks, J.; Richardson, J.C.; Conklin, V.; Cameransi, B.G.; et al. Stabilizing ER Ca2+ Channel Function as an Early Preventative Strategy for Alzheimer’s Disease. PLoS ONE 2012, 7, e52056. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, J.; Sun, S.; Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium Signaling, Excitability and Synaptic Plasticity Defects in a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 45, 561–580. [Google Scholar] [CrossRef]
- Ferreiro, E.; Resende, R.; Costa, R.; Oliveira, C.R.; Pereira, C.M.F. An Endoplasmic-Reticulum-Specific Apoptotic Pathway Is Involved in Prion and Amyloid-Beta Peptides Neurotoxicity. Neurobiol. Dis. 2006, 23, 669–678. [Google Scholar] [CrossRef]
- Rybalchenko, V.; Grillo, M.A.; Gastinger, M.J.; Rybalchenko, N.; Payne, A.J.; Koulen, P. The Unliganded Long Isoform of Estrogen Receptor Beta Stimulates Brain Ryanodine Receptor Single Channel Activity alongside with Cytosolic Ca2+. J. Recept. Signal Transduct. Res. 2009, 29, 326–341. [Google Scholar] [CrossRef]
- Duncan, R.S.; Hwang, S.Y.; Koulen, P. Differential Inositol 1,4,5-Trisphosphate Receptor Signaling in a Neuronal Cell Line. Int. J. Biochem. Cell Biol. 2007, 39, 1852–1862. [Google Scholar] [CrossRef]
- Blaustein, M.P.; Golovina, M.P. Structural Complexity and Functional Diversity of Endoplasmic Reticulum Ca2+ Stores. Trends Neurosci. 2001, 24, 602–608. [Google Scholar] [CrossRef]
- Putney, J.W. Capacitative Calcium Entry in the Nervous System. Cell Calcium 2003, 34, 339–344. [Google Scholar] [CrossRef]
- Putney, J.W.; Steinckwich-Besançon, N.; Numaga-Tomita, T.; Davis, F.M.; Desai, P.N.; D’Agostin, D.M.; Wu, S.; Bird, G.S. The Functions of Store-Operated Calcium Channels. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Serwach, K.; Gruszczynska-Biegala, J. Target Molecules of STIM Proteins in the Central Nervous System. Front. Mol. Neurosci. 2020, 13, 617422. [Google Scholar] [CrossRef] [PubMed]
- Keil, J.M.; Shen, Z.; Briggs, S.P.; Patrick, G.N. Regulation of STIM1 and SOCE by the Ubiquitin-Proteasome System (UPS). PLoS ONE 2010, 5, e13465. [Google Scholar] [CrossRef]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 Is a Feedback Regulator That Stabilizes Basal Cytosolic and Endoplasmic Reticulum Ca2+ Levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef]
- Berna-Erro, A.; Braun, A.; Kraft, R.; Kleinschnitz, C.; Schuhmann, M.K.; Stegner, D.; Wultsch, T.; Eilers, J.; Meuth, S.G.; Stoll, G.; et al. STIM2 Regulates Capacitative Ca2+ Entry in Neurons and Plays a Key Role in Hypoxic Neuronal Cell Death. Sci. Signal. 2009, 2, ra67. [Google Scholar] [CrossRef]
- Henke, N.; Albrecht, P.; Bouchachia, I.; Ryazantseva, M.; Knoll, K.; Lewerenz, J.; Kaznacheyeva, E.; Maher, P.; Methner, A. The Plasma Membrane Channel ORAI1 Mediates Detrimental Calcium Influx Caused by Endogenous Oxidative Stress. Cell Death Dis. 2013, 4, e470–e479. [Google Scholar] [CrossRef]
- Rao, W.; Zhang, L.; Su, N.; Wang, K.; Hui, H.; Wang, L.; Chen, T.; Luo, P.; Yang, Y.F.; Liu, Z.B.; et al. Blockade of SOCE Protects HT22 Cells from Hydrogen Peroxide-Induced Apoptosis. Biochem. Biophys. Res. Commun. 2013, 441, 351–356. [Google Scholar] [CrossRef]
- Rao, W.; Peng, C.; Zhang, L.; Su, N.; Wang, K.; Hui, H.; Dai, S.H.; Yang, Y.F.; Luo, P.; Fei, Z. Homer1a Attenuates Glutamate-Induced Oxidative Injury in HT-22 Cells through Regulation of Store-Operated Calcium Entry. Sci. Rep. 2016, 6, 33975. [Google Scholar] [CrossRef]
- Li, X.; Wu, G.; Yang, Y.; Fu, S.; Liu, X.; Kang, H.; Yang, X.; Su, X.C.; Shen, Y. Calmodulin dissociates the STIM1-Orai1 complex and STIM1 oligomers. Nat. Commun. 2017, 8, 1042. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, R.; Augustynek, B.S.; Ercan-Herbst, E.; Kandasamy, P.; Seedorf, M.; Peinelt, C.; Hediger, M.A. Ca2+/Calmodulin Binding to STIM1 Hydrophobic Residues Facilitates Slow Ca2+-Dependent Inactivation of the Orai1 Channel. Cell Physiol. Biochem. 2020, 54, 252–270. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Wang, M.; Zhao, Q.; Li, X.; Liu, P.; Ren, B.; Wu, C.; Du, X.; Li, N.; Liu, Q. Bis(Ethylmaltolato)Oxidovanadium (IV) Mitigates Neuronal Apoptosis Resulted from Amyloid-Beta Induced Endoplasmic Reticulum Stress through Activating Peroxisome Proliferator-Activated Receptor γ. J. Inorg. Biochem. 2020, 208, 111073. [Google Scholar] [CrossRef] [PubMed]
- Corbin, E.A.; Millet, L.J.; Keller, K.R.; King, W.P.; Bashir, R. Measuring Physical Properties of Neuronal and Glial Cells with Resonant Microsensors. Anal. Chem. 2014, 86, 4864–4872. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.M.; Soling, H.D. The Protein Disulphide-Isomerase Family: Unravelling a String of Folds. Biochem. J. 1999, 339, 1–10. [Google Scholar] [CrossRef]
- Ishikawa, J.; Ohga, K.; Yoshino, T.; Takezawa, R.; Ichikawa, A.; Kubota, H.; Yamada, T. A Pyrazole Derivative, YM-58483, Potently Inhibits Store-Operated Sustained Ca2+ Influx and IL-2 Production in T-Lymphocytes. J. Immunol. 2003, 170, 4441–4449. [Google Scholar] [CrossRef]
- Zitt, C.; Strauss, B.; Schwarz, E.C.; Spaeth, N.; Rast, G.; Hatzelmann, A.; Hoth, M. Potent Inhibition of Ca2+ Release-Activated Ca2+ Channels and T-Lymphocyte Activation by the Pyrazole Derivative BTP2. J. Biol. Chem. 2004, 279, 12427–12437. [Google Scholar] [CrossRef]
- Kandimalla, K.K.; Scott, O.G.; Fulzele, S.; Davidson, M.W.; Poduslo, J.F. Mechanism of Neuronal versus Endothelial Cell Uptake of Alzheimer’s Disease Amyloid β Protein. PLoS ONE 2009, 4, e4627. [Google Scholar] [CrossRef]
- Wesén, E.; Jeffries, G.D.M.; Dzebo, M.M.; Esbjörner, E.K. Endocytic Uptake of Monomeric Amyloid-β Peptides Is Clathrin-and Dynamin-Independent and Results in Selective Accumulation of Aβ(1-42) Compared to Aβ(1-40). Sci. Rep. 2017, 7, 2021. [Google Scholar] [CrossRef]
- Lai, A.Y.; McLaurin, J. Mechanisms of Amyloid-Beta Peptide Uptake by Neurons: The Role of Lipid Rafts and Lipid Raft-Associated Proteins. Int. J. Alzheimer’s Dis. 2011, 2011, 1–11. [Google Scholar] [CrossRef]
- Marques-da-Silva, D.; Gutierrez-Merino, C. Caveolin-Rich Lipid Rafts of the Plasma Membrane of Mature Cerebellar Granule Neurons Are Microcompartments for Calcium/Reactive Oxygen and Nitrogen Species Cross-Talk Signaling. Cell Calcium 2014, 56, 108–123. [Google Scholar] [CrossRef] [PubMed]
- Fortalezas, S.; Poejo, J.; Samhan-Arias, A.K.; Gutierrez-Merino, C. Cholesterol-Rich Plasma Membrane Submicrodomains Can Be a Major Extramitochondrial Source of Reactive Oxygen Species in Partially Depolarized Mature Cerebellar Granule Neurons in Culture. J. Neurophysiol. Neurol. Disord. 2019, 5, 1–22. [Google Scholar] [CrossRef]
- Poejo, J. Molecular Mechanisms of Early Cytosolic Calcium Dysregulation Induced by Amyloid-β Peptides and Neuroprotection Strategies. Ph.D. Thesis, Universidad de Extremadura, Badajoz, Spain, April 2022. [Google Scholar]
- El-Sayed, A.; Harashima, H. Endocytosis of Gene Delivery Vectors: From Clathrin-Dependent to Lipid Raft-Mediated Endocytosis. Mol. 2013, 21, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Honjo, Y.; Horibe, T.; Torisawa, A.; Ito, H.; Nakanishi, A.; Mori, H.I.; Komiya, T.; Takahashi, R.; Kawakami, K. Protein Disulfide Isomerase P5-Immunopositive Inclusions in Patients with Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 38, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Nomura, J.; Hosoi, T.; Kaneko, M.; Ozawa, K.; Nishi, A.; Nomura, Y. Neuroprotection by Endoplasmic Reticulum Stress-Induced HRD1 and Chaperones: Possible Therapeutic Targets for Alzheimer’s and Parkinson’s Disease. Med. Sci. 2016, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Raturi, A.; Mutus, B. Characterization of Redox State and Reductase Activity of Protein Disulfide Isomerase under Different Redox Environments Using a Sensitive Fluorescent Assay. Free Radic. Biol. Med. 2007, 43, 62–70. [Google Scholar] [CrossRef]
- Pascual-Caro, C.; Berrocal, M.; Lopez-Guerrero, A.M.; Alvarez-Barrientos, A.; Pozo-Guisado, E.; Gutierrez-Merino, C.; Mata, A.M.; Martin-Romero, F.J. STIM1 Deficiency Is Linked to Alzheimer’s Disease and Triggers Cell Death in SH-SY5Y Cells by Upregulation of L-Type Voltage-Operated Ca2+ Entry. J. Mol. Med. 2018, 96, 1061–1079. [Google Scholar] [CrossRef]
- Berrocal, M.; Marcos, D.; Sepúlveda, M.R.; Pérez, M.; Ávila, J.; Mata, A.M. Altered Ca2+ Dependence of Synaptosomal Plasma Membrane Ca2+ -ATPase in Human Brain Affected by Alzheimer’s Disease. FASEB J. 2009, 23, 1826–1834. [Google Scholar] [CrossRef]
- Paula-Lima, A.; Hidalgo, C. Amyloid SS-Peptide Oligomers, Ryanodine Receptor-Mediated Ca2+ Release and Wnt-5a/Ca2+ Signaling: Opposing Roles in Neuronal Mitochondrial Dynamics? Front. Cell. Neurosci. 2013, 7, 2011–2013. [Google Scholar] [CrossRef]
- Lanner, J.T.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine Receptors: Structure, Expression, Molecular Details, and Function in Calcium Release. Cold Spring Harb. Perspect. Biol. 2010, 2, a003996. [Google Scholar] [CrossRef]
- Pozo-Guisado, E.; Casas-Rua, V.; Tomas-Martin, P.; Lopez-Guerrero, A.M.; Alvarez-Barrientos, A.; Martin-Romero, F.J. Phosphorylation of STIM1 at ERK1/2 target sites regulates interaction with the microtubule plus-end binding protein EB1. J. Cell Sci. 2013, 126 Pt 14, 3170–3180. [Google Scholar] [CrossRef]
- Casas-Rua, V.; Alvarez, I.S.; Pozo-Guisado, E.; Martín-Romero, F.-J. Inhibition of STIM1 phosphorylation underlies resveratrol-induced inhibition of store-operated calcium entry. Biochem. Pharmacol. 2013, 86, 1555–1563. [Google Scholar] [CrossRef] [PubMed]
- Fortalezas, S.; Marques-da-Silva, D.; Gutierrez-Merino, C. Creatine Protects Against Cytosolic Calcium Dysregulation, Mitochondrial Depolarization and Increase of Reactive Oxygen Species Production in Rotenone-Induced Cell Death of Cerebellar Granule Neurons. Neurotox. Res. 2018, 34, 717–732. [Google Scholar] [CrossRef] [PubMed]
- Samhan-Arias, A.K.; Marques-da-Silva, D.; Yanamala, N.; Gutierrez-Merino, C. Stimulation and Clustering of Cytochrome b5 Reductase in Caveolin-Rich Lipid Microdomains Is an Early Event in Oxidative Stress-Mediated Apoptosis of Cerebellar Granule Neurons. J. Proteom. 2012, 75, 2934–2949. [Google Scholar] [CrossRef] [PubMed]
- Fortalezas, S.; Marques-da-Silva, D.; Gutierrez-Merino, C. Methyl-β-Cyclodextrin Impairs the Phosphorylation of the β2 Subunit of L-Type Calcium Channels and Cytosolic Calcium Homeostasis in Mature Cerebellar Granule Neurons. Int. J. Mol. Sci. 2018, 19, 3667. [Google Scholar] [CrossRef]
- Pozo-Guisado, E.; Campbell, D.G.; Deak, M.; Alvarez-Barrientos, A.; Morrice, N.A.; Alvarez, I.S.; Alessi, D.R.; Martín-Romero, F.J. Phosphorylation of STIM1 at ERK1/2 target sites modulates store-operated calcium entry. J. Cell Sci. 2010, 123 Pt 18, 3084–3093. [Google Scholar] [CrossRef]
- Purlyte, E.; Dhekne, H.S.; Sarhan, A.R.; Gomez, R.; Lis, P.; Wightman, M.; Martinez, T.N.; Tonelli, F.; Pfeffer, S.R.; Alessi, D.R. Rab29 activation of the Parkinson's disease-associated LRRK2 kinase. EMBO J. 2018, 37, 1–18. [Google Scholar] [CrossRef]
- Gutierrez-Merino, C.; Molina, A.; Escudero, B.; Diez, A.; Laynez, J. Interaction of the Local Anesthetics Dibucaine and Tetracaine with Sarcoplasmic Reticulum Membranes. Differential Scanning Calorimetry and Fluorescence Studies. Biochemistry 1989, 28, 3398–3406. [Google Scholar] [CrossRef]
- Centeno, F.; Gutierrez-Merino, C. Location of Functional Centers in the Microsomal Cytochrome P450 System. Biochemistry 1992, 31, 8473–8481. [Google Scholar] [CrossRef]
- Wall, K.P.; Dillon, R.; Knowles, M.K. Fluorescence Quantum Yield Measurements of Fluorescent Proteins: A Laboratory Experiment for a Biochemistry or Molecular Biophysics Laboratory Course. Biochem. Mol. Biol. Educ. 2015, 43, 52–59. [Google Scholar] [CrossRef]
- Stryer, L. Fluorescence Energy Transfer as a Spectroscopic Ruler. Annu. Rev. Biochem. 1978, 47, 819–846. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poejo, J.; Orantos-Aguilera, Y.; Martin-Romero, F.J.; Mata, A.M.; Gutierrez-Merino, C. Internalized Amyloid-β (1-42) Peptide Inhibits the Store-Operated Calcium Entry in HT-22 Cells. Int. J. Mol. Sci. 2022, 23, 12678. https://doi.org/10.3390/ijms232012678
Poejo J, Orantos-Aguilera Y, Martin-Romero FJ, Mata AM, Gutierrez-Merino C. Internalized Amyloid-β (1-42) Peptide Inhibits the Store-Operated Calcium Entry in HT-22 Cells. International Journal of Molecular Sciences. 2022; 23(20):12678. https://doi.org/10.3390/ijms232012678
Chicago/Turabian StylePoejo, Joana, Yolanda Orantos-Aguilera, Francisco Javier Martin-Romero, Ana Maria Mata, and Carlos Gutierrez-Merino. 2022. "Internalized Amyloid-β (1-42) Peptide Inhibits the Store-Operated Calcium Entry in HT-22 Cells" International Journal of Molecular Sciences 23, no. 20: 12678. https://doi.org/10.3390/ijms232012678
APA StylePoejo, J., Orantos-Aguilera, Y., Martin-Romero, F. J., Mata, A. M., & Gutierrez-Merino, C. (2022). Internalized Amyloid-β (1-42) Peptide Inhibits the Store-Operated Calcium Entry in HT-22 Cells. International Journal of Molecular Sciences, 23(20), 12678. https://doi.org/10.3390/ijms232012678