
Treadmill Exercise Reduces Neuroinflammation, Glial Cell Activation and Improves Synaptic Transmission in the Prefrontal Cortex in 3 × Tg-AD Mice
 , , and
, , and 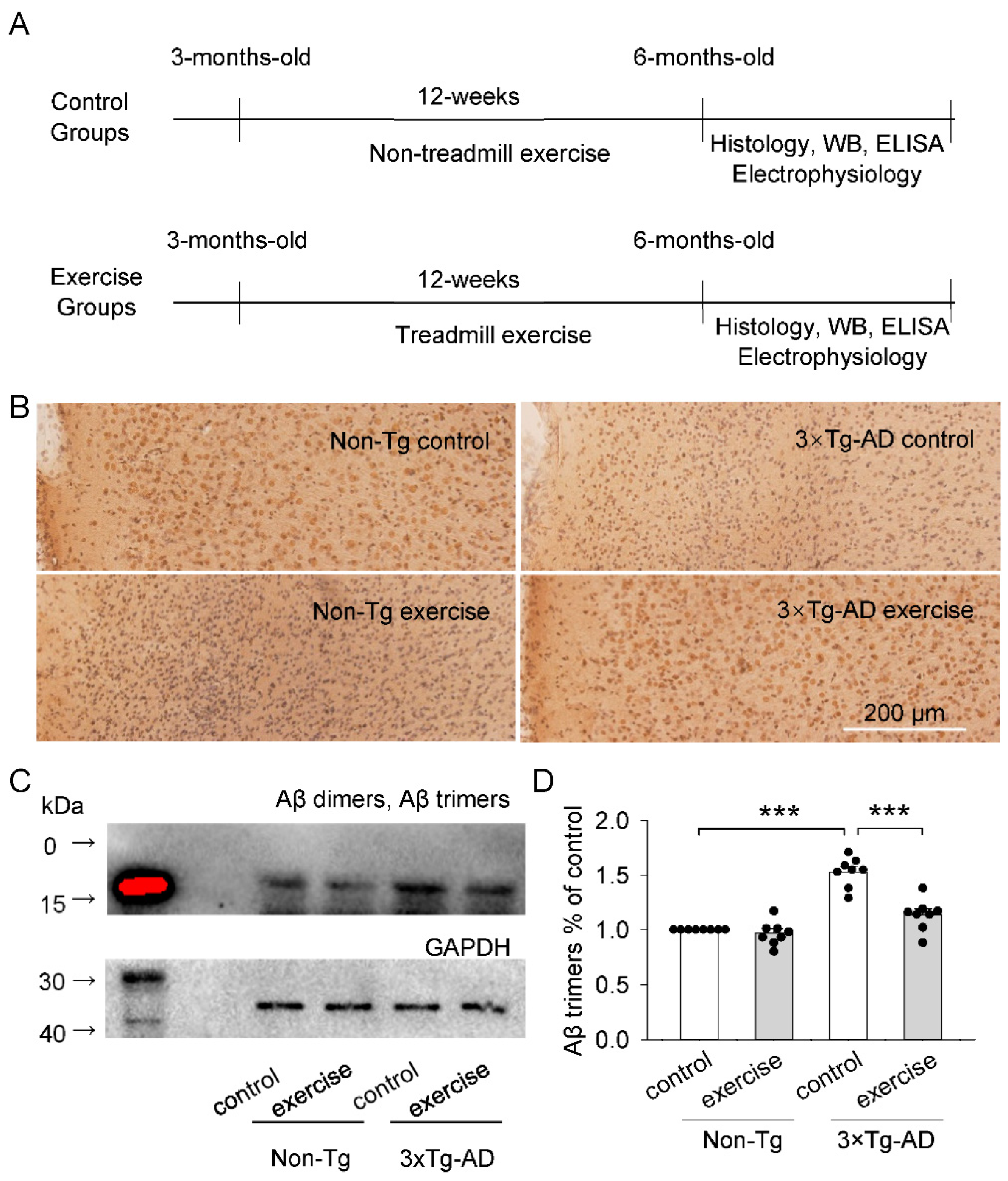{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Effects of Treadmill Exercise on Aβ Plaques and Aβ Oligomers (Aβ Dimers and Aβ Trimers) Levels of the Prefrontal Cortex in 3 × Tg-AD Mice
2.2. Treadmill Exercise Inhibited the Kinase Activity of GSK3β of the Prefrontal Cortex in 3 × Tg-AD Mice
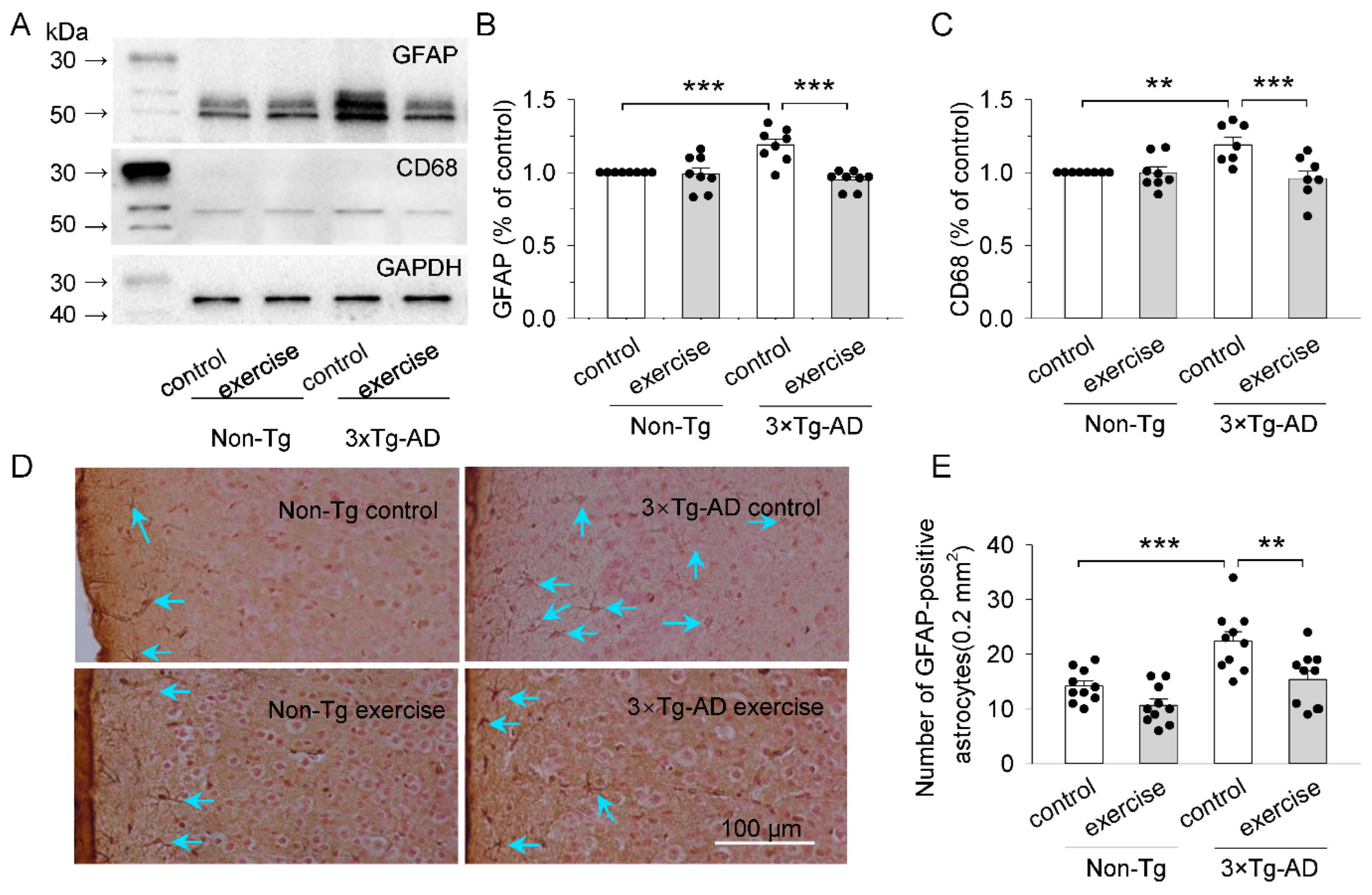
2.3. Treadmill Exercise Reduced the Activation of Microglia and Astrocytes of the Prefrontal Cortex in 3 × Tg-AD Mice
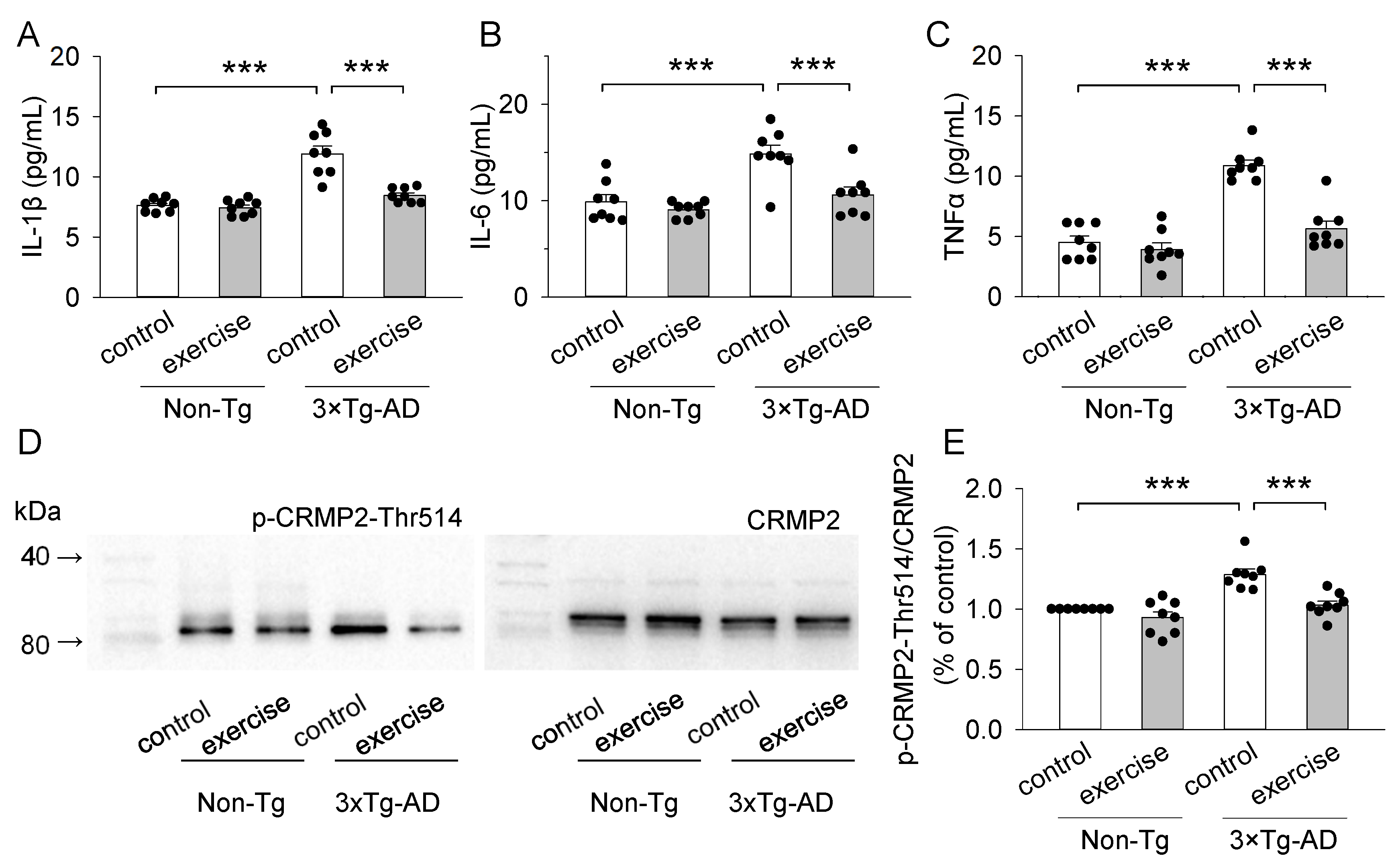
2.4. Treadmill Exercise Decreased the Concentration of Pro-Inflammatory Cytokines (IL-1β, IL-6, and TNFα) of the Prefrontal Cortex in 3 × Tg-AD Mice
2.5. Treadmill Exercise Decreased the Levels of Phosphorylation of CRMP2 at Thr514 of the Prefrontal Cortex in 3 × Tg-AD Mice
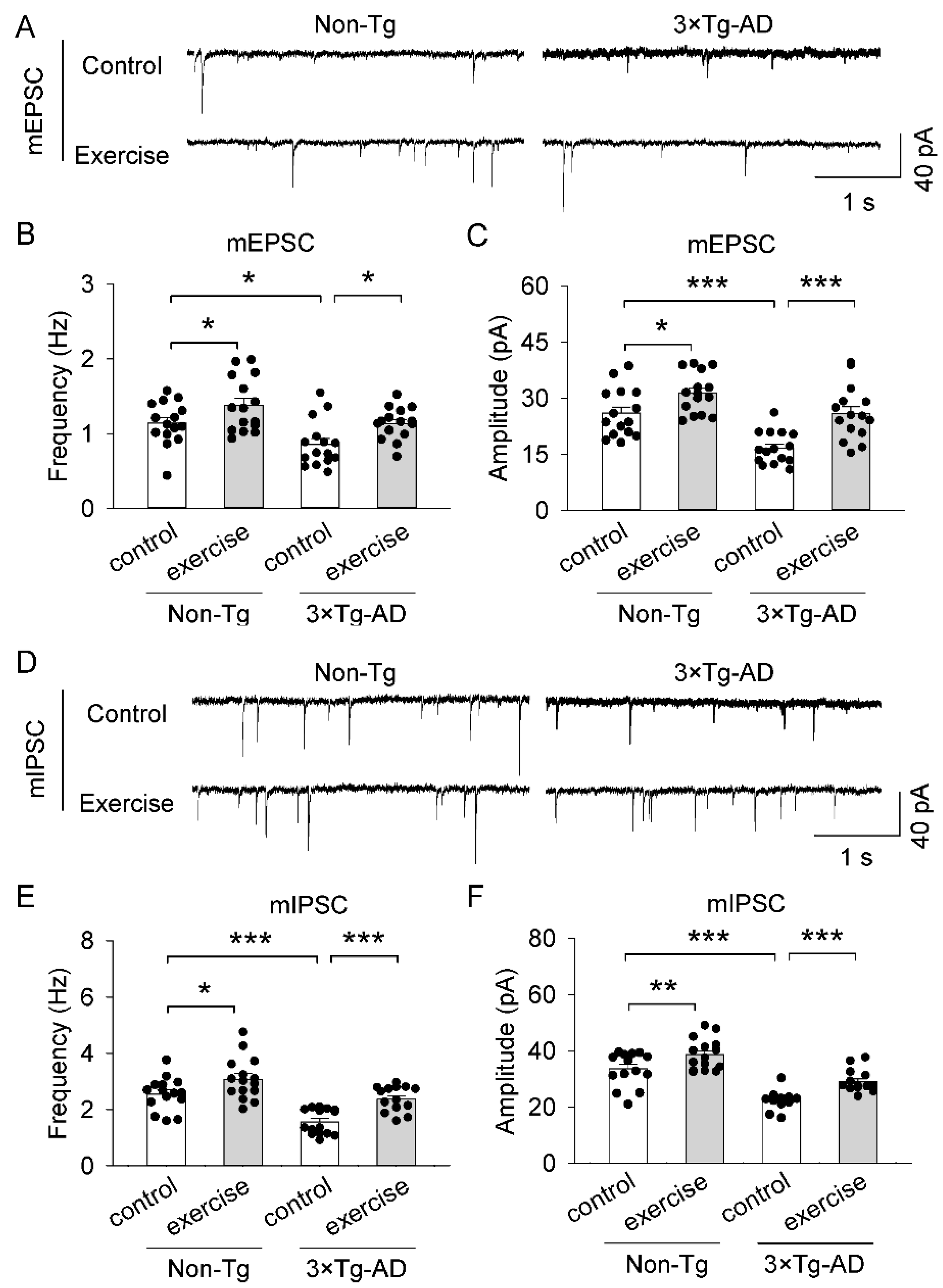
2.6. Treadmill Exercise Ameliorated Synaptic Transmissi on Dysfunction of the Prefrontal Cortex in 3 × Tg-AD Mice
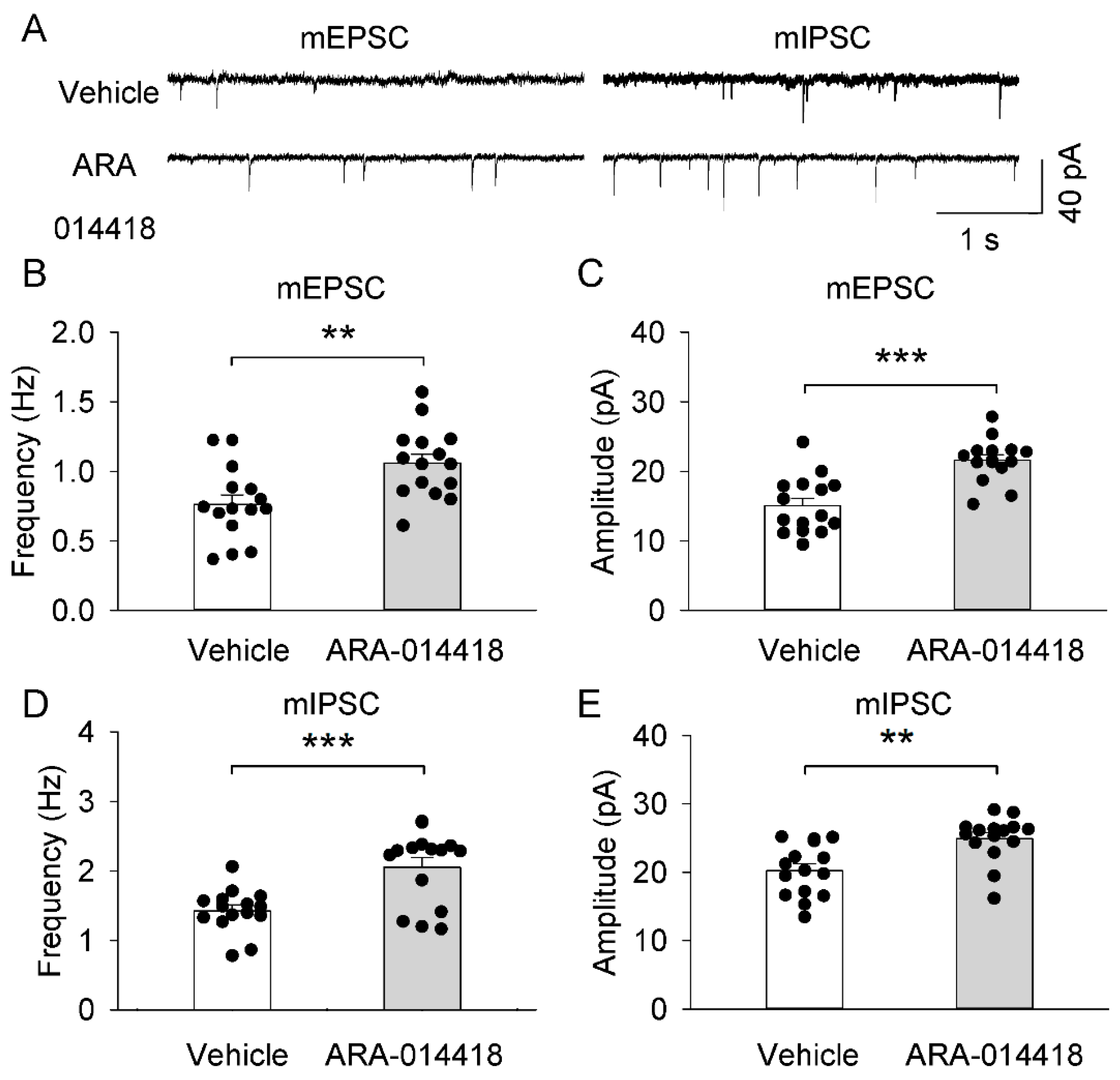
2.7. GSK3β Inhibitor (ARA-014418) Restored the Synaptic Transmission Dysfunction of the Prefrontal Cortex in 3 × Tg-AD Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Chemical Reagents
4.3. Treadmill Exercise Protocol
4.4. Animal Surgery and Microinjection Procedures
4.5. Immunohistochemistry Analysis
4.6. Western Blot
4.7. Enzyme-Linked Immunosorbent Assay (ELISA)
4.8. Slice Electrophysiology
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sheng, M.; Sabatini, B.L.; Südhof, T.C. Synapses and Alzheimer’s disease. Cold Spring Harb. Perspect. Biol. 2012, 4, a005777. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.B.; Cao, Q.; Yan, Z. Transcriptomic analysis of human brains with Alzheimer’s disease reveals the altered expression of synaptic genes linked to cognitive deficits. Brain Commun. 2021, 3, fcab123. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Sastre, M.; Klockgether, T.; Heneka, M.T. Contribution of inflammatory processes to Alzheimer’s disease: Molecular mechanisms. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2006, 24, 167–176. [Google Scholar] [CrossRef]
- Hopperton, K.E.; Mohammad, D.; Trépanier, M.O.; Giuliano, V.; Bazinet, R.P. Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: A systematic review. Mol. Psychiatry 2018, 23, 177–198. [Google Scholar] [CrossRef]
- Abbas, N.; Bednar, I.; Mix, E.; Marie, S.; Paterson, D.; Ljungberg, A.; Morris, C.; Winblad, B.; Nordberg, A.; Zhu, J. Up-regulation of the inflammatory cytokines IFN-gamma and IL-12 and down-regulation of IL-4 in cerebral cortex regions of APP(SWE) transgenic mice. J. Neuroimmunol. 2002, 126, 50–57. [Google Scholar] [CrossRef]
- Liu, L.; Liu, Y.; Li, N.; Huang, R.; Zheng, X.; Huang, L.; Hou, S.; Yuan, Q. Multiple inflammatory profiles of microglia and altered neuroimages in APP/PS1 transgenic AD mice. Brain Res. Bull. 2020, 156, 86–104. [Google Scholar] [CrossRef] [PubMed]
- Ano, Y.; Ohya, R.; Yamazaki, T.; Takahashi, C.; Taniguchi, Y.; Kondo, K.; Takashima, A.; Uchida, K.; Nakayama, H. Hop bitter acids containing a β-carbonyl moiety prevent inflammation-induced cognitive decline via the vagus nerve and noradrenergic system. Sci. Rep. 2020, 10, 20028. [Google Scholar] [CrossRef]
- Yang, J.T.; Wang, Z.J.; Cai, H.Y.; Yuan, L.; Hu, M.M.; Wu, M.N.; Qi, J.S. Sex Differences in Neuropathology and Cognitive Behavior in APP/PS1/tau Triple-Transgenic Mouse Model of Alzheimer’s Disease. Neurosci. Bull. 2018, 34, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, R.; Rodin, A.; Ferreira, E.; Velazquez, R.; Branca, C.; Caccamo, A.; Oddo, S. Temporal and regional progression of Alzheimer’s disease-like pathology in 3xTg-AD mice. Aging Cell 2019, 18, e12873. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Matsuyama, S.; Iso, H.; Umeda, T.; Takuma, H.; Ohnishi, K.; Ishibashi, K.; Teraoka, R.; Sakama, N.; Yamashita, T.; et al. A mouse model of amyloid beta oligomers: Their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 4845–4856. [Google Scholar] [CrossRef] [PubMed]
- White, J.A.; Manelli, A.M.; Holmberg, K.H.; Van Eldik, L.J.; Ladu, M.J. Differential effects of oligomeric and fibrillar amyloid-beta 1-42 on astrocyte-mediated inflammation. Neurobiol. Dis. 2005, 18, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Michaud, J.P.; Bellavance, M.A.; Préfontaine, P.; Rivest, S. Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid beta. Cell Rep. 2013, 5, 646–653. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef]
- Zaheer, A.; Zaheer, S.; Thangavel, R.; Wu, Y.; Sahu, S.K.; Yang, B. Glia maturation factor modulates beta-amyloid-induced glial activation, inflammatory cytokine/chemokine production and neuronal damage. Brain Res. 2008, 1208, 192–203. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef]
- Giuliani, F.; Vernay, A.; Leuba, G.; Schenk, F. Decreased behavioral impairments in an Alzheimer mice model by interfering with TNF-alpha metabolism. Brain Res. Bull. 2009, 80, 302–308. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, F.E.; Lee, J.K.; Harms, A.S.; Ruhn, K.A.; Blurton-Jones, M.; Hong, J.; Das, P.; Golde, T.E.; LaFerla, F.M.; Oddo, S.; et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol. Dis. 2009, 34, 163–177. [Google Scholar] [CrossRef]
- Koistinaho, J.; Malm, T.; Goldsteins, G. Glycogen synthase kinase-3β: A mediator of inflammation in Alzheimer’s disease? Int. J. Alzheimers Dis. 2011, 2011, 129753. [Google Scholar] [CrossRef] [PubMed]
- Yuskaitis, C.J.; Jope, R.S. Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell. Signal. 2009, 21, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Renault-Mihara, F.; Katoh, H.; Ikegami, T.; Iwanami, A.; Mukaino, M.; Yasuda, A.; Nori, S.; Mabuchi, Y.; Tada, H.; Shibata, S.; et al. Beneficial compaction of spinal cord lesion by migrating astrocytes through glycogen synthase kinase-3 inhibition. EMBO Mol. Med. 2011, 3, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Yamashita, N.; Kimura, A.; Kimura, Y.; Hirano, H.; Makihara, H.; Kawamoto, Y.; Jitsuki-Takahashi, A.; Yonezaki, K.; Takase, K.; et al. Comprehensive behavioral study and proteomic analyses of CRMP2-deficient mice. Genes Cells Devoted Mol. Cell. Mech. 2016, 21, 1059–1079. [Google Scholar] [CrossRef]
- Kadoyama, K.; Matsuura, K.; Nakamura-Hirota, T.; Takano, M.; Otani, M.; Matsuyama, S. Changes in the expression of collapsin response mediator protein-2 during synaptic plasticity in the mouse hippocampus. J. Neurosci. Res. 2015, 93, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T.; Kawano, Y.; Arimura, N.; Kawabata, S.; Kikuchi, A.; Kaibuchi, K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 2005, 120, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.R.; Noble, W.; van Aalten, L.; Plattner, F.; Meimaridou, R.; Hogan, D.; Taylor, M.; LaFrancois, J.; Gunn-Moore, F.; Verkhratsky, A.; et al. Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer’s disease progression. J. Neurochem. 2007, 103, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Ikezu, S.; Ingraham Dixie, K.L.; Koro, L.; Watanabe, T.; Kaibuchi, K.; Ikezu, T. Tau-tubulin kinase 1 and amyloid-β peptide induce phosphorylation of collapsin response mediator protein-2 and enhance neurite degeneration in Alzheimer disease mouse models. Acta Neuropathol. Commun. 2020, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.; Cai, J.; Gu, B.; Yu, L.; Li, C.; Liu, Q.S.; Zhao, L. Treadmill Exercise Prevents Decline in Spatial Learning and Memory in 3×Tg-AD Mice through Enhancement of Structural Synaptic Plasticity of the Hippocampus and Prefrontal Cortex. Cells 2022, 11, 244. [Google Scholar] [CrossRef]
- Zhao, G.; Liu, H.L.; Zhang, H.; Tong, X.J. Treadmill exercise enhances synaptic plasticity, but does not alter β-amyloid deposition in hippocampi of aged APP/PS1 transgenic mice. Neuroscience 2015, 298, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.W.; Tsai, S.F.; Kuo, Y.M. Physical Exercise Enhances Neuroplasticity and Delays Alzheimer’s Disease. Brain Plast. (Amst. Neth.) 2018, 4, 95–110. [Google Scholar] [CrossRef]
- Liu, Y.; Chu, J.M.T.; Yan, T.; Zhang, Y.; Chen, Y.; Chang, R.C.C.; Wong, G.T.C. Short-term resistance exercise inhibits neuroinflammation and attenuates neuropathological changes in 3xTg Alzheimer’s disease mice. J. Neuroinflam. 2020, 17, 4. [Google Scholar] [CrossRef]
- Kim, Y.M.; Song, I.; Seo, Y.H.; Yoon, G. Glycogen Synthase Kinase 3 Inactivation Induces Cell Senescence through Sterol Regulatory Element Binding Protein 1-Mediated Lipogenesis in Chang Cells. Endocrinol. Metab. 2013, 28, 297–308. [Google Scholar] [CrossRef]
- Hu, S.; Begum, A.N.; Jones, M.R.; Oh, M.S.; Beech, W.K.; Beech, B.H.; Yang, F.; Chen, P.; Ubeda, O.J.; Kim, P.C.; et al. GSK3 inhibitors show benefits in an Alzheimer’s disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol. Dis. 2009, 33, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Morales-García, J.A.; Susín, C.; Alonso-Gil, S.; Pérez, D.I.; Palomo, V.; Pérez, C.; Conde, S.; Santos, A.; Gil, C.; Martínez, A.; et al. Glycogen synthase kinase-3 inhibitors as potent therapeutic agents for the treatment of Parkinson disease. ACS Chem. Neurosci. 2013, 4, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.E.; Lesné, S.E. Soluble Aβ oligomer production and toxicity. J. Neurochem. 2012, 120 (Suppl. S1), 125–139. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Amar, F.; Sherman, M.A.; Rush, T.; Larson, M.; Boyle, G.; Chang, L.; Götz, J.; Buisson, A.; Lesné, S.E. The amyloid-β oligomer Aβ*56 induces specific alterations in neuronal signaling that lead to tau phosphorylation and aggregation. Sci. Signal. 2017, 10, eaal2021. [Google Scholar] [CrossRef]
- Reddy, P.H. Amyloid beta-induced glycogen synthase kinase 3β phosphorylated VDAC1 in Alzheimer’s disease: Implications for synaptic dysfunction and neuronal damage. Biochim. Biophys. Acta 2013, 1832, 1913–1921. [Google Scholar] [CrossRef]
- Krishnankutty, A.; Kimura, T.; Saito, T.; Aoyagi, K.; Asada, A.; Takahashi, S.I.; Ando, K.; Ohara-Imaizumi, M.; Ishiguro, K.; Hisanaga, S.I. In vivo regulation of glycogen synthase kinase 3β activity in neurons and brains. Sci. Rep. 2017, 7, 8602. [Google Scholar] [CrossRef]
- Taipa, R.; Ferreira, V.; Brochado, P.; Robinson, A.; Reis, I.; Marques, F.; Mann, D.M.; Melo-Pires, M.; Sousa, N. Inflammatory pathology markers (activated microglia and reactive astrocytes) in early and late onset Alzheimer disease: A post mortem study. Neuropathol. Appl. Neurobiol. 2018, 44, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.R.; Liu, R.T. The Toxicity and Polymorphism of β-Amyloid Oligomers. Int. J. Mol. Sci. 2020, 21, 4477. [Google Scholar] [CrossRef] [PubMed]
- Jana, M.K.; Cappai, R.; Pham, C.L.; Ciccotosto, G.D. Membrane-bound tetramer and trimer Aβ oligomeric species correlate with toxicity towards cultured neurons. J. Neurochem. 2016, 136, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Kawarabayashi, T.; Shoji, M.; Younkin, L.H.; Wen-Lang, L.; Dickson, D.W.; Murakami, T.; Matsubara, E.; Abe, K.; Ashe, K.H.; Younkin, S.G. Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 2004, 24, 3801–3809. [Google Scholar] [CrossRef]
- Meilandt, W.J.; Cisse, M.; Ho, K.; Wu, T.; Esposito, L.A.; Scearce-Levie, K.; Cheng, I.H.; Yu, G.Q.; Mucke, L. Neprilysin overexpression inhibits plaque formation but fails to reduce pathogenic Abeta oligomers and associated cognitive deficits in human amyloid precursor protein transgenic mice. J. Neurosci. 2009, 29, 1977–1986. [Google Scholar] [CrossRef]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef]
- Zhang, F.; Gannon, M.; Chen, Y.; Yan, S.; Zhang, S.; Feng, W.; Tao, J.; Sha, B.; Liu, Z.; Saito, T.; et al. β-amyloid redirects norepinephrine signaling to activate the pathogenic GSK3β/tau cascade. Sci. Transl. Med. 2020, 12, eaay6931. [Google Scholar] [CrossRef]
- Resende, R.; Ferreiro, E.; Pereira, C.; Oliveira, C.R. ER stress is involved in Abeta-induced GSK-3beta activation and tau phosphorylation. J. Neurosci. Res. 2008, 86, 2091–2099. [Google Scholar] [CrossRef]
- Fang, X.; Yu, S.X.; Lu, Y.; Bast, R.C., Jr.; Woodgett, J.R.; Mills, G.B. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. USA 2000, 97, 11960–11965. [Google Scholar] [CrossRef]
- Peineau, S.; Bradley, C.; Taghibiglou, C.; Doherty, A.; Bortolotto, Z.A.; Wang, Y.T.; Collingridge, G.L. The role of GSK-3 in synaptic plasticity. Br. J. Pharmacol. 2008, 153 (Suppl. S1), S428–S437. [Google Scholar] [CrossRef]
- Zhang, Z.; Ma, Z.; Zou, W.; Guo, H.; Liu, M.; Ma, Y.; Zhang, L. The Appropriate Marker for Astrocytes: Comparing the Distribution and Expression of Three Astrocytic Markers in Different Mouse Cerebral Regions. BioMed Res. Int. 2019, 2019, 9605265. [Google Scholar] [CrossRef] [PubMed]
- Yeo, H.G.; Hong, J.J.; Lee, Y.; Yi, K.S.; Jeon, C.Y.; Park, J.; Won, J.; Seo, J.; Ahn, Y.J.; Kim, K.; et al. Increased CD68/TGFβ Co-expressing Microglia/Macrophages after Transient Middle Cerebral Artery Occlusion in Rhesus Monkeys. Exp. Neurobiol. 2019, 28, 458–473. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.R.; Liu, J.C.; Bao, J.S.; Bai, Q.Q.; Wang, G.Q. Interaction of Microglia and Astrocytes in the Neurovascular Unit. Front. Immunol. 2020, 11, 1024. [Google Scholar] [CrossRef] [PubMed]
- Green, H.F.; Nolan, Y.M. Unlocking mechanisms in interleukin-1β-induced changes in hippocampal neurogenesis—A role for GSK-3β and TLX. Transl. Psychiatry 2012, 2, e194. [Google Scholar] [CrossRef] [PubMed]
- Edara, V.V.; Nooka, S.; Proulx, J.; Stacy, S.; Ghorpade, A.; Borgmann, K. β-Catenin Regulates Wound Healing and IL-6 Expression in Activated Human Astrocytes. Biomedicines 2020, 8, 479. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Wang, L.; Yang, B.; Zeng, J.; Zhang, Q.; Lei, H.; Xu, S. Protective effect of pretreatment with propofol against tumor necrosis factor-α-induced hepatic insulin resistance. Exp. Ther. Med. 2015, 10, 289–294. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xing, H.; Lim, Y.A.; Chong, J.R.; Lee, J.H.; Aarsland, D.; Ballard, C.G.; Francis, P.T.; Chen, C.P.; Lai, M.K. Increased phosphorylation of collapsin response mediator protein-2 at Thr514 correlates with β-amyloid burden and synaptic deficits in Lewy body dementias. Mol. Brain 2016, 9, 84. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Wang, Y.; Feng, Q.; Yang, P.; Qin, L. Intracerebroventricular administration of lupus serum induces microglia activation and leukocyte adhesion in the cerebromicrovasculature of mice. J. Neuroimmunol. 2019, 334, 576994. [Google Scholar] [CrossRef]
- Hironaka, K.; Yamazaki, Y.; Hirai, Y.; Yamamoto, M.; Miyake, N.; Miyake, K.; Okada, T.; Morita, A.; Shimada, T. Enzyme replacement in the CSF to treat metachromatic leukodystrophy in mouse model using single intracerebroventricular injection of self-complementary AAV1 vector. Sci. Rep. 2015, 5, 13104. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, L.; Li, X.; Run, X.; Liang, Z.; Li, Y.; Liu, Y.; Lee, M.H.; Grundke-Iqbal, I.; Iqbal, K.; et al. Differential effects of an O-GlcNAcase inhibitor on tau phosphorylation. PLoS ONE 2012, 7, e35277. [Google Scholar] [CrossRef]
- Azim, K.; Butt, A.M. GSK3β negatively regulates oligodendrocyte differentiation and myelination in vivo. Glia 2011, 59, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Vickstrom, C.R.; Liu, X.; Zhang, Y.; Mu, L.; Kelly, T.J.; Yan, X.; Hu, M.M.; Snarrenberg, S.T.; Liu, Q.S. T-Type Calcium Channels Contribute to Burst Firing in a Subpopulation of Medial Habenula Neurons. eNeuro 2020, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Vickstrom, C.R.; Yu, H.; Liu, S.; Snarrenberg, S.T.; Friedman, V.; Mu, L.; Chen, B.; Kelly, T.J.; Baker, D.A.; et al. Epac2 in midbrain dopamine neurons contributes to cocaine reinforcement via enhancement of dopamine release. Elife 2022, 11, e80747. [Google Scholar] [CrossRef] [PubMed]
- Ting, J.T.; Lee, B.R.; Chong, P.; Soler-Llavina, G.; Cobbs, C.; Koch, C.; Zeng, H.; Lein, E. Preparation of Acute Brain Slices Using an Optimized N-Methyl-D-glucamine Protective Recovery Method. J. Vis. Exp. JoVE 2018, 26, e53825. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Liu, X.; Vickstrom, C.; Li, Y.; Yu, L.; Lu, Y.; Smrcka, A.V.; Liu, Q.S. The Epac-Phospholipase Cepsilon Pathway Regulates Endocannabinoid Signaling and Cocaine-Induced Disinhibition of Ventral Tegmental Area Dopamine Neurons. J. Neurosci. 2017, 37, 3030–3044. [Google Scholar] [CrossRef]
- Liu, X.; Chen, Y.; Tong, J.; Reynolds, A.M.; Proudfoot, S.C.; Qi, J.; Penzes, P.; Lu, Y.; Liu, Q.S. Epac Signaling Is Required for Cocaine-Induced Change in AMPA Receptor Subunit Composition in the Ventral Tegmental Area. J. Neurosci. 2016, 36, 4802–4815. [Google Scholar] [CrossRef]
- Gao, X.P.; Liu, Q.S.; Liu, Q.; Wong-Riley, M.T. Excitatory-inhibitory imbalance in hypoglossal neurons during the critical period of postnatal development in the rat. J. Physiol. 2011, 589, 1991–2006. [Google Scholar] [CrossRef]
- Liu, X.; Zhong, P.; Vickstrom, C.; Li, Y.; Liu, Q.S. PDE4 Inhibition Restores the Balance Between Excitation and Inhibition in VTA Dopamine Neurons Disrupted by Repeated In Vivo Cocaine Exposure. Neuropsychopharmacology 2017, 42, 1991–1999. [Google Scholar] [CrossRef]
- Zhong, P.; Wang, W.; Yu, F.; Nazari, M.; Liu, X.; Liu, Q.S. Phosphodiesterase 4 inhibition impairs cocaine-induced inhibitory synaptic plasticity and conditioned place preference. Neuropsychopharmacology 2012, 37, 2377–2387. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mu, L.; Xia, D.; Cai, J.; Gu, B.; Liu, X.; Friedman, V.; Liu, Q.-S.; Zhao, L. Treadmill Exercise Reduces Neuroinflammation, Glial Cell Activation and Improves Synaptic Transmission in the Prefrontal Cortex in 3 × Tg-AD Mice. Int. J. Mol. Sci. 2022, 23, 12655. https://doi.org/10.3390/ijms232012655
Mu L, Xia D, Cai J, Gu B, Liu X, Friedman V, Liu Q-S, Zhao L. Treadmill Exercise Reduces Neuroinflammation, Glial Cell Activation and Improves Synaptic Transmission in the Prefrontal Cortex in 3 × Tg-AD Mice. International Journal of Molecular Sciences. 2022; 23(20):12655. https://doi.org/10.3390/ijms232012655
Chicago/Turabian StyleMu, Lianwei, Dongdong Xia, Jiajia Cai, Boya Gu, Xiaojie Liu, Vladislav Friedman, Qing-Song Liu, and Li Zhao. 2022. "Treadmill Exercise Reduces Neuroinflammation, Glial Cell Activation and Improves Synaptic Transmission in the Prefrontal Cortex in 3 × Tg-AD Mice" International Journal of Molecular Sciences 23, no. 20: 12655. https://doi.org/10.3390/ijms232012655
APA StyleMu, L., Xia, D., Cai, J., Gu, B., Liu, X., Friedman, V., Liu, Q.-S., & Zhao, L. (2022). Treadmill Exercise Reduces Neuroinflammation, Glial Cell Activation and Improves Synaptic Transmission in the Prefrontal Cortex in 3 × Tg-AD Mice. International Journal of Molecular Sciences, 23(20), 12655. https://doi.org/10.3390/ijms232012655