


Transcriptome Analysis of Developing Grains from Wheat Cultivars TAM 111 and TAM 112 Reveal Cultivar-Specific Regulatory Networks

 , , , and
, , , and
Abstract
1. Introduction
2. Results and Discussion
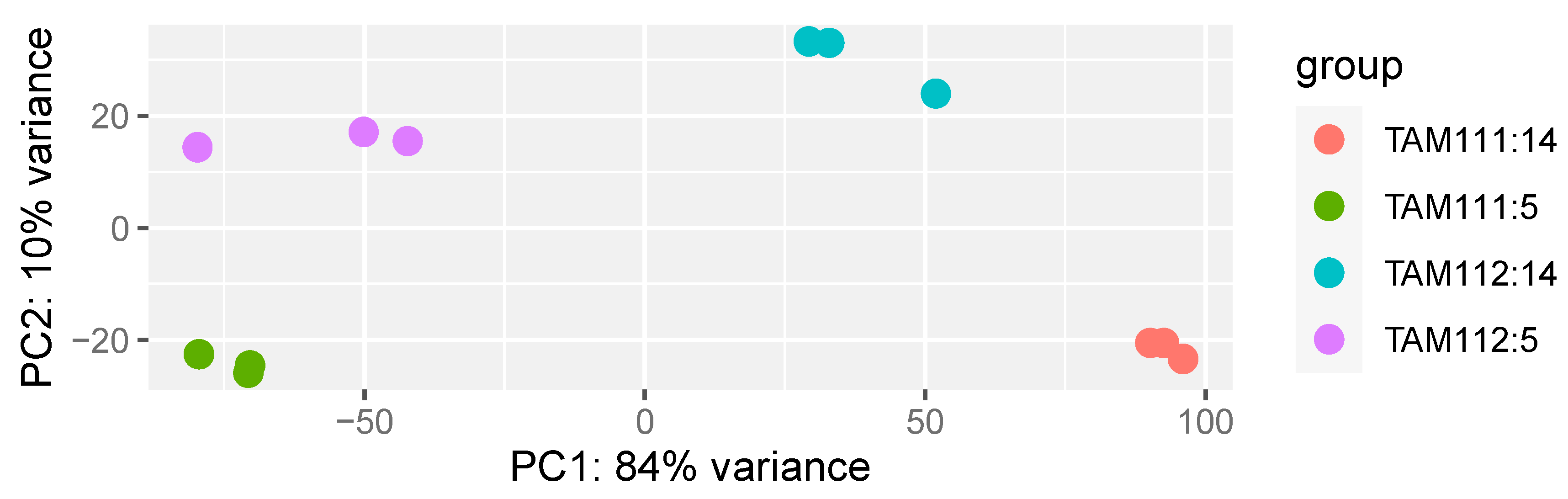
2.1. RNAseq Data Structure
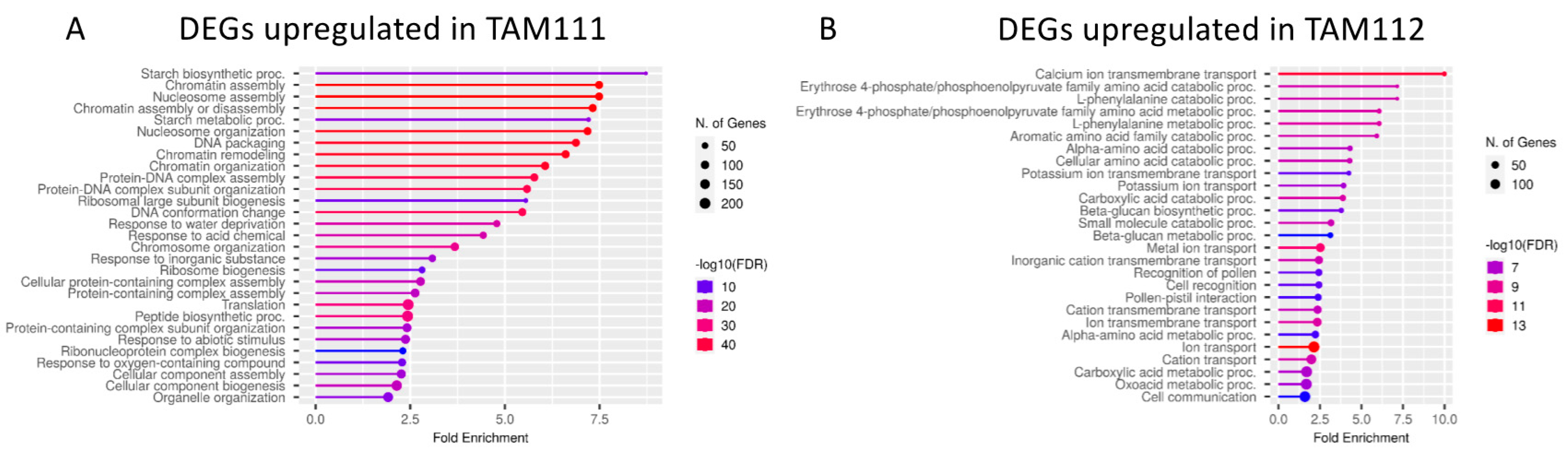
2.2. DEGs between TAM 111 and TAM 112 at Each Time Points Show Different Pathways Over-Represented
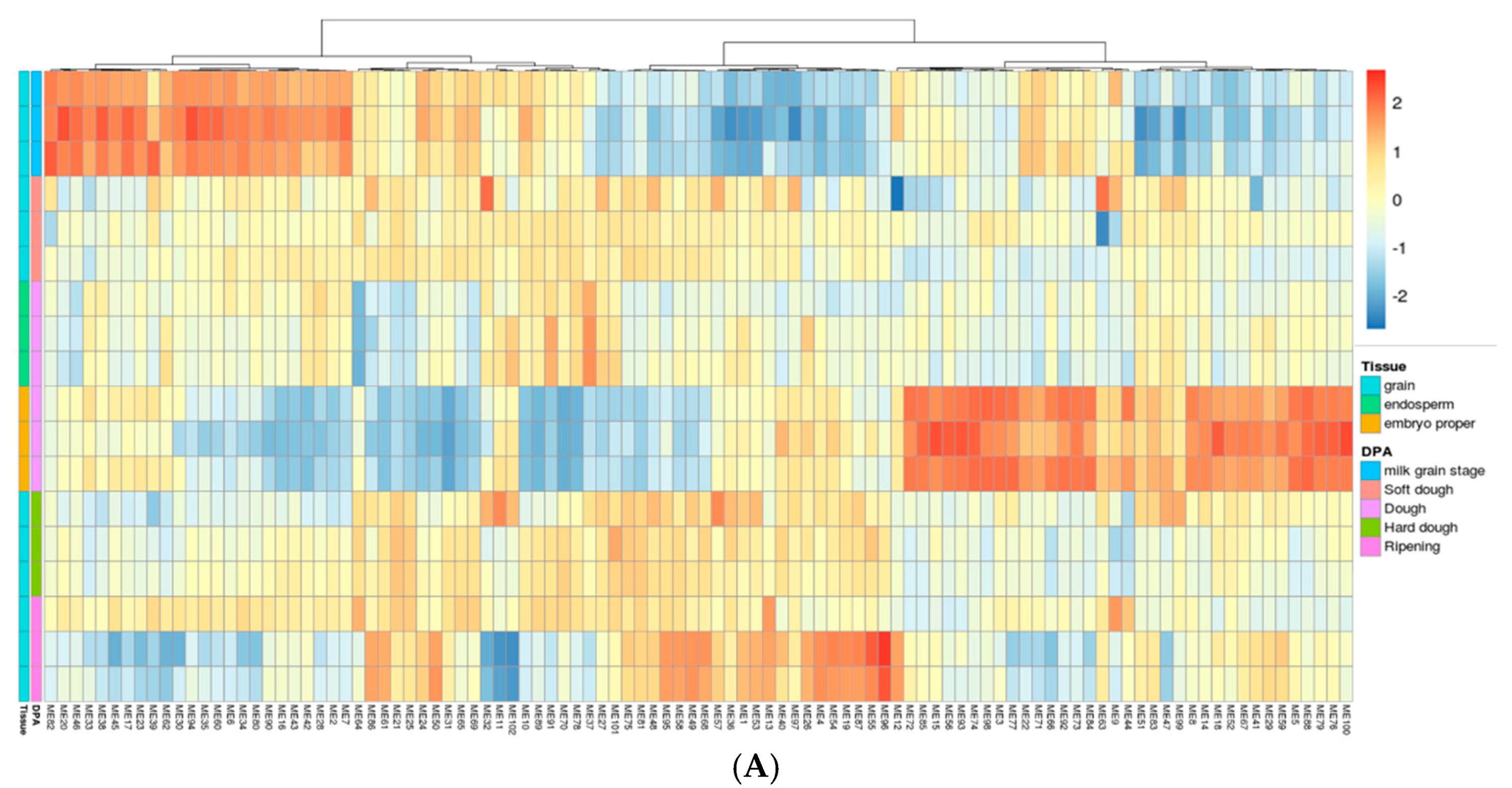
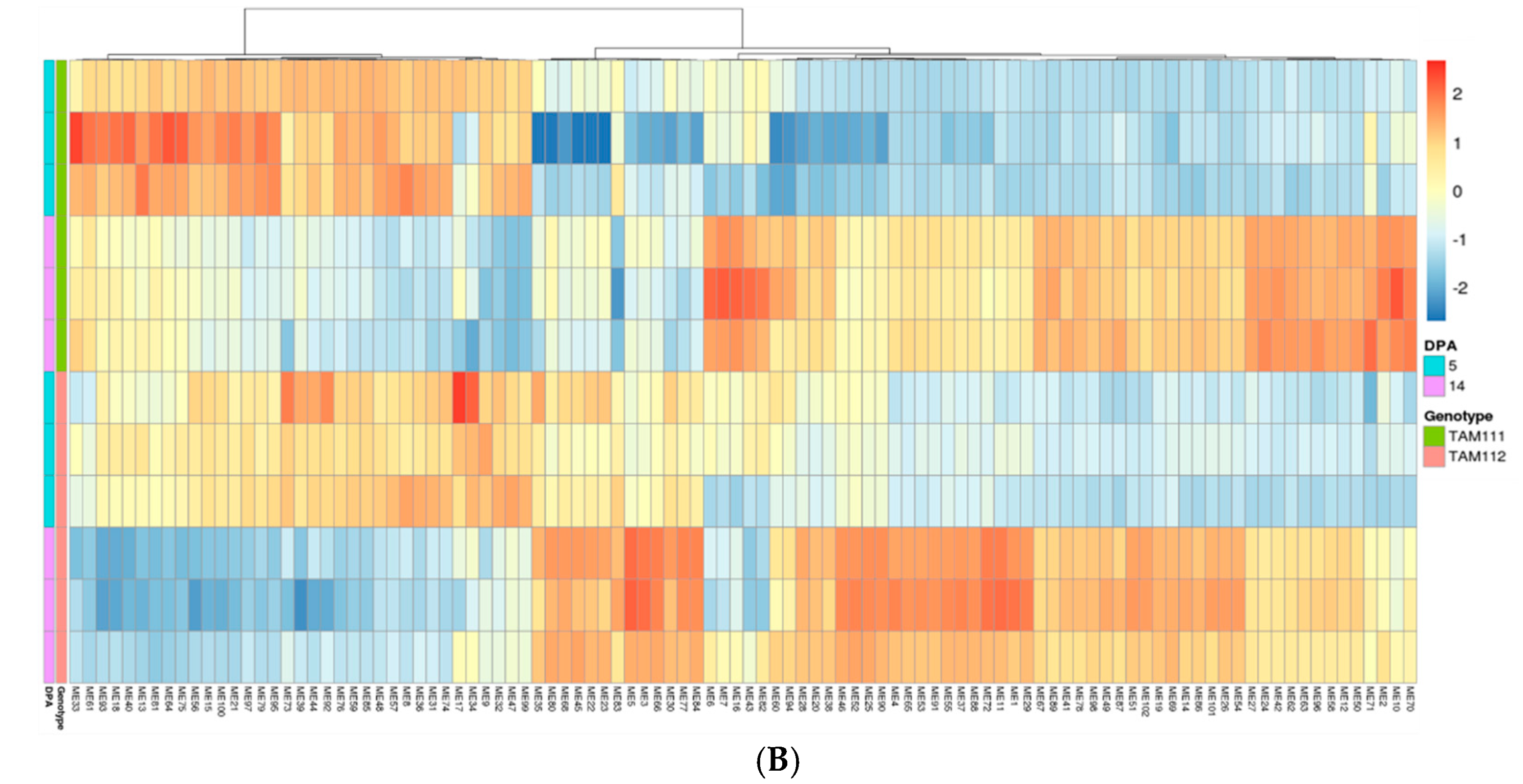
2.3. Gene Network Analysis to Identify Modules Enriched in DEGs
2.4. Transcription Factors Identified by GLASSO to Be Central to Starch Synthesis
3. Materials and Methods
3.1. Plant Materials
3.2. RNA Extraction and Sequencing
3.3. RNAseq Data Processing
3.4. Differential Expressed Genes (DEG) and Gene Ontology Enrichment (GO) Analysis
3.5. Constructing Gene Network Analysis
3.6. GLASSO Analysis
3.7. Phytogenic Tree Analysis of NF-YC and NF-YB
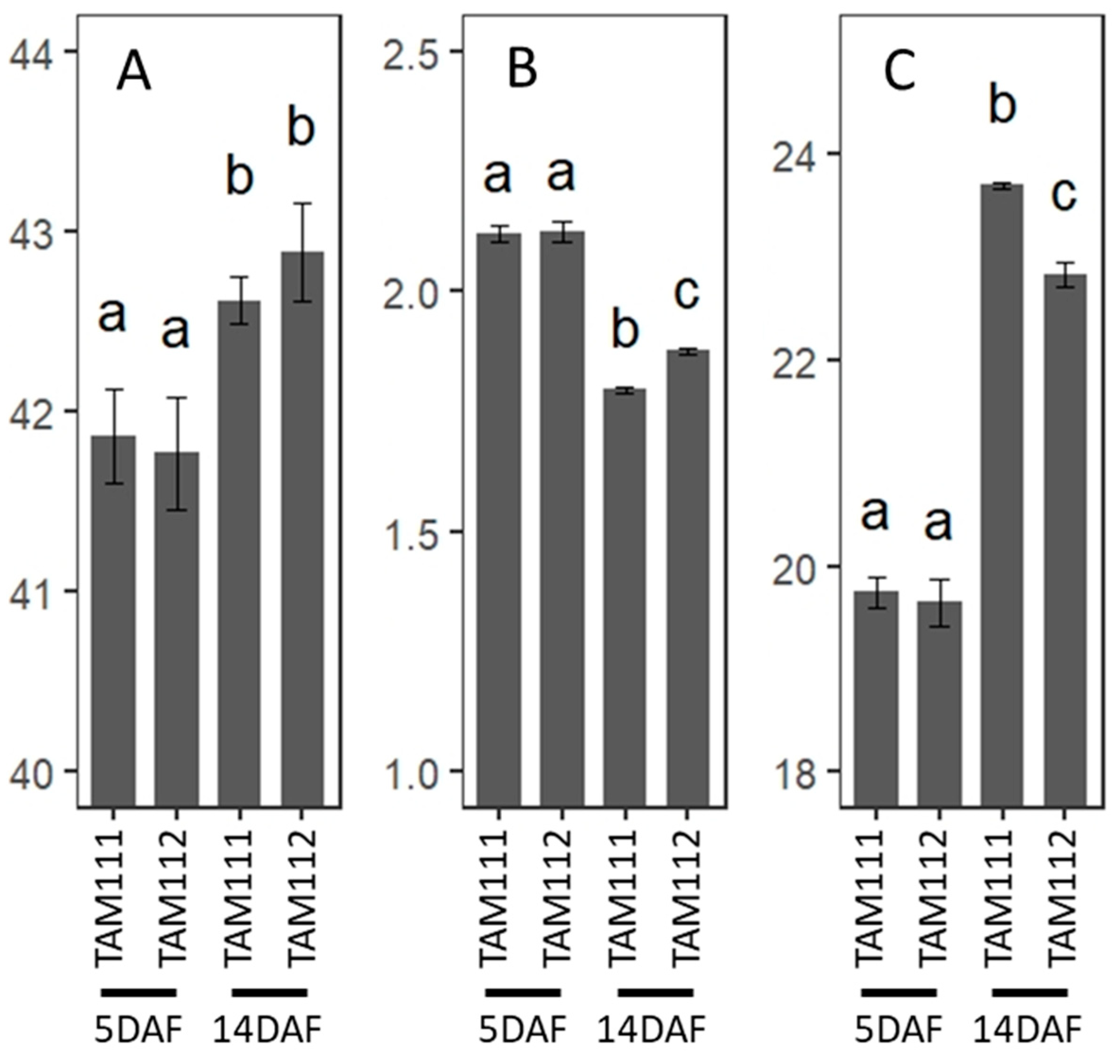
3.8. C and N Analysis of Wheat Grains
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sowell, B.S.A.; Huntchins, C. Situation and Outlook Report (Wheat) in Economic Research Service; USDA: Washington, DC, USA, 2022. [Google Scholar]
- Kang, G.; Liu, G.; Peng, X.; Wei, L.; Wang, C.; Zhu, Y.; Ma, Y.; Jiang, Y.; Guo, T. Increasing the starch content and grain weight of common wheat by overexpression of the cytosolic AGPase large subunit gene. Plant Physiol. Biochem. 2013, 73, 93–98. [Google Scholar] [CrossRef]
- Peng, B.; Kong, H.; Li, Y.; Wang, L.; Zhong, M.; Sun, L.; Gao, G.; Zhang, Q.; Luo, L.; Wang, G. OsAAP6 functions as an important regulator of grain protein content and nutritional quality in rice. Nat. Commun. 2014, 5, 4847. [Google Scholar] [CrossRef] [PubMed]
- Perchlik, M.; Tegeder, M. Improving plant nitrogen use efficiency through alteration of amino acid transport processes. Plant Physiol. 2017, 175, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; An, K.; Guo, W.; Chen, Y.; Zhang, R.; Zhang, X.; Chang, S.; Rossi, V.; Jin, F.; Cao, X. The endosperm-specific transcription factor TaNAC019 regulates glutenin and starch accumulation and its elite allele improves wheat grain quality. Plant Cell 2021, 33, 603–622. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Su, Q.; Nian, J.; Zhang, J.; Guo, M.; Dong, G.; Hu, J.; Wang, R.; Wei, C.; Li, G.; et al. The Ghd7 transcription factor represses ARE1 expression to enhance nitrogen utilization and grain yield in rice. Mol. Plant 2021, 14, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.S.; Hotta, C.T.; Poelking, V.G.D.C.; Leite, D.C.C.; Buckeridge, M.S.; Loureiro, M.E.; Barbosa, M.H.P.; Carneiro, M.S.; Souza, G.M. Co-expression network analysis reveals transcription factors associated to cell wall biosynthesis in sugarcane. Plant Mol. Biol. 2016, 91, 15–35. [Google Scholar] [CrossRef]
- Yousefirad, S.; Soltanloo, H.; Ramezanpour, S.S.; Nezhad, K.Z.; Shariati, V. The RNA-seq transcriptomic analysis reveals genes mediating salt tolerance through rapid triggering of ion transporters in a mutant barley. PLoS ONE 2020, 15, e0229513. [Google Scholar] [CrossRef]
- Zhu, Q.; Gao, S.; Zhang, W. Identification of Key Transcription Factors Related to Bacterial Spot Resistance in Pepper through Regulatory Network Analyses. Genes 2021, 12, 1351. [Google Scholar] [CrossRef]
- Goh, K.-I.; Cusick, M.E.; Valle, D.; Childs, B.; Vidal, M.; Barabási, A.-L. The human disease network. Proc. Natl. Acad. Sci. USA 2007, 104, 8685–8690. [Google Scholar] [CrossRef]
- Horvath, S.; Dong, J. Geometric interpretation of gene coexpression network analysis. PLoS Comput. Biol. 2008, 4, e1000117. [Google Scholar] [CrossRef] [PubMed]
- Borrill, P.; Harrington, S.A.; Simmonds, J.; Uauy, C. Identification of transcription factors regulating senescence in wheat through gene regulatory network modelling. Plant Physiol. 2019, 180, 1740–1755. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Qin, P.; Hu, L.; Zhan, S.; Wang, S.; Gao, P.; Li, J.; Jin, M.; Xu, Z.; Gao, Q.; et al. OsSPL18 controls grain weight and grain number in rice. J. Genet. Genom. 2019, 46, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Huang, Z.; Jiang, H.; Wang, Z.; Wu, F.; Xiong, Y.; Yao, J. A heat stress responsive NAC transcription factor heterodimer plays key roles in rice grain filling. J. Exp. Bot. 2021, 72, 2947–2964. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Wang, S.; Paetzold, L.; Wang, Z.; Hui, K.; Rudd, J.C.; Xue, Q.; Ibrahim, A.M.; Metz, R.; Johnson, C.D. RNA-seq analysis reveals different drought tolerance mechanisms in two broadly adapted wheat cultivars ‘TAM 111′ and ‘TAM 112′. Sci. Rep. 2021, 11, 4301. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.K.; Liu, S.; Rudd, J.C.; Xue, Q.; Payton, P.; Finlayson, S.A.; Mahan, J.; Akhunova, A.; Holalu, S.V.; Lu, N. Physiology and transcriptomics of water-deficit stress responses in wheat cultivars TAM 111 and TAM 112. J. Plant Physiol. 2014, 171, 1289–1298. [Google Scholar] [CrossRef]
- Dhakal, S.; Liu, X.; Girard, A.; Chu, C.; Yang, Y.; Wang, S.; Xue, Q.; Rudd, J.C.; Ibrahim, A.M.; Awika, J.M. Genetic dissection of end-use quality traits in two widely adapted wheat cultivars ‘TAM 111′and ‘TAM 112′. Crop Sci. 2021, 61, 1944–1959. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Fahy, B.; Siddiqui, H.; David, L.C.; Powers, S.J.; Borrill, P.; Uauy, C.; Smith, A.M. Final grain weight is not limited by the activity of key starchsynthesising enzymes during grain filling in wheat. J. Exp. Bot. 2019, 70, 367. [Google Scholar] [CrossRef]
- Gillies, S.A.; Futardo, A.; Henry, R.J. Gene expression in the developing aleurone and starchy endosperm of wheat. Plant Biotechnol. J. 2012, 10, 668–679. [Google Scholar] [CrossRef]
- Ramírez-González, R.H.; Borrill, P.; Lang, D.; Harrington, S.A.; Brinton, J.; Venturini, L.; Davey, M.; Jacobs, J.; Van Ex, F.; Pasha, A.; et al. The transcriptional landscape of polyploid wheat. Science 2018, 361, eaar6089. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Chen, Y.; Zhang, M.; Cai, R.; Cheng, B.; Ma, Q.; Zhao, Y. A novel GRAS transcription factor, ZmGRAS20, regulates starch biosynthesis in rice endosperm. Physiol. Mol. Biol. Plants 2017, 23, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Bello, B.K.; Hou, Y.; Zhao, J.; Jiao, G.; Wu, Y.; Li, Z.; Wang, Y.; Tong, X.; Wang, W.; Yuan, W. NF-YB 1-YC 12-bHLH 144 complex directly activates Wx to regulate grain quality in rice (Oryza sativa L.). Plant Biotechnol. J. 2019, 17, 1222–1235. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Li, X.; Li, H.; Hu, Y.; Liu, H.; Wen, S.; Li, Y.; Liu, Y.; Huang, H.; Yu, G.; et al. Gibberellin induced transcription factor bZIP53 regulates CesA1 expression in maize kernels. PLoS ONE 2021, 16, e0244591. [Google Scholar] [CrossRef] [PubMed]
- Borrill, P.; Harrington, S.A.; Uauy, C. Applying the latest advances in genomics and phenomics for trait discovery in polyploid wheat. Plant J. 2019, 97, 56–72. [Google Scholar] [CrossRef]
- Xu, J.-J.; Zhang, X.-F.; Xue, H.-W. Rice aleurone layer specific OsNF-YB1 regulates grain filling and endosperm development by interacting with an ERF transcription factor. J. Exp. Bot. 2016, 67, erw409. [Google Scholar] [CrossRef]
- Li, S.; Chen, N.; Li, F.; Mei, F.; Wang, Z.; Cheng, X.; Kang, Z.; Mao, H. Characterization of wheat homeodomain-leucine zipper family genes and functional analysis of TaHDZ5-6A in drought tolerance in transgenic Arabidopsis. BMC Plant Biol. 2020, 20, 50. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.C.; Hrmova, M.; Lopato, S.; Langridge, P. Modulation of plant growth by HD-Zip class I and II transcription factors in response to environmental stimuli. New Phytol. 2011, 190, 823–837. [Google Scholar] [CrossRef]
- Zhang, S.; Haider, I.; Kohlen, W.; Jiang, L.; Bouwmeester, H.; Meijer, A.H.; Schluepmann, H.; Liu, C.-M.; Ouwerkerk, P.B. Function of the HD-Zip I gene Oshox22 in ABA-mediated drought and salt tolerances in rice. Plant Mol. Biol. 2012, 80, 571–585. [Google Scholar] [CrossRef]
- Turchi, L.; Baima, S.; Morelli, G.; Ruberti, I. Interplay of HD-Zip II and III transcription factors in auxin-regulated plant development. J. Exp. Bot. 2015, 66, 5043–5053. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Ding, Y.; Hu, S.; Ding, L.; Chen, Z.; Zhu, C. The role of HD-Zip class I transcription factors in plant response to abiotic stresses. Physiol. Plant. 2019, 167, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Sharif, R.; Raza, A.; Chen, P.; Li, Y.; El-Ballat, E.M.; Rauf, A.; Hano, C.; El-Esawi, M.A. HD-ZIP gene family: Potential roles in improving plant growth and regulating stress-responsive mechanisms in plants. Genes 2021, 12, 1256. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Luo, T.; Zhao, H.; Su, Y.; Ji, W.; Li, H. Identification of wheat DREB genes and functional characterization of TaDREB3 in response to abiotic stresses. Gene 2020, 740, 144514. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.T.; Kapoor, R.; Datta, A.; Okumoto, S. Tissue specific expression of UMAMIT amino acid transporters in wheat. Sci. Rep. 2022, 12, 348. [Google Scholar] [PubMed]
- Li, Z.; Trick, H.N. Rapid method for high-quality RNA isolation from seed endosperm containing high levels of starch. Biotechniques 2005, 38, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. In Babraham Bioinformatics; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from highthroughput sequencing reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Ignatiadis, N.; Klaus, B.; Zaugg, J.B.; Huber, W. Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat. Methods 2016, 13, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Botía, J.A.; Vandrovcova, J.; Forabosco, P.; Guelfi, S.; D’Sa, K.; Hardy, J.; Lewis, C.M.; Ryten, M.; Weale, M.E. An additional k-means clustering step improves the biological features of WGCNA gene co-expression networks. BMC Syst. Biol. 2017, 11, 47. [Google Scholar] [CrossRef] [PubMed]
- Danaher, P.; Wang, P.; Witten, D.M. The joint graphical lasso for inverse covariance estimation across multiple classes. J. R. Stat. Soc. Ser. B 2014, 76, 373–397. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef] [PubMed]
- Bogard, M.; Allard, V.; Brancourt-Hulmel, M.; Heumez, E.; Machet, J.M.; Jeuffroy, M.H.; Gate, P.; Martre, P.; Le Gouis, J. Deviation from the grain protein concentration-grain yield negative relationship is highly correlated to post-anthesis N uptake in winter wheat. J. Exp. Bot. 2010, 61, 4303–4312. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A. DEG Analysis of TAM111 vs. TAM112 | |||
|---|---|---|---|
| Total DEGs | Upregulated in TAM111 | Upregulated in TAM112 | |
| 5 DAF | 2629 | 1414 | 1215 |
| 14 DAF | 9368 | 4868 | 4500 |
| Shared | 1937 | 1081 | 856 |
| B. DEG Analysis of 5 vs. 14 DAF | |||
| Total DEGs | Upregulated in 5 DAF | Upregulated in 14 DAF | |
| In TAM111 | 14,745 | 6370 | 8375 |
| In TAM112 | 13,061 | 5481 | 7580 |
| Shared | 8044 | 3248 | 4754 |
| A. GLASSO TAM111A | ||||
|---|---|---|---|---|
| Gene | Transcription Factor Family | Edges in TAM111 | Edges in TAM112 | Difference |
| TraesCS7B02G248300 | Nuclear Factor-Y | 208 | 36 | 172 |
| TraesCS5B02G043100 | MYB-related | 39 | 0 | 39 |
| TraesCS5A02G245900 | NAC | 48 | 20 | 28 |
| TraesCS7D02G196300 | NAC | 35 | 12 | 23 |
| TraesCS7B02G056300 | NAC | 33 | 12 | 21 |
| TraesCS7A02G336700 | Nuclear Factor-Y | 46 | 26 | 20 |
| TraesCS3A02G368700 | MYB-related | 23 | 3 | 20 |
| TraesCS7B02G100300 | NAC | 37 | 19 | 18 |
| TraesCS7D02G344400 | Nuclear Factor-Y | 58 | 41 | 17 |
| TraesCS5D02G161000 | C2C2_Dof | 26 | 9 | 17 |
| B. GLASSO TAM111B | ||||
| Gene | Transcription Factor Family | Edges in TAM111 | Edges in TAM112 | Difference |
| TraesCS7B02G248300 | Nuclear Factor-Y | 343 | 16 | 327 |
| TraesCS7D02G344400 | Nuclear Factor-Y | 125 | 16 | 109 |
| TraesCS7A02G336700 | Nuclear Factor-Y | 97 | 9 | 88 |
| TraesCS5B02G043100 | MYB-related | 68 | 0 | 68 |
| TraesCS7D02G216600 | CCAAT_Dr1 | 36 | 9 | 27 |
| TraesCS5A02G155900 | C2C2_Dof | 40 | 21 | 19 |
| TraesCS2D02G234000 | MYB-related | 22 | 5 | 17 |
| TraesCS7A02G194700 | NAC | 55 | 39 | 16 |
| TraesCS5B02G154100 | C2C2_Dof | 35 | 19 | 16 |
| TraesCS4B02G299000 | MYB-related | 18 | 2 | 16 |
| C. GLASSO TAM111C | ||||
| Gene | Transcription Factor Family | Edges in TAM111 | Edges in TAM112 | Difference |
| TraesCS7B02G248300 | Nuclear Factor-Y | 448 | 39 | 409 |
| TraesCS5B02G043100 | MYB-related | 80 | 0 | 80 |
| TraesCS5A02G245900 | NAC | 43 | 8 | 35 |
| TraesCS3A02G077900 | NAC | 35 | 6 | 29 |
| TraesCS7D02G154200 | NAC | 34 | 8 | 26 |
| TraesCS7B02G056300 | NAC | 32 | 10 | 22 |
| TraesCS7A02G569300 | NAC | 23 | 1 | 22 |
| TraesCS7B02G489500 | NAC | 30 | 10 | 20 |
| TraesCS7D02G344400 | Nuclear Factor-Y | 69 | 52 | 17 |
| TraesCS7A02G336700 | Nuclear Factor-Y | 47 | 30 | 17 |
| A. GLASSO TAM112A | ||||
|---|---|---|---|---|
| Gene | Transcription Factor Family | Edges in TAM111 | Edges in TAM112 | Difference |
| TraesCS4A02G231300 | HD-Zip_IV | 96 | 2325 | 2229 |
| TraesCS1B02G256000 | AP2/EREBP | 63 | 258 | 195 |
| TraesCS3A02G325800 | HD-Zip_IV | 3 | 214 | 211 |
| TraesCS2A02G389400 | HD-Zip_I_II | 2 | 172 | 170 |
| TraesCS4A02G131000 | NAC | 11 | 140 | 129 |
| TraesCS1D02G244500 | AP2/EREBP | 60 | 116 | 56 |
| TraesCS1A02G244800 | AP2/EREBP | 60 | 109 | 49 |
| TraesCS3B02G354900 | HD-Zip_IV | 3 | 97 | 94 |
| TraesCS7B02G112400 | MYB-related | 68 | 88 | 20 |
| TraesCS4A02G263300 | bHLH | 51 | 85 | 34 |
| B. GLASSO TAM112B | ||||
| Gene | Transcription Factor Family | Edges in TAM111 | Edges in TAM112 | Difference |
| TraesCS4A02G231300 | HD-Zip_IV | 104 | 253 | 149 |
| TraesCS2A02G389400 | HD-Zip_I_II | 3 | 68 | 65 |
| TraesCS5A02G155900 | C2C2_Dof | 31 | 83 | 52 |
| TraesCS4A02G131000 | NAC | 10 | 60 | 50 |
| TraesCS3A02G325800 | HD-Zip_IV | 3 | 52 | 49 |
| TraesCS7A02G189200 | NAC | 20 | 55 | 35 |
| TraesCS2B02G119100 | NAC | 4 | 39 | 35 |
| TraesCS3B02G354900 | HD-Zip_IV | 1 | 36 | 35 |
| TraesCS4A02G006100 | MYB-related | 13 | 46 | 33 |
| TraesCS4D02G051600 | bHLH | 25 | 52 | 27 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, Z.-T.; Kapoor, R.; Datta, A.; Liu, S.; Stull, M.A.; Seitz, P.G.; Johnson, C.D.; Okumoto, S. Transcriptome Analysis of Developing Grains from Wheat Cultivars TAM 111 and TAM 112 Reveal Cultivar-Specific Regulatory Networks. Int. J. Mol. Sci. 2022, 23, 12660. https://doi.org/10.3390/ijms232012660
Fang Z-T, Kapoor R, Datta A, Liu S, Stull MA, Seitz PG, Johnson CD, Okumoto S. Transcriptome Analysis of Developing Grains from Wheat Cultivars TAM 111 and TAM 112 Reveal Cultivar-Specific Regulatory Networks. International Journal of Molecular Sciences. 2022; 23(20):12660. https://doi.org/10.3390/ijms232012660
Chicago/Turabian StyleFang, Ze-Tian, Rajan Kapoor, Aniruddha Datta, Shuyu Liu, Matthew A. Stull, Paige G. Seitz, Charles D. Johnson, and Sakiko Okumoto. 2022. "Transcriptome Analysis of Developing Grains from Wheat Cultivars TAM 111 and TAM 112 Reveal Cultivar-Specific Regulatory Networks" International Journal of Molecular Sciences 23, no. 20: 12660. https://doi.org/10.3390/ijms232012660
APA StyleFang, Z.-T., Kapoor, R., Datta, A., Liu, S., Stull, M. A., Seitz, P. G., Johnson, C. D., & Okumoto, S. (2022). Transcriptome Analysis of Developing Grains from Wheat Cultivars TAM 111 and TAM 112 Reveal Cultivar-Specific Regulatory Networks. International Journal of Molecular Sciences, 23(20), 12660. https://doi.org/10.3390/ijms232012660