
Role of Macrophages in Air Pollution Exposure Related Asthma

 , and
, and
Abstract
1. Introduction
2. Origins of Lung Macrophages
3. Macrophage Polarization in Asthma
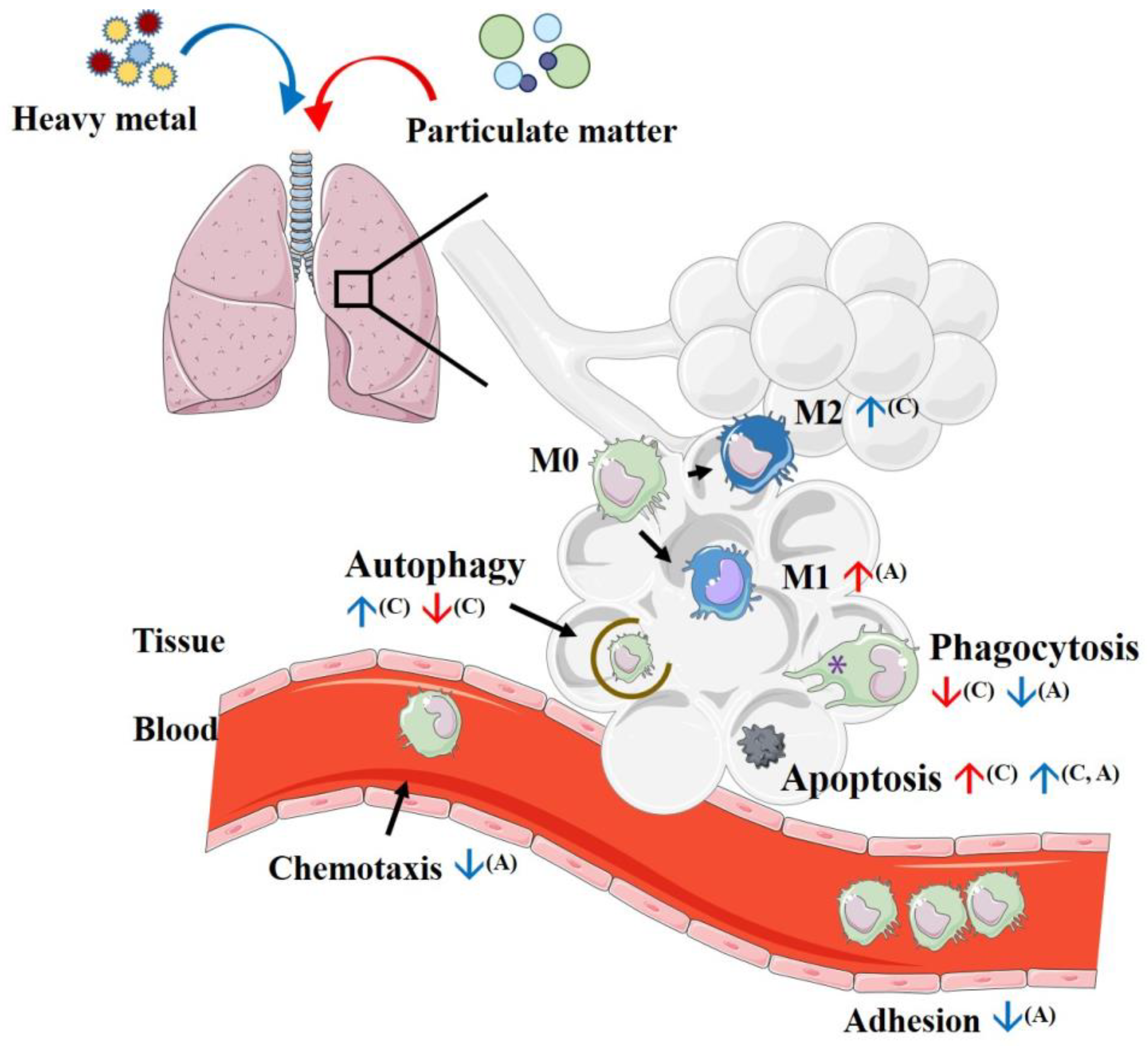
4. Initial Process Reactions after Introducing Particulate Matter to Macrophages
4.1. Phagocytosis
4.2. Efferocytosis
4.3. Autophagy
4.4. Apoptosis
5. Immune Reaction after Exposure to Particulate Matter
5.1. Toll-like Receptor (TLR) Responses
5.2. Reactive Oxygen Species (ROS) Reaction
5.3. Polyaromatic Hydrocarbon (PAH)
6. Inflammatory Mediator Production after Exposure to Particulate Matter
6.1. Proinflammatory Cytokines
6.2. The Effect of PM Exposure on Macrophage Polarization
7. Effects of Heavy Metals on Macrophage Polarization
7.1. Lead (Pb)
7.2. Vanadium (V)
7.3. Arsenic (As)
7.4. Manganese (Mn)
7.5. Nickel (Ni)
7.6. Cadmium (Cd)
7.7. Chromium (Cr(VI))
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Diseases, G.B.D.; Injuries, C. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar]
- Ambient (Outdoor) Air Pollution. Available online: https://www.who.int/news-room/fact-sheets/detail/ambient-(outdoor)-air-quality-and-health (accessed on 22 September 2021).
- Attademo, L.; Bernardini, F.; Garinella, R.; Compton, M.T. Environmental pollution and risk of psychotic disorders: A review of the science to date. Schizophr. Res. 2017, 181, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Kabir, E.; Kabir, S. A review on the human health impact of airborne particulate matter. Environ. Int. 2015, 74, 136–143. [Google Scholar] [CrossRef]
- Favarato, G.; Anderson, H.R.; Atkinson, R.; Fuller, G.; Mills, I.; Walton, H. Traffic-related pollution and asthma prevalence in children. Quantification of associations with nitrogen dioxide. Air Qual. Atmos. Health 2014, 7, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Stanek, L.W.; Brown, J.S.; Stanek, J.; Gift, J.; Costa, D.L. Air pollution toxicology—A brief review of the role of the science in shaping the current understanding of air pollution health risks. Toxicol. Sci. 2011, 120 (Suppl. 1), S8–S27. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Gordon, T.; Price, O.; Asgharian, B. Thoracic and respirable particle definitions for human health risk assessment. Part. Fibre Toxicol. 2013, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Melgert, B.N.; ten Hacken, N.H.; Rutgers, B.; Timens, W.; Postma, D.S.; Hylkema, M.N. More alternative activation of macrophages in lungs of asthmatic patients. J. Allergy Clin. Immunol. 2011, 127, 831–833. [Google Scholar] [CrossRef] [PubMed]
- Draijer, C.; Boorsma, C.E.; Robbe, P.; Timens, W.; Hylkema, M.N.; Ten Hacken, N.H.; van den Berge, M.; Postma, D.S.; Melgert, B.N. Human asthma is characterized by more IRF5+ M1 and CD206+ M2 macrophages and less IL-10+ M2-like macrophages around airways compared with healthy airways. J. Allergy Clin. Immunol. 2017, 140, 280–283.e3. [Google Scholar] [CrossRef]
- Hogg, J.C.; van Eeden, S. Pulmonary and systemic response to atmospheric pollution. Respirology 2009, 14, 336–346. [Google Scholar] [CrossRef]
- Chitu, V.; Stanley, E.R. Regulation of Embryonic and Postnatal Development by the CSF-1 Receptor. Curr. Top. Dev. Biol. 2017, 123, 229–275. [Google Scholar] [PubMed]
- Kopf, M.; Schneider, C.; Nobs, S.P. The development and function of lung-resident macrophages and dendritic cells. Nat. Immunol. 2015, 16, 36–44. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; De Kleer, I.; Henri, S.; Post, S.; Vanhoutte, L.; De Prijck, S.; Deswarte, K.; Malissen, B.; Hammad, H.; Lambrecht, B.N. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 2013, 210, 1977–1992. [Google Scholar] [CrossRef] [PubMed]
- Yona, S.; Kim, K.W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Tsou, C.L.; Peters, W.; Si, Y.; Slaymaker, S.; Aslanian, A.M.; Weisberg, S.P.; Mack, M.; Charo, I.F. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J. Clin. Investig. 2007, 117, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Gibbings, S.L.; Thomas, S.M.; Atif, S.M.; McCubbrey, A.L.; Desch, A.N.; Danhorn, T.; Leach, S.M.; Bratton, D.L.; Henson, P.M.; Janssen, W.J.; et al. Three Unique Interstitial Macrophages in the Murine Lung at Steady State. Am. J. Respir. Cell Mol. Biol. 2017, 57, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.R.; Hotten, D.F.; Malakhau, Y.; Volker, E.; Ghio, A.J.; Noble, P.W.; Kraft, M.; Hollingsworth, J.W.; Gunn, M.D.; Tighe, R.M. Flow Cytometric Analysis of Myeloid Cells in Human Blood, Bronchoalveolar Lavage, and Lung Tissues. Am. J. Respir. Cell Mol. Biol. 2016, 54, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.J.; Ma, Y.Q.; Zhou, X.; Zhang, Y.D.; Lu, R.Y.; Sun, H.Y.; Guo, Z.Z.; Zhang, Z.; Li, Y.M.; Wei, L.Q. Temporal and spatial characterization of mononuclear phagocytes in circulating, lung alveolar and interstitial compartments in a mouse model of bleomycin-induced pulmonary injury. J. Immunol. Methods 2014, 403, 7–16. [Google Scholar] [CrossRef]
- Desch, A.N.; Gibbings, S.L.; Goyal, R.; Kolde, R.; Bednarek, J.; Bruno, T.; Slansky, J.E.; Jacobelli, J.; Mason, R.; Ito, Y.; et al. Flow Cytometric Analysis of Mononuclear Phagocytes in Nondiseased Human Lung and Lung-Draining Lymph Nodes. Am. J. Respir. Crit. Care Med. 2016, 193, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Diette, G.B.; McCormack, M.C.; Hansel, N.N.; Breysse, P.N.; Matsui, E.C. Environmental issues in managing asthma. Respir. Care 2008, 53, 602–615; discussion 616–617. [Google Scholar] [PubMed]
- Yang, J.; Scicluna, B.P.; van Engelen, T.S.R.; Bonta, P.I.; Majoor, C.J.; Van’t Veer, C.; de Vos, A.F.; Bel, E.H.; van der Poll, T. Transcriptional changes in alveolar macrophages from adults with asthma after allergen challenge. Allergy 2021, 76, 2218–2222. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska, M.; Chen, L.Y.; Liu, Y.; Martinez-Anton, A.; Logun, C.; Alsaaty, S.; Cuento, R.A.; Cai, R.; Sun, J.; Quehenberger, O.; et al. Dysregulation of lipidomic profile and antiviral immunity in response to hyaluronan in patients with severe asthma. J. Allergy Clin. Immunol. 2017, 139, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Zaslona, Z.; Przybranowski, S.; Wilke, C.; van Rooijen, N.; Teitz-Tennenbaum, S.; Osterholzer, J.J.; Wilkinson, J.E.; Moore, B.B.; Peters-Golden, M. Resident alveolar macrophages suppress, whereas recruited monocytes promote, allergic lung inflammation in murine models of asthma. J. Immunol. 2014, 193, 4245–4253. [Google Scholar] [CrossRef] [PubMed]
- Komlosi, Z.I.; van de Veen, W.; Kovacs, N.; Szucs, G.; Sokolowska, M.; O’Mahony, L.; Akdis, M.; Akdis, C.A. Cellular and molecular mechanisms of allergic asthma. Mol. Asp. Med. 2022, 85, 100995. [Google Scholar] [CrossRef] [PubMed]
- Lauzon-Joset, J.F.; Marsolais, D.; Langlois, A.; Bissonnette, E.Y. Dysregulation of alveolar macrophages unleashes dendritic cell-mediated mechanisms of allergic airway inflammation. Mucosal Immunol. 2014, 7, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Moniuszko, M.; Bodzenta-Lukaszyk, A.; Kowal, K.; Dabrowska, M. Bronchial macrophages in asthmatics reveal decreased CD16 expression and substantial levels of receptors for IL-10, but not IL-4 and IL-7. Folia Histochem. Cytobiol. 2007, 45, 181–189. [Google Scholar]
- Lambrecht, B.N.; Hammad, H. The immunology of the allergy epidemic and the hygiene hypothesis. Nat. Immunol. 2017, 18, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Luo, L.; Lei, Z.; Li, B.; Liang, Z.; Liu, G.; Li, D.; Zhang, G.; Huang, B.; Feng, Z.H. IL-17-producing alveolar macrophages mediate allergic lung inflammation related to asthma. J. Immunol. 2008, 181, 6117–6124. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.H.; Wang, C.C.; Suen, J.L.; Sheu, C.C.; Kuo, C.H.; Liao, W.T.; Yang, Y.H.; Wu, C.C.; Leung, S.Y.; Lai, R.S.; et al. Altered pattern of monocyte differentiation and monocyte-derived TGF-beta1 in severe asthma. Sci. Rep. 2018, 8, 919. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.H.; Tsai, M.L.; Li, C.H.; Hsiao, H.P.; Chao, M.C.; Lee, M.S.; Lin, Y.C.; Hung, C.H. Altered Pattern of Macrophage Polarization as a Biomarker for Severity of Childhood Asthma. J. Inflamm. Res. 2021, 14, 6011–6023. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.J.; Peters, K.N.; Behar, S.M. Macrophages clean up: Efferocytosis and microbial control. Curr. Opin. Microbiol. 2014, 17, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Goodridge, H.S. Information processing during phagocytosis. Nat. Rev. Immunol. 2012, 12, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Beck-Speier, I.; Karg, E.; Behrendt, H.; Stoeger, T.; Alessandrini, F. Ultrafine particles affect the balance of endogenous pro- and anti-inflammatory lipid mediators in the lung: In-vitro and in-vivo studies. Part. Fibre Toxicol. 2012, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.J.; Héliot, A.; Trémolet, G.; Landkocz, Y.; Dewaele, D.; Cazier, F.; Ledoux, F.; Courcot, D. Cellular response and extracellular vesicles characterization of human macrophages exposed to fine atmospheric particulate matter. Environ. Pollut. 2019, 254 Pt A, 112933. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, X.; An, X.; Zhang, L.; Li, X.; Wang, L.; Zhu, G. Continuous exposure of PM2.5 exacerbates ovalbumin-induced asthma in mouse lung via a JAK-STAT6 signaling pathway. Adv. Clin. Exp. Med. 2020, 29, 825–832. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Ichinose, T.; Yoshida, S.; Shiba, F.; Arashidani, K.; Takano, H.; Sun, G.; Shibamoto, T. Differences in allergic inflammatory responses in murine lungs: Comparison of PM2.5 and coarse PM collected during the hazy events in a Chinese city. Inhal. Toxicol. 2016, 28, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhong, W.; Meng, Q.; Lin, Q.; Fang, C.; Huang, X.; Li, C.; Huang, Y.; Tan, J. Ambient PM2.5 exposure exacerbates severity of allergic asthma in previously sensitized mice. J. Asthma 2015, 52, 785–794. [Google Scholar] [PubMed]
- Rahmani, H.; Sadeghi, S.; Taghipour, N.; Roshani, M.; Amani, D.; Ghazanfari, T.; Mosaffa, N. The Effects of Particulate Matter on C57BL/6 Peritoneal and Alveolar Macrophages. Iran. J. Allergy Asthma Immunol. 2020, 19, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Shahbaz, M.A.; Martikainen, M.V.; Rönkkö, T.J.; Komppula, M.; Jalava, P.I.; Roponen, M. Urban air PM modifies differently immune defense responses against bacterial and viral infections in vitro. Environ. Res. 2021, 192, 110244. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Liu, X.; Li, W.; Zu, Y.; Zhou, F.; Shou, Q.; Ding, Z. PM2.5 Exposure Induces Inflammatory Response in Macrophages via the TLR4/COX-2/NF-κB Pathway. Inflammation 2020, 43, 1948–1958. [Google Scholar] [CrossRef]
- Schneider, J.C.; Card, G.L.; Pfau, J.C.; Holian, A. Air pollution particulate SRM 1648 causes oxidative stress in RAW 264.7 macrophages leading to production of prostaglandin E2, a potential Th2 mediator. Inhal. Toxicol. 2005, 17, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.L.; Cheng, W.L.; Lee, C.T.; Huang, H.C.; Chan, C.C. Contribution of endotoxin in macrophage cytokine response to ambient particles in vitro. J. Toxicol. Environ. Health A 2002, 65, 1261–1272. [Google Scholar] [CrossRef] [PubMed]
- Walters, D.M.; Breysse, P.N.; Wills-Karp, M. Ambient urban Baltimore particulate-induced airway hyperresponsiveness and inflammation in mice. Am. J. Respir. Crit. Care Med. 2001, 164, 1438–1443. [Google Scholar] [CrossRef]
- Paplinska-Goryca, M.; Misiukiewicz-Stepien, P.; Proboszcz, M.; Nejman-Gryz, P.; Gorska, K.; Zajusz-Zubek, E.; Krenke, R. Interactions of nasal epithelium with macrophages and dendritic cells variously alter urban PM-induced inflammation in healthy, asthma and COPD. Sci. Rep. 2021, 11, 13259. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.L.; Wang, B.; Chen, H.; Ho, K.F.; Cao, J.; Hai, G.; Jalaludin, B.; Herbert, C.; Thomas, P.S.; Saad, S.; et al. Pulmonary inflammation induced by low-dose particulate matter exposure in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L424–L430. [Google Scholar] [CrossRef] [PubMed]
- Selley, L.; Schuster, L.; Marbach, H.; Forsthuber, T.; Forbes, B.; Gant, T.W.; Sandström, T.; Camiña, N.; Athersuch, T.J.; Mudway, I.; et al. Brake dust exposure exacerbates inflammation and transiently compromises phagocytosis in macrophages. Metallomics 2020, 12, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Monn, C.; Becker, S. Cytotoxicity and induction of proinflammatory cytokines from human monocytes exposed to fine (PM2.5) and coarse particles (PM10-2.5) in outdoor and indoor air. Toxicol. Appl. Pharmacol. 1999, 155, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Gawda, A.; Majka, G.; Nowak, B.; Śróttek, M.; Walczewska, M.; Marcinkiewicz, J. Air particulate matter SRM 1648a primes macrophages to hyperinflammatory response after LPS stimulation. Inflamm. Res. 2018, 67, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Terashima, T.; Wiggs, B.; English, D.; Hogg, J.C.; van Eeden, S.F. Phagocytosis of small carbon particles (PM10) by alveolar macrophages stimulates the release of polymorphonuclear leukocytes from bone marrow. Am. J. Respir. Crit. Care Med. 1997, 155, 1441–1447. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Brugha, R.E.; Jacobs, L.; Grigg, J.; Nawrot, T.S.; Nemery, B. Carbon loading in airway macrophages as a biomarker for individual exposure to particulate matter air pollution-A critical review. Environ. Int. 2015, 74, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Morrow, P.E. Possible mechanisms to explain dust overloading of the lungs. Fundam. Appl. Toxicol. 1988, 10, 369–384. [Google Scholar] [CrossRef]
- Kulkarni, N.; Pierse, N.; Rushton, L.; Grigg, J. Carbon in airway macrophages and lung function in children. N. Engl. J. Med. 2006, 355, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Soukup, J.M.; Becker, S. Human alveolar macrophage responses to air pollution particulates are associated with insoluble components of coarse material, including particulate endotoxin. Toxicol. Appl. Pharmacol. 2001, 171, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Pu, W.; Niu, M.; Wazir, J.; Song, S.; Wei, L.; Li, L.; Su, Z.; Wang, H. Respiratory exposure to PM2.5 soluble extract induced chronic lung injury by disturbing the phagocytosis function of macrophage. Environ. Sci. Pollut. Res. Int. 2022, 29, 13983–13997. [Google Scholar] [CrossRef]
- Li, Y.; Yong, Y.L.; Yang, M.; Wang, W.; Qu, X.; Dang, X.; Shang, D.; Shao, Y.; Liu, J.; Chang, Y. Fine particulate matter inhibits phagocytosis of macrophages by disturbing autophagy. FASEB J. 2020, 34, 16716–16735. [Google Scholar] [CrossRef]
- Chen, Y.W.; Huang, M.Z.; Chen, C.L.; Kuo, C.Y.; Yang, C.Y.; Chiang-Ni, C.; Chen, Y.M.; Hsieh, C.M.; Wu, H.Y.; Kuo, M.L.; et al. PM(2.5) impairs macrophage functions to exacerbate pneumococcus-induced pulmonary pathogenesis. Part. Fibre Toxicol. 2020, 17, 37. [Google Scholar] [CrossRef]
- Doran, A.C.; Yurdagul, A., Jr.; Tabas, I. Efferocytosis in health and disease. Nat. Rev. Immunol. 2020, 20, 254–267. [Google Scholar] [CrossRef]
- Grabiec, A.M.; Denny, N.; Doherty, J.A.; Happonen, K.E.; Hankinson, J.; Connolly, E.; Fife, M.E.; Fujimori, T.; Fujino, N.; Goenka, A.; et al. Diminished airway macrophage expression of the Axl receptor tyrosine kinase is associated with defective efferocytosis in asthma. J. Allergy Clin. Immunol. 2017, 140, 1144–1146.e4. [Google Scholar] [CrossRef] [PubMed]
- de Souza Xavier Costa, N.; Ribeiro Júnior, G.; Dos Santos Alemany, A.A.; Belotti, L.; Schalch, A.S.; Cavalcante, M.F.; Ribeiro, S.; Veras, M.M.; Kallás, E.G.; Saldiva, P.H.N.; et al. Air pollution impairs recovery and tissue remodeling in a murine model of acute lung injury. Sci. Rep. 2020, 10, 15314. [Google Scholar] [CrossRef] [PubMed]
- Hodge, M.X.; Reece, S.W.; Madenspacher, J.H.; Gowdy, K.M. In Vivo Assessment of Alveolar Macrophage Efferocytosis Following Ozone Exposure. J. Vis. Exp. 2019, 152, e60109. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Stern, S.T.; Adiseshaiah, P.P.; Crist, R.M. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part. Fibre Toxicol. 2012, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.C.; Li, Z.; Harder, S.D.; Soukup, J.M. Apoptotic and inflammatory effects induced by different particles in human alveolar macrophages. Inhal. Toxicol. 2004, 16, 863–878. [Google Scholar] [CrossRef]
- Obot, C.J.; Morandi, M.T.; Beebe, T.P.; Hamilton, R.F.; Holian, A. Surface components of airborne particulate matter induce macrophage apoptosis through scavenger receptors. Toxicol. Appl. Pharmacol. 2002, 184, 98–106. [Google Scholar] [CrossRef]
- Xiong, Q.; Ru, Q.; Chen, L.; Yue, K.; Tian, X.; Ma, B.; Liu, L.; Wu, R.; Xu, C.; Pi, M.; et al. Combined effects of fine particulate matter and lipopolysaccharide on apoptotic responses in NR8383 macrophages. J. Toxicol. Environ. Health A 2015, 78, 443–452. [Google Scholar] [CrossRef]
- Muralidharan, S.; Mandrekar, P. Cellular stress response and innate immune signaling: Integrating pathways in host defense and inflammation. J. Leukoc. Biol. 2013, 94, 1167–1184. [Google Scholar] [CrossRef]
- Glencross, D.A.; Ho, T.R.; Camiña, N.; Hawrylowicz, C.M.; Pfeffer, P.E. Air pollution and its effects on the immune system. Free Radic. Biol. Med. 2020, 151, 56–68. [Google Scholar] [CrossRef]
- Guttenberg, M.A.; Vose, A.T.; Tighe, R.M. Role of Innate Immune System in Environmental Lung Diseases. Curr. Allergy Asthma Rep. 2021, 21, 34. [Google Scholar] [CrossRef] [PubMed]
- Oakes, J.L.; O’Connor, B.P.; Warg, L.A.; Burton, R.; Hock, A.; Loader, J.; Laflamme, D.; Jing, J.; Hui, L.; Schwartz, D.A.; et al. Ozone enhances pulmonary innate immune response to a Toll-like receptor-2 agonist. Am. J. Respir. Cell Mol. Biol. 2013, 48, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Potts, E.N.; Piantadosi, C.A.; Foster, W.M.; Hollingsworth, J.W. Hyaluronan fragments contribute to the ozone-primed immune response to lipopolysaccharide. J. Immunol. 2010, 185, 6891–6898. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, J.W.; Maruoka, S.; Li, Z.; Potts, E.N.; Brass, D.M.; Garantziotis, S.; Fong, A.; Foster, W.M.; Schwartz, D.A. Ambient ozone primes pulmonary innate immunity in mice. J. Immunol. 2007, 179, 4367–4375. [Google Scholar] [CrossRef]
- He, M.; Ichinose, T.; Ren, Y.; Song, Y.; Yoshida, Y.; Arashidani, K.; Yoshida, S.; Nishikawa, M.; Takano, H.; Sun, G. PM2.5-rich dust collected from the air in Fukuoka, Kyushu, Japan, can exacerbate murine lung eosinophilia. Inhal. Toxicol. 2015, 27, 287–299. [Google Scholar] [CrossRef]
- Bernstein, J.A.; Alexis, N.; Barnes, C.; Bernstein, I.L.; Bernstein, J.A.; Nel, A.; Peden, D.; Diaz-Sanchez, D.; Tarlo, S.M.; Williams, P.B. Health effects of air pollution. J. Allergy Clin. Immunol. 2004, 114, 1116–1123. [Google Scholar] [CrossRef]
- Frush, B.W.; Li, Z.; Stiles, J.V.; Cotter, S.F.; Shofer, S.L.; Foster, W.M.; Hollingsworth, J.W.; Tighe, R.M. Ozone primes alveolar macrophage-derived innate immunity in healthy human subjects. J. Allergy Clin. Immunol. 2016, 138, 1213–1215.e1. [Google Scholar] [CrossRef]
- Hussain, S.; Johnson, C.G.; Sciurba, J.; Meng, X.; Stober, V.P.; Liu, C.; Cyphert-Daly, J.M.; Bulek, K.; Qian, W.; Solis, A.; et al. TLR5 participates in the TLR4 receptor complex and promotes MyD88-dependent signaling in environmental lung injury. eLife 2020, 9, e50458. [Google Scholar] [CrossRef]
- Ghio, A.J.; Carraway, M.S.; Madden, M.C. Composition of air pollution particles and oxidative stress in cells, tissues, and living systems. J. Toxicol. Environ. Health B Crit. Rev. 2012, 15, 1–21. [Google Scholar] [CrossRef]
- Scandalios, J.G. Oxidative stress: Molecular perception and transduction of signals triggering antioxidant gene defenses. Braz. J. Med. Biol. Res. 2005, 38, 995–1014. [Google Scholar] [CrossRef]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed]
- Hirota, J.A.; Hirota, S.A.; Warner, S.M.; Stefanowicz, D.; Shaheen, F.; Beck, P.L.; Macdonald, J.A.; Hackett, T.L.; Sin, D.D.; Van Eeden, S.; et al. The airway epithelium nucleotide-binding domain and leucine-rich repeat protein 3 inflammasome is activated by urban particulate matter. J. Allergy Clin. Immunol. 2012, 129, 1116–1125.e6. [Google Scholar] [CrossRef]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef] [PubMed]
- den Hartigh, L.J.; Lamé, M.W.; Ham, W.; Kleeman, M.J.; Tablin, F.; Wilson, D.W. Endotoxin and polycyclic aromatic hydrocarbons in ambient fine particulate matter from Fresno, California initiate human monocyte inflammatory responses mediated by reactive oxygen species. Toxicol. In Vitro 2010, 24, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Harb, H.; Saffari, A.; Sioutas, C.; Chatila, T.A. A Jagged 1-Notch 4 molecular switch mediates airway inflammation induced by ultrafine particles. J. Allergy Clin. Immunol. 2018, 142, 1243–1256.e17. [Google Scholar] [CrossRef] [PubMed]
- Diesel exhaust particles induce the over expression of tumor necrosis factor-alpha (TNF-alpha) gene in alveolar macrophages and failed to induce apoptosis through activation of nuclear factor-kappaB (NF-kappaB). Int. J. Environ. Res. Public Health 2005, 2, 107–113. [CrossRef]
- Mundandhara, S.D.; Becker, S.; Madden, M.C. Effects of diesel exhaust particles on human alveolar macrophage ability to secrete inflammatory mediators in response to lipopolysaccharide. Toxicol. In Vitro 2006, 20, 614–624. [Google Scholar] [CrossRef]
- van Eeden, S.F.; Hogg, J.C. Systemic inflammatory response induced by particulate matter air pollution: The importance of bone-marrow stimulation. J. Toxicol. Environ. Health A 2002, 65, 1597–1613. [Google Scholar] [CrossRef]
- Fujii, T.; Hayashi, S.; Hogg, J.C.; Mukae, H.; Suwa, T.; Goto, Y.; Vincent, R.; van Eeden, S.F. Interaction of alveolar macrophages and airway epithelial cells following exposure to particulate matter produces mediators that stimulate the bone marrow. Am. J. Respir. Cell Mol. Biol. 2002, 27, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Ishii, H.; Hayashi, S.; Hogg, J.C.; Fujii, T.; Goto, Y.; Sakamoto, N.; Mukae, H.; Vincent, R.; van Eeden, S.F. Alveolar macrophage-epithelial cell interaction following exposure to atmospheric particles induces the release of mediators involved in monocyte mobilization and recruitment. Respir. Res. 2005, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Hiraiwa, K.; van Eeden, S.F. Contribution of lung macrophages to the inflammatory responses induced by exposure to air pollutants. Mediat. Inflamm. 2013, 2013, 619523. [Google Scholar] [CrossRef] [PubMed]
- Wooding, D.J.; Ryu, M.H.; Hüls, A.; Lee, A.D.; Lin, D.T.S.; Rider, C.F.; Yuen, A.C.Y.; Carlsten, C. Particle Depletion Does Not Remediate Acute Effects of Traffic-related Air Pollution and Allergen. A Randomized, Double-Blind Crossover Study. Am. J. Respir. Crit. Care Med. 2019, 200, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Bollati, V.; Marinelli, B.; Apostoli, P.; Bonzini, M.; Nordio, F.; Hoxha, M.; Pegoraro, V.; Motta, V.; Tarantini, L.; Cantone, L.; et al. Exposure to metal-rich particulate matter modifies the expression of candidate microRNAs in peripheral blood leukocytes. Environ. Health Perspect. 2010, 118, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wu, Y.; Ge, X.; Nie, D.; Wang, M.; Zhou, H.; Chen, M. In vitro toxicity evaluation of heavy metals in urban air particulate matter on human lung epithelial cells. Sci. Total Environ. 2019, 678, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, S.; Sun, C.; Qi, M.; Yu, X.; Zhao, W.; Li, X. Pollution Level and Health Risk Assessment of PM2.5-Bound Metals in Baoding City Before and After the Heating Period. Int. J. Environ. Res. Public Health 2018, 15, 2286. [Google Scholar] [CrossRef] [PubMed]
- Health impact Assessment: Main Concepts and Suggested Approach. The Gothenburg Consensus Paper; WHO Regional Office for Europe: Copenhagen, Denmark, 2000. [Google Scholar]
- Metryka, E.; Kupnicka, P.; Kapczuk, P.; Siminska, D.; Tarnowski, M.; Goschorska, M.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Lead (Pb) as a Factor Initiating and Potentiating Inflammation in Human THP-1 Macrophages. Int. J. Mol. Sci. 2020, 21, 2254. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Robles, C.D.; Cortes-Montoya, V.; Reyes-Aguilar, S.S.; Albores, A.; Calderon-Aranda, E.S. Low concentrations of lead disturb phenotypical markers of the inflammatory and the anti-inflammatory profile of bone marrow-derived macrophages from BALB/c mice. Toxicology 2021, 460, 152885. [Google Scholar] [CrossRef]
- Shimizu, K.; Horie, M.; Tabei, Y.; Kashiwada, S. Proinflammatory response caused by lead nanoparticles triggered by engulfed nanoparticles. Environ. Toxicol. 2021, 36, 2040–2050. [Google Scholar] [CrossRef]
- Kasten-Jolly, J.; Lawrence, D.A. Lead modulation of macrophages causes multiorgan detrimental health effects. J. Biochem. Mol. Toxicol. 2014, 28, 355–372. [Google Scholar] [CrossRef]
- Chong, I.W.; Lin, S.R.; Hwang, J.J.; Huang, M.S.; Wang, T.H.; Tsai, M.S.; Hou, J.J.; Paulauskis, J.D. Expression and regulation of macrophage inflammatory protein-2 gene by vanadium in mouse macrophages. Inflammation 2000, 24, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.D.; Parsons, E.; Schlesinger, R.B.; Zelikoff, J.T. Immunotoxicity of in vitro vanadium exposures: Effects on interleukin-1, tumor necrosis factor-alpha, and prostaglandin E2 production by WEHI-3 macrophages. Int. J. Immunopharmacol. 1993, 15, 437–446. [Google Scholar] [CrossRef]
- Cohen, M.D.; Becker, S.; Devlin, R.; Schlesinger, R.B.; Zelikoff, J.T. Effects of vanadium upon polyl:C-induced responses in rat lung and alveolar macrophages. J. Toxicol. Environ. Health 1997, 51, 591–608. [Google Scholar] [CrossRef] [PubMed]
- Lemarie, A.; Morzadec, C.; Bourdonnay, E.; Fardel, O.; Vernhet, L. Human macrophages constitute targets for immunotoxic inorganic arsenic. J. Immunol. 2006, 177, 3019–3027. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Xu, W.; Chen, J.; Li, H.; Dai, L.; Frank, J.A.; Peng, S.; Wang, S.; Chen, G. M2 polarization of macrophages facilitates arsenic-induced cell transformation of lung epithelial cells. Oncotarget 2017, 8, 21398–21409. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Ohta, T.; Tomita, N.; Kojima, C.; Hariya, Y.; Mizukami, A.; Fujiwara, K. Evaluation of immunotoxic and immunodisruptive effects of inorganic arsenite on human monocytes/macrophages. Int. Immunopharmacol. 2006, 6, 304–315. [Google Scholar] [CrossRef]
- Bourdonnay, E.; Morzadec, C.; Sparfel, L.; Galibert, M.D.; Jouneau, S.; Martin-Chouly, C.; Fardel, O.; Vernhet, L. Global effects of inorganic arsenic on gene expression profile in human macrophages. Mol. Immunol. 2009, 46, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Bourdonnay, E.; Morzadec, C.; Fardel, O.; Vernhet, L. Redox-sensitive regulation of gene expression in human primary macrophages exposed to inorganic arsenic. J. Cell. Biochem. 2009, 107, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Mokgobu, M.I.; Cholo, M.C.; Anderson, R.; Steel, H.C.; Motheo, M.P.; Hlatshwayo, T.N.; Tintinger, G.R.; Theron, A.J. Oxidative induction of pro-inflammatory cytokine formation by human monocyte-derived macrophages following exposure to manganese in vitro. J. Immunotoxicol. 2015, 12, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Roach, K.A.; Anderson, S.E.; Stefaniak, A.B.; Shane, H.L.; Kodali, V.; Kashon, M.; Roberts, J.R. Surface area- and mass-based comparison of fine and ultrafine nickel oxide lung toxicity and augmentation of allergic response in an ovalbumin asthma model. Inhal. Toxicol. 2019, 31, 299–324. [Google Scholar] [CrossRef]
- Glista-Baker, E.E.; Taylor, A.J.; Sayers, B.C.; Thompson, E.A.; Bonner, J.C. Nickel nanoparticles cause exaggerated lung and airway remodeling in mice lacking the T-box transcription factor, TBX21 (T-bet). Part. Fibre Toxicol. 2014, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Lag, M.; Rodionov, D.; Ovrevik, J.; Bakke, O.; Schwarze, P.E.; Refsnes, M. Cadmium-induced inflammatory responses in cells relevant for lung toxicity: Expression and release of cytokines in fibroblasts, epithelial cells and macrophages. Toxicol. Lett. 2010, 193, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.N.; Rahman, M.A.; Bao, S.; Liu, M.; Wheeler, S.E.; Knoell, D.L. Cadmium attenuates the macrophage response to LPS through inhibition of the NF-kappaB pathway. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L754–L765. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.K.; Kannan, K. Chromium chloride inhibits oxidative stress and TNF-alpha secretion caused by exposure to high glucose in cultured U937 monocytes. Biochem. Biophys. Res. Commun. 2001, 289, 687–691. [Google Scholar] [CrossRef] [PubMed]
- Boscolo, P.; Di Gioacchino, M.; Sabbioni, E.; Di Giacomo, F.; Reale, M.; Volpe, A.R.; Di Sciascio, M.B.; Conti, P.; Giuliano, G. Lymphocyte subpopulations, cytokines and trace elements in asymptomatic atopic women exposed to an urban environment. Life Sci. 2000, 67, 1119–1126. [Google Scholar] [CrossRef]
- Lead Air Pollution. Available online: https://www.epa.gov/lead-air-pollution (accessed on 8 July 2022).
- Rojas-Lemus, M.; Lopez-Valdez, N.; Bizarro-Nevares, P.; Gonzalez-Villalva, A.; Ustarroz-Cano, M.; Zepeda-Rodriguez, A.; Pasos-Najera, F.; Garcia-Pelaez, I.; Rivera-Fernandez, N.; Fortoul, T.I. Toxic Effects of Inhaled Vanadium Attached to Particulate Matter: A Literature Review. Int. J. Environ. Res. Public Health 2021, 18, 8457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gao, Y.; Wu, S.; Zhang, S.; Smith, K.R.; Yao, X.; Gao, H. Global impact of atmospheric arsenic on health risk: 2005 to 2015. Proc. Natl. Acad. Sci. USA 2020, 117, 13975–13982. [Google Scholar] [CrossRef] [PubMed]
- Bishayi, B.; Sengupta, M. Intracellular survival of Staphylococcus aureus due to alteration of cellular activity in arsenic and lead intoxicated mature Swiss albino mice. Toxicology 2003, 184, 31–39. [Google Scholar] [CrossRef]
- Bowler, R.M.; Beseler, C.L.; Gocheva, V.V.; Colledge, M.; Kornblith, E.S.; Julian, J.R.; Kim, Y.; Bollweg, G.; Lobdell, D.T. Environmental exposure to manganese in air: Associations with tremor and motor function. Sci. Total Environ. 2016, 541, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Genchi, G.; Carocci, A.; Lauria, G.; Sinicropi, M.S.; Catalano, A. Nickel: Human Health and Environmental Toxicology. Int. J. Environ. Res. Public Health 2020, 17, 679. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Plant, J.A.; Voulvoulis, N.; Oates, C.J.; Ihlenfeld, C. Cadmium levels in Europe: Implications for human health. Environ. Geochem. Health 2010, 32, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Lawrence, D.A. Metal-induced modulation of nitric oxide production in vitro by murine macrophages: Lead, nickel, and cobalt utilize different mechanisms. Toxicol. Appl. Pharmacol. 1996, 141, 540–547. [Google Scholar] [CrossRef] [PubMed]
- De Guise, S.; Bernier, J.; Lapierre, P.; Dufresne, M.M.; Dubreuil, P.; Fournier, M. Immune function of bovine leukocytes after in vitro exposure to selected heavy metals. Am. J. Vet. Res. 2000, 61, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, A.; Shelburne, C.E.; Gibbons, D.F. Cellular proliferation and cytokine responses of murine macrophage cell line J774A.1 to polymethylmethacrylate and cobalt-chrome alloy particles. J. Biomed. Mater. Res. 1998, 42, 655–663. [Google Scholar] [CrossRef]
- Berman, R.; Kopf, K.W.; Min, E.; Huang, J.; Downey, G.P.; Alam, R.; Chu, H.W.; Day, B.J. IL-33/ST2 signaling modulates Afghanistan particulate matter induced airway hyperresponsiveness in mice. Toxicol. Appl. Pharmacol. 2020, 404, 115186. [Google Scholar] [CrossRef]


{kind=link}
| Pollutants | M1 Phenotype | M2 Phenotype |
|---|---|---|
| ultrafine particles (UFP) | Leukotriene B4 ↑ (A) [37] | Prostaglandin E2 (PGE-2) ↑ (A) [37] |
| 8-isoprostane ↑ (A) [37] | 15(S)-HETE ↑ (A) [37] | |
| Lipoxin A4 ↑ (A) [37] | ||
| PM 2.5 | IL-1β ↑ (H) [38], ↑ (A) [39,40] | IL-10 ↑ (A) [40,41], ↓ (A) [42] |
| IL-6 ↑ (H) [38,43], ↑ (A) [40,44] | Prostaglandin D2 ↑ (A) [39] | |
| IL-8 ↑ (H) [38,43] | PGE-2 ↑ (C) [45] | |
| IL-12 ↑ (A) [39,40] | Arginase ↓ (C) [42] | |
| TNF-α↑ (H) [38,43], ↑ (A) [41,42,44,46] | ||
| IFN-γ ↑ (A) [47], ↓ (A) [41] | ||
| CCL-2 ↑ (A) [39,44] | ||
| CCL-3 ↑ (A) [40] | ||
| iNOS ↑ (A) [40,42] | ||
| PM 10 | IL-1β ↑ (H) [48], ↑ (A) [40,49] | IL-10 ↑ (H) [50], ↑ (A) [40] |
| IL-6 ↑ (H) [51], ↑ (A) [52] | PGE-2 ↑ (A) [52] | |
| IL-8 ↑ (H) [50,51] | ||
| IL-12 ↑ (A) [40,52] | ||
| TNF-α ↑ (H) [50], ↑ (A) [40,46,52] | ||
| CCL-2 ↑ (A) [40] | ||
| CCL-3 ↑ (A) [40] |
| Pollutants | M1 Phenotype | M2 Phenotype |
|---|---|---|
| Lead (Pb) | IL-1β ↑ (C) [98] | CD206 ↑ (A) [99] |
| IL-6 ↑ (C) [98] | TGF-β1 ↑ (A) [99] | |
| MHC class II ↑ (A) [99] | ||
| IL-8 ↑ (C) [100] | ||
| IFN-γ ↓ (C,A) [101] | ||
| IL-12p40 ↓ (A) [101] | ||
| Vanadium (V) | CXCL-2 ↑ (C) [102] | |
| TNF-α ↓ (C) [103] | ||
| IL-6 ↑ (A) [104] | ||
| IFN-γ ↑ (A) [104] | ||
| Arsenic (As) | TNF-α ↑ (C) [105] | CD206 ↑ (C) [106] |
| IL-1α ↑ (C) [107] | CD163 ↑ (C) [106] | |
| IL-8 ↑ (C) [105] | IL-10 ↑ (C) [106] | |
| IL-12 ↑ (C) [107] | CCL-18 ↑ (C) [106] | |
| CXCL2 ↑ (C) [108,109] | TGF-β1 ↑ (C) [106] | |
| CD86 ↓ (C) [107] | CCL-22 ↑ (C) [108,109] | |
| Manganese (Mn) | IL-1β ↑ (C) [110] | |
| IL-6 ↑ (C) [110] | ||
| IL-8 ↑ (C) [110] | ||
| TNF-α ↑ (C) [110] | ||
| IFN-γ ↑ (C) [110] | ||
| Nickel (Ni) | IL-2 ↑ (A) [111] | |
| IL-6 ↑ (A) [111] | ||
| IL-12 ↑ (A) [111] | ||
| IFN-γ ↑ (A) [111] | ||
| TNF-α ↑ (A) [111] | ||
| CCL-2 ↑ (A) [112] | ||
| Cadmium (Cd) | IL-1β ↑ (C) [113] | |
| TNF-α ↑ (C) [113], ↓ (C,A) [114] | ||
| CXCL2 ↑ (C) [113], ↓ (A) [114] | ||
| IL-6 ↓ (C,A) [114] | ||
| IL-8 ↓ (C) [114] | ||
| CXCL-9 ↓ (C) [114] | ||
| CXCL-10 ↓ (C) [114] | ||
| CXCL-11 ↓ (C) [114] | ||
| CCL-5 ↓ (C) [114] | ||
| Chromium (Cr)(VI) | TNF-α ↓ (C) [115] | |
| IFN-γ ↑ (H) [116] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.-H.; Tsai, M.-L.; Chiou, H.-Y.; Lin, Y.-C.; Liao, W.-T.; Hung, C.-H. Role of Macrophages in Air Pollution Exposure Related Asthma. Int. J. Mol. Sci. 2022, 23, 12337. https://doi.org/10.3390/ijms232012337
Li C-H, Tsai M-L, Chiou H-Y, Lin Y-C, Liao W-T, Hung C-H. Role of Macrophages in Air Pollution Exposure Related Asthma. International Journal of Molecular Sciences. 2022; 23(20):12337. https://doi.org/10.3390/ijms232012337
Chicago/Turabian StyleLi, Chung-Hsiang, Mei-Lan Tsai, Hsin-Ying (Clair) Chiou, Yi-Ching Lin, Wei-Ting Liao, and Chih-Hsing Hung. 2022. "Role of Macrophages in Air Pollution Exposure Related Asthma" International Journal of Molecular Sciences 23, no. 20: 12337. https://doi.org/10.3390/ijms232012337
APA StyleLi, C.-H., Tsai, M.-L., Chiou, H.-Y., Lin, Y.-C., Liao, W.-T., & Hung, C.-H. (2022). Role of Macrophages in Air Pollution Exposure Related Asthma. International Journal of Molecular Sciences, 23(20), 12337. https://doi.org/10.3390/ijms232012337